Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(16):2916-2929. doi:10.7150/jca.24415 This issue Cite
Research Paper
Stromal mesenchymal stem cells facilitate pancreatic cancer progression by regulating specific secretory molecules through mutual cellular interaction
Ken Saito1, Masakiyo Sakaguchi2, Satoshi Maruyama3, Hidekazu Iioka1, Endy Widya Putranto5, I Wayan Sumardika2,6, Nahoko Tomonobu2, Takashi Kawasaki4, Keiichi Homma4, Eisaku Kondo1 ![]()
1. Division of Molecular and Cellular Pathology, Niigata University Graduate School of Medical and Dental Sciences, 1-757 Asahimachi-dori, Chuo-ku, Niigata 951-8510, Japan
2. Department of Cell Biology, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, 2-5-1 Shikata-cho, Kita-ku, Okayama 700-8558, Japan
3. Oral Pathology Section, Department of Surgical Pathology, Niigata University Hospital, 2-5274 Gakkoucho-dori, Chuo-ku, Niigata 951-8514, Japan
4. Department of Pathology, Niigata Cancer Center Hospital, 2-15-3 Kawagishi-cho, Chuo-ku, Niigata 951-8133, Japan
5. Faculty of Medicine, Universitas Gadjah Mada, Yogyakarta 55281
6. Faculty of Medicine, Udayana University, Denpasar 80232, Bali, Indonesia
Received 2017-12-17; Accepted 2018-7-3; Published 2018-7-30
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is currently one of the most intractable malignancies with a typical scirrhous pattern in histology. Due to its abundant tumor stroma and scant vascularization, chemotherapeutic agents are considered inefficiently permeable to cancer nests, making it highly difficult to cure the patients with PDAC. However, PDAC is also considered to owe its intractability to other critical factors such as cellular interaction between tumor cells and tumor microenvironment as well as architectural barriers, which increases in therapeutic resistance. Here, we report a specific cellular interaction between PDAC cells and mesenchymal stem cells (MSCs) intermingled in PDAC stroma, which facilitates cancer invasion. Secretory phenotype profiling revealed that production of Amphiregulin (AREG) and MMP-3 were specifically upregulated under the coexistence of BxPC3 cells with human MSCs (approximately four to ten folds in AREG, and twenty to sixty-folds in MMP-3 compared to that of BxPC3 cells alone), whereas MMP-9 expression was decreased (less than one-tenth comparing with that of BxPC3 cells alone). Blockage of AREG production by its specific siRNA removed MSC-mediated driving force of BxPC3 invasiveness. Immunohistochemical analysis of tissue samples obtained both from PDAC patients and PDAC imitating mouse xenografted models revealed that significant coexpression of AREG and its receptor EGFR were detected on the cancer cells at invasive front. These results strongly suggested that cellular interaction between cancer cells and MSCs in the PDAC stroma might be critical to cancer progression, especially in the process of local invasion and the early stage development of metastasis.
Keywords: MSC, PDAC, microenvironment, stroma, AREG, MMP
Introduction
Pancreatic ductal adenocarcinoma (PDAC) occupies 90% of all pancreatic cancers, displaying features of local invasion and metastasis to distant organs even at early stages. Owing to its aggressiveness, the overall survival rate is growing extremely low [1-3]. In addition, dense stroma was histologically one of the most prominent characteristics of PDAC, acting as barriers and protecting the cancer cells during chemotherapy. These all contribute to the difficulties in curing PDAC patients. Hence, the development of an advanced and effective approach to treat PDAC is a high priority. In establishing research strategies, a study of not only PDAC cell itself but also of the surrounding stromal cell as a tumor microenvironment was essential at a molecular level to obtain a clue for the human body therapeutics.
The tumor stroma is composed of multiple cells; including cancer-associated fibroblasts (CAFs), inflammatory immune cells and vascular cells. Among these, CAFs are especially notable, as they provide cancer cells with extracelluler matrixes and soluble proteins such as cytokines and growth factors. Previous basic and clinical studies in PDAC have suggested that CAFs promote pancreatic cancer growth and metastasis [4-8]. Taking into consideration of CAF's unique role in PDAC, we should not rule out the presence of CAFs' origin, mesenchymal stem cells (MSCs) because recently some progress has been made about biological role of MSC in cancers of several origins. Previous studies reported that bone marrow-derived MSCs were preferentially accumulated to colon cancers [9], breast cancers [10], and pancreatic cancers in vivo [11] to promote tumor growth, metastasis, angiogenesis, and so on. Moreover, Kabashima-Niibe et al. reported that MSCs play a pivotal role in the induction of epithelial-mesenchymal transition (EMT) of PDACs [12]. However, MSCs role in malignant conversion may possibly not be limited to EMT transition alone. The presence of other diverse mechanisms that coordinately influence and function with EMT in PDACs is needed for the level of aggressiveness it exhibits at such an early stage.
In the present study, we therefore pursue other important roles of MSCs on PDAC progression. Our studies revealed that MSCs are present at a small population in the tumor stroma of PDACs. Co-culture of pancreatic cancer BxPC3 cells and MSCs uncovered a dramatic change of secretory phenotype compared to those from each single culture. Among the altered candidates, we found that cancer-induced Amphiregurin (AREG), which is significantly enhanced through MSCs interaction, plays a critical role in cancer invasion. In mouse xenograft models transplanted with both BxPC3 and MSCs and in clinical PDAC tissue sections, we further found a strong co-localization of the soluble ligand AREG and its receptor EGFR in the invasive front of PDACs where MSCs are present. These results provide us with a novel role of MSCs in PDACs; which enhances induction of AREG soluble factor in pancreatic cancer cells, promoting PDAC local invasion potential through an autocrine mechanism. Thus, AREG targeting might prove critical in the prevention of earlier metastasis of the stroma enriched PDACs.
Materials and Methods
Cells and antibodies. BxPC3 cells (ATCC, Manassas, VA) and their GFP expressing subline in a stable manner, named BxPC3-GFP cells, which we established by using GFP expression vector (pEF/myc/cyto/GFP; Thermo Fisher Scientific, Waltham, MA) were cultivated in RPMI medium (Wako Laboratory Chemicals, Japan) supplemented with 10% FBS. The different batches of human bone marrow-derived MSCs (hMSC; KURABO, Osaka, Japan, and UE6E7T-12; JCRB Cell Bank, Osaka, Japan) were grown in mesenchymal stem cell basal medium (MSCBM; Lonza, Walkersville, MD) containing 10% FBS, FGF-B (5 ng/ml), EGF (10 ng/ml), hydrocortisone (1 μg/ml), and heparin (10 μg/ml), and were subjected to the analysis. Normal human dermal fibroblasts (NHDFs; NHDF-neo; Lonza, Walkersville, MD, or NHDF from adult donor (PromoCell GmbH, Heidelberg, Germany) were grown in RPMI medium (Wako Laboratory Chemicals, Japan) supplemented with 10% FBS. hMSCs from KURABO and NHDFs from Lonza at three to six passages were mainly used in this study. UE6E7T-12 and NHDF from PromoCell which showed similar origins were only used for confirmation of immunophenotype as a human bone marrow-derived MSC or a human dermal fibroblast, respectively (data was seen as a supplementary information). Human PDAC lines, AsPC1, MiaPaCa-2, and Panc-1, all derived from the patients with pancreatic invasive ductal adenocarcinoma were obtained from ATCC (Manassas, VA) and maintained with RPMI medium (Wako Laboratory Chemicals, Japan) supplemented with 10% FBS for the analysis.
Antibodies used in the present study were as follows: mouse anti-human CD105 antibody (Sigma-Aldrich, St. Louis, MO), rabbit anti-human CD73 antibody (NT5E/CD73 (D7F9A); Cell Signaling Technology Japan; cross-reactive to mouse, rat), rabbit anti-human CD29 antibody (Cell Signaling Technology Japan), mouse anti-human αSMA antibody (Dako, Agilent Technologies, Japan), mouse anti-human CD34 antibody (Dako, Agilent Technologies, Japan), mouse anti-human CD45, LCA antibody (Dako, Agilent Technologies, Japan), rabbit anti-human MMP-3 antibody (Sigma-Aldrich, St. Louis, MO), rabbit anti-human MMP-9 antibody (Cell Signaling Technology Japan), mouse anti-human AREG antibody (R&D Systems, Minneapolis, MN), mouse anti-human EGFR antibody (NICHIREI BIOSCIENCES INC, Japan), sheep anti-human Fibroblast activation protein alpha (FAPa) antibody (R&D systems, Minneapolis, MN).
Flow Cytometric Analysis.The trypsinized floating MSCs were collected, washed with PBS containing 1% BSA, and fixed with 4% paraformaldehyde for 10 min at RT. The fixed cells were then incubated with primary antibodies (anti-human CD34 mouse mAb, anti-human CD45 mouse mAb, anti-human CD73 rabbit mAb and anti-human CD105 rabbit mAb, respectively) for 60 min at RT and subsequent secondary antibody (either Alexa fluor 488 goat anti-mouse or anti-rabbit IgG antibody) for 30 min at RT. After washing, the stained cells were analyzed by using the FACS Aria flow cytometer (BD Bioscience). Each cell was determined by FSC-A and SSC-A. The gated populations with positive signals were identified and calculated for their positive-cell populations in comparison to the data of reference cells stained with the IgG isotype controls (mouse IgG for mouse monoclonal antibody or rabbit IgG for rabbit monoclonal antibody, respectively). Mean fluorescence intensity (MFI) values were also calculated by FlowJo software (BD Biosciences Japan, Tokyo, Japan).
Invasion assay. Invasion assay was performed by Boyden chambers, using the transwell culture inserts sealed with Matrigel (Trevigen, Gaithersburg, MD). Cells were seeded in the upper chamber in serum free medium, and the lower chamber was filled with either serum free or serum-containing medium (0.5% and 10% FBS). After 12 hours, invading cells with GFP on the underside of inserts were imaged under a fluorescent microscope (BZ-9000, KEYENCE, Tokyo, Japan). All the invaded GFP-positive cells were measured by fluorescence-based scanning (Fluoroskan ASCENT FL, Thermo Fisher Scientific, Waltham, MA). Panc-1 cells were labeled with PKH26 (PKH26-GL; MERCK, Germany) and seeded to upper transwell chambers.
Protein array. The conditioned media were prepared from three different cultures, BxPC3 cells alone, hMSCs alone, BxPC3 cells + hMSCs (ratio 1:1), 120-hours after replacements with the serum-free RPMI media. The prepared media were then subjected to analysis using three kinds of protein profiling arrays (Human Chemokine Array, Human Soluble Receptor Array and Human Protease Array; R&D Systems, Minneapolis, MN). The detection procedures for these arrays were following the manufacture's instructions. After blocking the array membranes, they were treated with the prepared media (1.5 ml) for two hours at RT and a series of the provided reagents (biotin-labeled antibodies and HRP-labeled streptavidin) in each kit one after another. Lastly, the washed membranes were all subjected to chemiluminescence detection equipped in the kits.
RNA Interference. Validated siRNAs for human MMP-3 (ID No. L-005968-00-0010) and for human AREG (ID No. L-017435-00-0010) and control siRNA (ON-TARGETplus Non-targeting Control siRNA; ID No. D-001810-01-20) were purchased from Dharmacon, GE Healthcare (Japan). siRNAs were transfected using Lipofectamin RNAiMAX reagent (Thermo Fisher Scientific, Waltham, MA).
Quantitative real-time PCR. Total RNA was extracted from cultured cells with ISOGEN (Nippon Gene, Tokyo, Japan) according to the manufacturer's instructions. The cDNA was reverse-transcribed with SuperScriptTM II (Thermo Fisher Scientific, Waltham, MA). Real-time RT-PCR was performed on a rapid thermal cycler system (STEP OnePlus, ABI,Fostercity, CA) using a Power SYBR Green Master Mix (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. Forward and reverse primer pairs used (5' to 3') are listed in below. MMP-3:Fw ACCCTTTTGATGGACCTGGAA, Rv ATCATCTTGAGACAGGCGGAA, MMP-9: Fw CTTTTCTTCTTCTCTGGGCGC, Rv TTCTCTCGGTACTGGAAGACG, AREG: Fw TTATGCTGCTGGATTGGACCT, Rv ACTACCTGTTCAACTCTGACTGA, ADAMTS1: Fw TTTCTTGCCATCAAAGCTGCT, Rv GACTTAGAACATTCGCCCCAC.
Mouse PDAC model. Animal experiments were approved by the animal care committee of Niigata University and performed in accordance with the guidelines. Six-weeks old NOD/SCID (CLEA Japan Inc, Japan) were housed under pathogen-free conditions. The mixed cells of BxPC3 cells (1 x 105 cells) with hMSCs (1 x 105 cells) were subcutaneously injected into the mice. Six weeks after cell injections, mice were euthanized with CO2 inhalation and the developed tumor tissues were processed in histological evaluations. All animal experiment in this study was approved by the local ethics committee of Niigata University Graduate School of Medical and Dental Sciences (#27-112-3, 27-112-4).
Magnetic beads separation and characterization of cellular phenotyping of mouse BxPC3-xenografted abdominal tumor in vivo. Intraabdominal tumor tissue grown in the human BxPC3 cells (1x106)-xenografted mouse was dissociated into individual cell by gentleMACSOcto separator using Tumor Tissue Dissociation Kit (Miltenyi Biotec, Germany) thirty days after intraperitoneal injection. Cells were then separated into two groups of murine and human cells by autoMACS Pro Separator using mouse cell depletion kit (Miltenyi Biotec, Germany). Cells from Positive fraction (mouse cells) were stained both with APC-conjugated anti-human CD324 (E-Cadherin) and Alexa488-conjugated anti-mouse CD73 and Propidium Iodide (Miltenyi Biotec, Germany), and live cell fraction (PI-negative cells) was subjected to FACS analysis (MACSQuant Analyzer; Miltenyi Biotec, Germany).
Immunohistochemistry. Human PDAC tissues surgically obtained from the patients were fixed in 10% buffered formalin solution. Paraffin-embedded sections were deparaffinized and antigen retrieval was performed by autocleavage (Pascal DakoCytomation S2800) in sodium citrate buffer, furthermore, endogenous peroxide activity was quenched by incubation in 3% hydrogen peroxide (KANTO CHEMICAL Co., INC) for 10 min at room temperature. Slides were incubated using the antibodies of our interests as a primary antibody overnight at 4°C. After the slides were washed with 0.05% Tween-20 in PBS (-), incubated with either AEC (red color) or DAB (brown color) detection system (Envision+Kit / HRP Dako North America, Inc, Carpinteria, CA). The study using patient-derived tissues was approved both by the research ethics committees of Niigata University (#2015-2334) and Niigata Cancer Center Hospital (#2017-20). Informed consent was also obtained from each patient for the use of biological materials at Niigata Cancer Center Hospital.
For the immunofluorescence staining, we used either Alexa488 (green) or Alexa555 (red)-conjugated goat anti-mouse IgG antibody and goat anti-rabbit IgG antibody (Thermo Fisher Scientific, Waltham, MA) as secondary antibodies, respectively. Cell nuclei were counterstained with 4′, 6-diamidino-2-phenylindole (DAPI, blue).
Statistical analysis. Data are expressed as means ± SD. We employed simple pair-wise comparison with Student's t-test (two-tailed distribution with two-sample equal variance). p <0.05 was considered significant.
Results
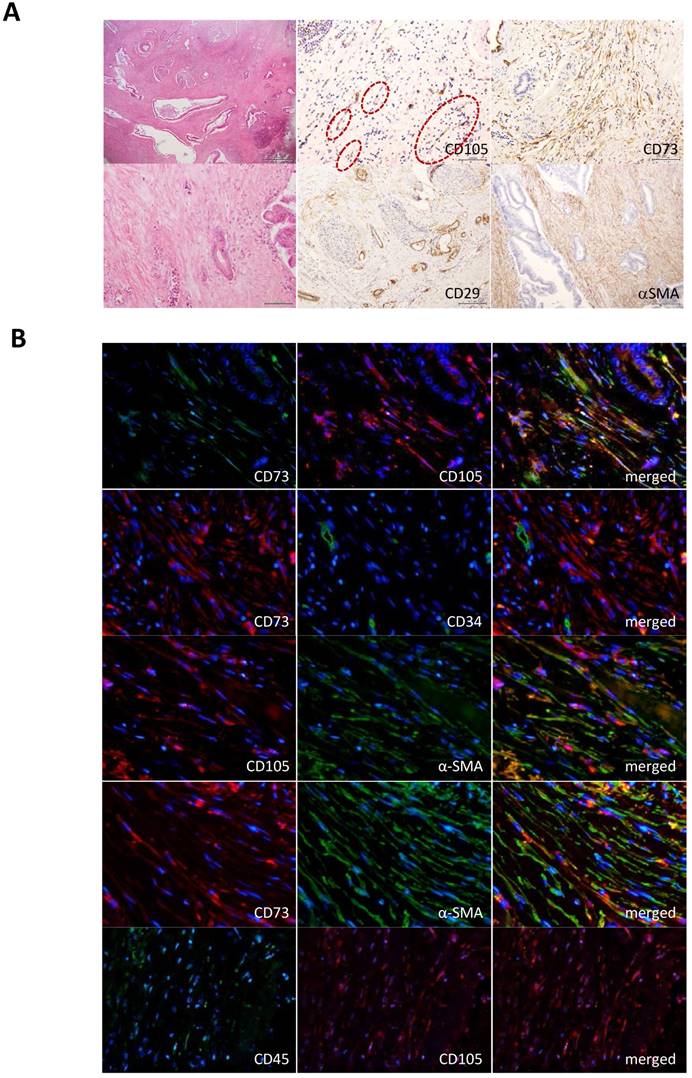
Detection of MSCs in tumor stroma of PDACs. To reveal the novel role of MSCs in progression of human pancreatic adenocarcinoma, we first attempted to examine the presence of bone marrow-derived MSCs in the stroma of PDACs. Immunohistochemistry using clinical specimens from PDAC patients showed that mesenchymal cells corresponding to the bone marrow-derived MSC markers (CD105+, CD73+, CD29+ αSMA+, CD34- and CD45-) sparsely appeared in parts of the PDAC stroma (Fig. 1A). To further define the presence of MSC, we performed double-immunofluorescence staining of the PDAC-derived tissue sections with different combination of the MSC markers and found that the procedure led consistent overlapping patterns in all cases of the MSC markers used (CD73 and CD105, CD105 and αSMA, CD73 and αSMA). CD73 and CD107 never co-localized with hematopoietic progenitor cell antigens, CD34 and CD45 (Fig. 1B). The staining profile provided us with firm belief of the presence of certain MSCs in the PDAC stroma.
Immunohistological detection of MSCs in PDAC stroma. A. Antibodies for representative MSC markers, CD105, CD73, CD29 and αSMA were used to detect MSCs in PDAC stroma prepared from clinical specimens. Magnifications of the stained images were all x10. Left panels show hematoxylin and eosin (HE) staining (top x4; bottom x20). Circle shows a presence of fibroblast-like cells in the CD105 positive cells. B. Double immunofluorescence staining was performed in PDAC tissue sections for the different combinations as indicated in the images. MSC markers: CD105, CD73 and αSMA. Hematopoietic stem cell markers: CD34 and CD45.
Characterization of MSCs. To find a unique interaction between MSC and PDAC cells, we employed in vitro coculture system of human bone marrow-derived MSC with human PDAC cells. First, to confirm whether the used human MSCs in culture display similar characteristics to that of the MSCs in PDAC stroma patients, cultured MSCs were stained with a series of representative MSC markers. Since the single cellular marker restricted to bone marrow-derived MSC have been not identified yet, phenotyping is usually evaluated by combined pattern of several antigens expression such as CD73+, CD105+, alpha-SMA+, CD34-, CD45-. As shown by FACS analysis in Figure 2A, MSCs in culture showed significant expression of CD73, CD105, while CD34 and CD45 were negative, which is consistent with immunophenotype of conventional human MSC and they exhibited similar patterns to that of the clinical specimens of PDAC. It was also confirmed by immunofluorescence staining (Supplementary Fig. 1A). Especially, increased frequency and intensity of CD73 antigen expression on MSCs discriminated between these cells and dermal fibroblasts in FACS analysis (Supplementary Fig. 1B and 1C). The similar result was obtained by using another batch of hMSC and NHDF, which suggested high and frequent expression of CD73 may be one of the unique characteristic of bone marrow-derived MSC (Supplementary Fig. 1D and 1E). Noticeably, coexistence of MSC with PDAC cells during coculture for 120 hours revealed augmented expression of Fibroblast activation protein alpha (FAPα) on stromal cells, which was accompanied by the increase in CD73. This implies that MSC immunophenotypically differentiated to cancer-associated fibroblast (CAF) in the presence of PDAC cells (Supplementary Fig.2A). When BxPC3-xenografted intraperitoneal tumor was subjected to FACS analysis after dissociation to single cell, CD73-positive murine mesenchymal cells (human E-Cadherin-negative fraction) were detected (supplementary Fig. 2B). It suggests MSCs mgiht be actively homing to PDAC tumor as their physiological property and eventually turn into CAFs in vivo, at least in part.
Immunophenotypic characterization of human MSC and MSC-mediated increase in invasiveness of pancreatic cancer cells. A. FACS analysis of human MSC using anti-human CD34, CD45, CD73, and CD105. Mouse IgG as classmatch control for mouse monoclonal CD34, CD45, rabbit IgG for rabbit monoclonal CD73 and CD105. Indirect immunofluorescence using Alexa488-conjugated secondary antibodies. B and C. BxPC3 invasiveness was assessed under the co-culture condition with hMSCs by employing Boyden chamber method. Filter membrane set in the top chamber was sealed with Matrigel to close the micro pore. The mixed cells (BxPC3-GFP cells and hMSCs, ratio 1:1) were placed in the top chamber filled with serum free medium and serum was added or not added to the bottom well to be final concentrations 0.5% and 10%. After incubation for 12 hours, passed BxPC3-GFP cells through the filter membrane were observed by the fluorescence microscope (A) and their fluorescence intensities were measured and quantified (B). Data from A are means ± SD, *P<0.05 and **P<0.01. C and D. The used cultured hMSCs were characterized in comparison to the cultured normal human dermal fibroblasts (NHDFs) by immnufluorescence staining with the representative MSC markers (CD105, CD73, CD29 and αSMA) that showed all highly positive in MSCs detected in clinical PDAC tissues. The staining results were shown in the images (C) and evaluated them by the table (D).
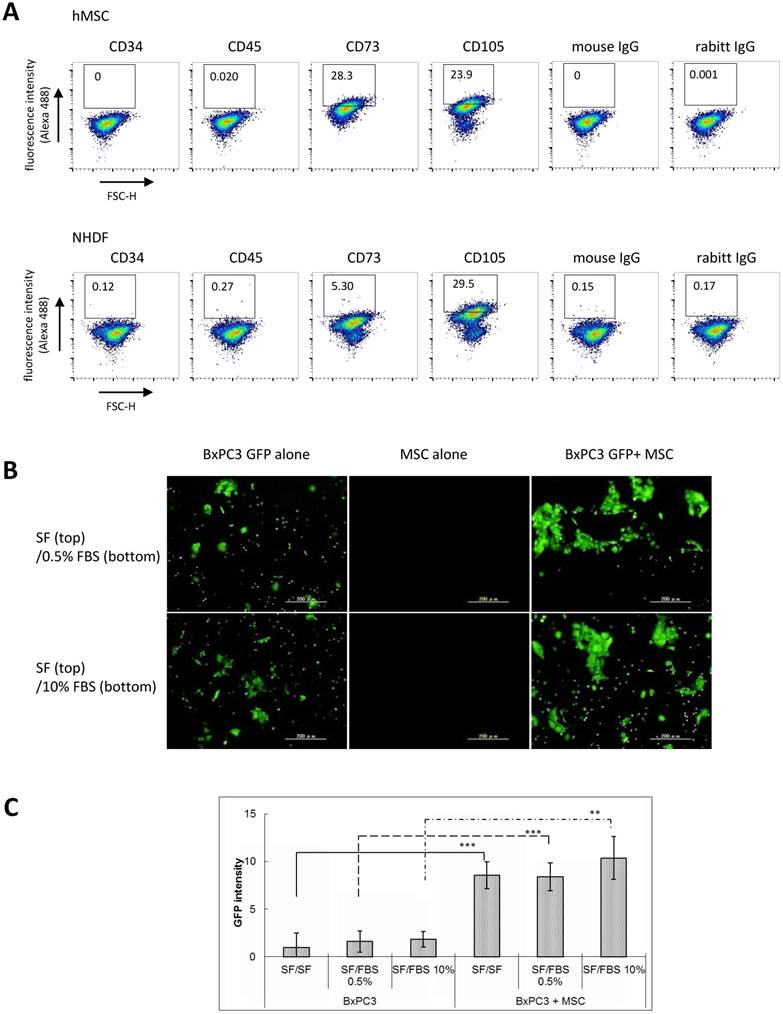
Significance of MSCs on cancer invasion. Based on the result of MSC characterization, we next examined the biological contribution of MSCs on PDAC invasiveness, namely, an invasion assay by employing the Boyden chamber method, in which the partitioned microporous membrane was sealed with matrigel and MSCs and GFP-expressed pancreatic BxPC3 cells (BxPC3-GFP) were set together in the same top compartment. After incubation, GFP fluorescence expression was observed on the lower-side of the membrane to visualize invasion of the mixed BxPC3-GFP cells but not that of MSCs. As a result, we found that MSCs have a unique ability to enhance invasiveness of BxPC3 cells irrespective to the serum contents in the bottom compartment (Fig. 2B and 2C). Similar phenomena were observed in other three PDAC line-MSC coculture system that presence of hMSC most significantly facilitated invasion of AsPC1, MiaPaCa-2 and Panc-1 cells, in comparison with NHDF or without any mesenchymal cells (Supplementary Fig. 3A and 3B). These results suggest that MSCs largely contribute to the PDAC local invasion in the physiological setting.
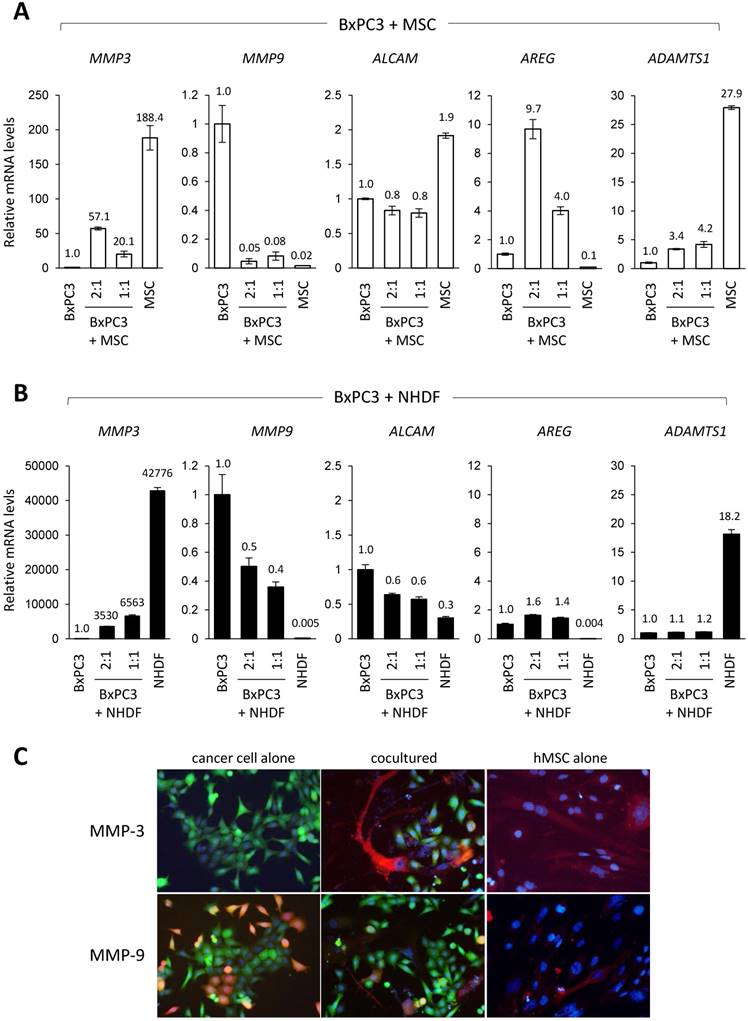
Study of a secretory phenotype in the stroma enriched PDAC microenvironment. To investigate further into the function of MSCs in cancer invasion at molecular levels, we turned our attention to the secretory proteins. We collected the conditioned media from either single cultures of BxPC3 cells and MSCs or mixed culture of BxPC3 cells with MSCs (Fig. 3A) and analyzed them for secretory soluble proteins by three kinds of detection arrays, cytokine-related molecules (Fig. 3B), soluble receptors and ligands (Fig. 3C) and soluble protease-related molecules (Fig. 3D). Through this approach, we found that the co-culture alters the secretory phenotype from each single culture. Many upregulated proteins appeared after co-cultivation, i.e., MCP-2 (CCL7), NAP-2 (CXCL7) and the soluble form of gp130 (CD130) (Fig. 3B); soluble form of ALCAM, AREG and soluble forms of integrin β1 and Transferrin R (Fig. 3C); soluble form of ADAMTS1, Cathepsin C and MMP-3 (Stromelysin-1) (Fig. 3D). Unexpectedly, MMP-9 (Gelatinase B) alone was downregulated by the co-culture (Fig. 3D).
Array-based profiling of secretory proteins in co-culture of pancreatic cancer cells with MSCs. A. Cell conditions at single- or mix-culture were observed by the inverted microscope. Left: BxPC3 cells alone, middle: BxPC3 cells and hMSCs, right: hMSCs alone. B, C and D. The conditioned media in each culture as indicated in A were prepared and subjected them to the protein arrays, cytokine -related molecules (A), soluble receptors and ligands (B) and soluble protease-related molecules (C), to look into secretory alterations of proteins. Upregulated and downregulated proteins were summarized in the tables displaying below each array data.

In our effort to determine which soluble proteins truly contribute to PDAC invasion among all detected candidates, we focused on AREG, ADAMTS1, MMP-3 and MMP-9 as they featured great potential in the invasion terms of PDAC, and therefore studied them for the gene expression. Through the quantitative PCR (Q-PCR) analysis, we found that basal expression of MMP-3 and ADAMTS1 was significantly high in both MSCs (Fig. 4A) and NHDFs (Fig. 4B), while MMP-9 was observed in BxPC3 cells at a noticeable level (Fig. 4A and 4B). After co-cultivation of MSCs cells and BxPC3 cells in different proportions (1:1 and 2:1), we surprisingly found that only AREG was markedly elevated by the co-existence of MSCs in a cell number dependent manner (Fig. 4A). This data was well inline with the protein array data (Fig. 3C). Interestingly, the increase was not observed in the case of NHDF co-cultivation (Fig. 4B). Through the Q-PCR analysis, cell proportion dependent upregulation of gene expression was also observed in case of MMP-3. This specifically was observed in MSCs (Fig. 4A) but not NHDFs (Fig. 4B), however the level was significantly lower than that of MSCs alone. In consistent to the array data, MMP-9 level in BxPC3 cells was largely downregulated by the co-cultivation with either MSCs (Fig. 4A) or NHDFs (Fig. 4B). ADAMTS1, which showed extremely high levels in both MSCs and NHDFs, exhibited lower levels in the co-culture system, even after increased proportion in both cell types, MSCs and NHDFs. Specific upregulation of AREG in PDAC line was also corroborated in Capan-2 cells only when they cocultuterd with MSC as well as in BxPC3 cells (Supplementary Fig. 4A and 4B).
Gene expression profiling of the selected secretory candidates in co-culture of pancreatic cancer cells with MSCs. A and B. Total RNAs prepared from indicated cell cultures in the panel A (BxPC3 cells alone, hMSCs alone, BxPC3 cells and hMSCs (ratio 1:1 and 1:2)) and the panel B (BxPC3 cells alone, NHDFs alone, BxPC3 cells and NHDFs (ratio 1:1 and 1:2)) were analyzed for expression of the selected genes of our interest by quantitative real-time PCR. GAPDH was used as control for the analysis. Relative expression of each culture was calculated and shown by ratio (aimed genes/GADH) against that of BxPC3 cells as a standard. C. MMP-3 and MMP-9 was evaluated by the immunofluorescence staining in the co-culture system with BxPC3-GFP cells and hMSCs. BxPC3-GFP cells were detected by their own GFP green fluorescence color, while MMP-3 and MMP-9 were detected by red fluorescence colors after the immunostaining.
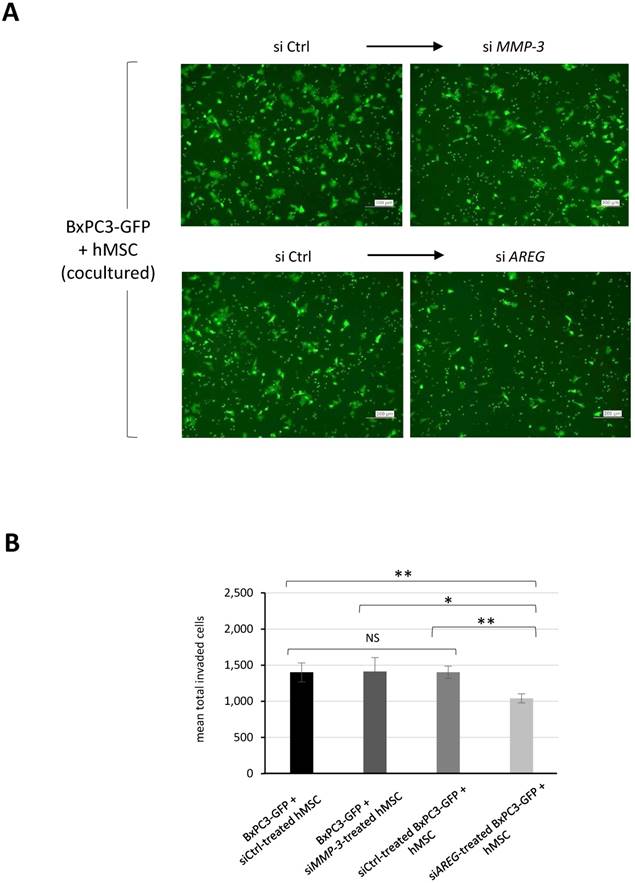
MMP-3 and MMP-9 are widely known secretory proteases directly linked to cancer invasion. We therefore performed additional evaluations by protein expression using immunofluorescence staining. Figure 4C revealed that co-culture-induced upregulation of MMP-3 occurred in MSCs but not BxPC3 cells. While MMP-9 positive population in BxPC3 cells was notably reduced by co-existence with MSCs. To determine which component was in charge for cancer invasion in the co-culture system, we tried to suppress either MMP-3 in MSCs or AREG in BxPC3-GFP cells and evaluated the invasion ability of BxPC3-GFP cells after co-culture. Using a similar approach as shown in Figure 2C and 2D, we found that the invasiveness of BxPC3-GFP cells was mitigated by the AREG suppression but not MMP-3 reduction in MSCs (Fig. 5A and 5B). From these data, we finally figured out the importance of AREG in pancreatic cancer cells to induce their invasion and AREG expression was potently enhanced by the surrounding MSCs.
Effect of siRNA-mediated gene suppression on pancreatic cancer invasiveness in co-culture with MSCs. A and B. BxPC3-GFP cells were mixed with either siControl (siCtrl) or siMMP-3 treated hMSCs, while BxPC3-GFP cells treated with either siCtrl or siAREG were mixed with hMSCs, and then they were placed on the upper transwell chambers and the invasiveness of placed BxPC3-GFP cells in each group was evaluated according to the similar procedure as indicated in Fig. 2A and 2B. The membrane-passed BxPC3-GFP cells were shown in images (A) and their fluorescence intensities were measured and quantified (B). Data from A are means ± SD, *P<0.05 and **P<0.01.
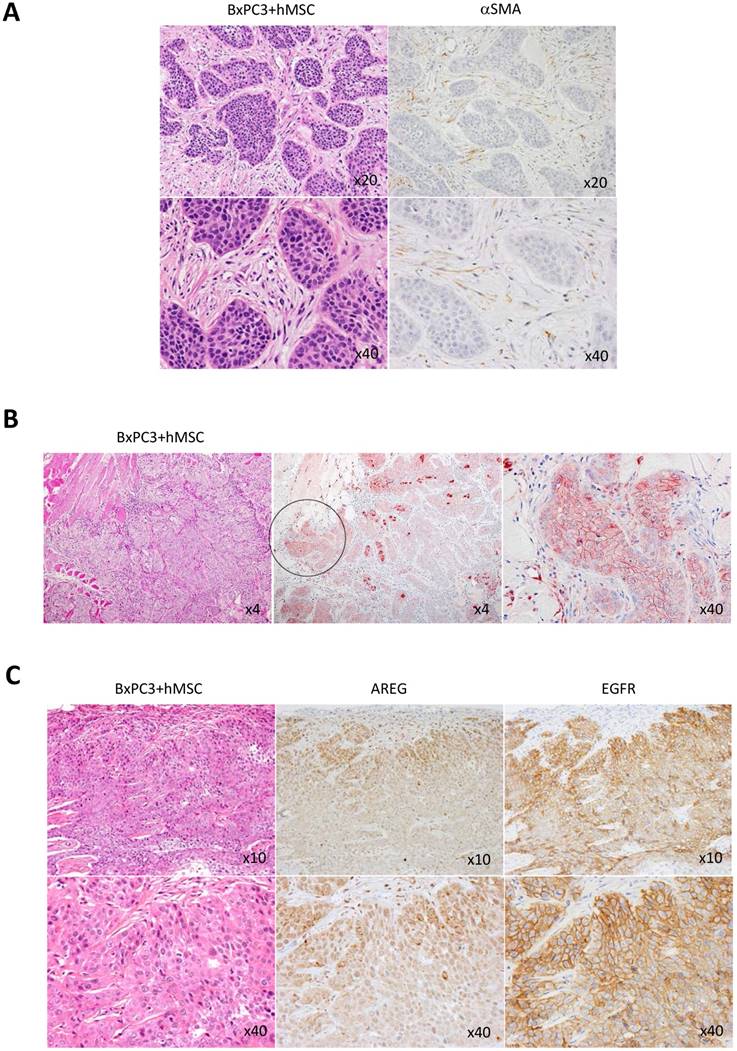
High expression of soluble ligand AREG and its receptor EGFR at the invasive cancer rim in PDACs in vivo. Knowing the importance of AREG in pancreatic cancer invasion with the help of MSCs in vitro, we next investigated the distribution of AREG receptor, EGFR in PDACs in vivo. To make the secreted AREG function properly with pancreatic cancer cells using autocrine mechanism, its receptor EGFR should express on the same cells. To condition this, we first evaluated the mouse subcutaneous transplantation model with BxPC3 cells and hMSCs. In this transplantation model, we first confirmed the presence of the transplanted human MSCs in part with a few populations in the tumor stroma area, which was detected by anti-human αSMC antibody (Fig. 6A), suggesting that artificial stroma includes not only hMSCs but also gathered mouse-derived MSCs and fibroblasts, and the hMSCs-differentiated fibroblasts. We found that the artificial stroma patterns in the mice were very similar to those in human PDACs in histology (Fig. 1A). Under the PDAC mimic setting, we detected strong positive signal of AREG in the invasive area of the BxPC3 tumor (Fig. 6B). Interestingly, EGFR was also potently expressed in the local invasive front (Fig. 6C).
Immunohistological analysis of AREG and EGFR expression in PDAC-imitated tissue environment in mouse model. BxPC3 cells mixed with hMSCs were subcutaneously injected into nude mice to built a copy of human PDAC tissue. A. Histologically, abundant and dense stroma appending to the BxPC3-derived cancer was appeared (left panel: HE staining, top x20 and bottom x40 in magnification). In the formed stroma area, αSMA positive cells were detected (right panel: αSMA immunostaining, top x20 and bottom x40 in magnification). B. AREG was detected at high level in the invasive front of the transplanted BxPC3 cells as shown by circle (middle panel, magnification x4). The circle region was stretched (right panel, magnification x 40). Left panels show HE staining (magnification x4). C. Immunnostainings of AREG and EGFR were performed in the human PDAC-mimic tissue. Left: HE staining, middle: AREG detection, right: EGFR detection. Magnifications of top and bottom images are x10 and x40, respectively.
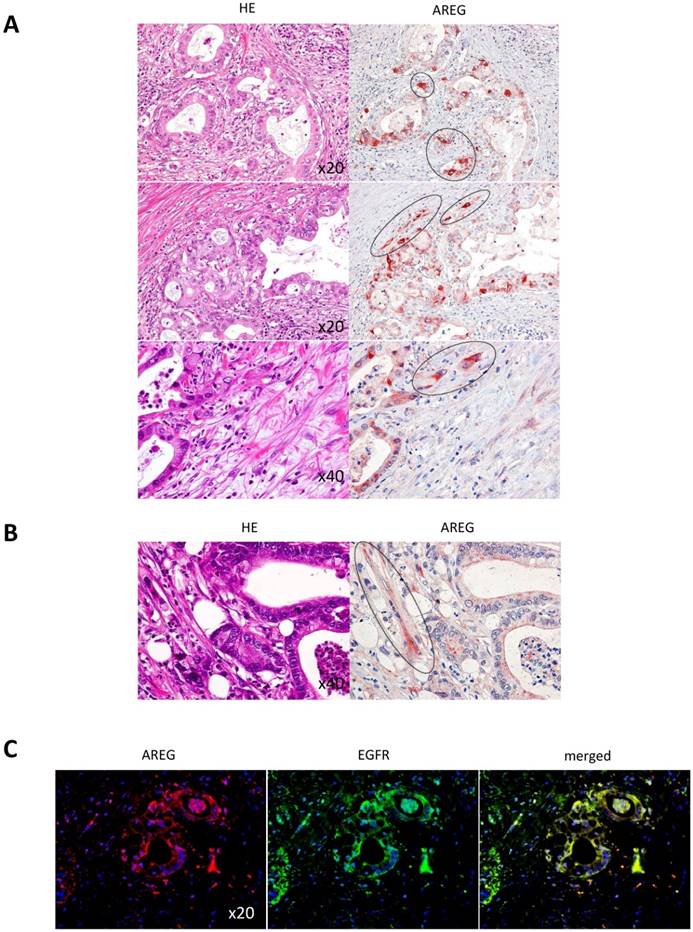
Finally, we assessed whether the strong appearance of AREG and EGFR in the invasive tumor rim observed in the artificial PDAC copy also mirrors in clinical cases. Figure 7A shows that a thick signal of AREG did appear in the invasive front of the PDAC cells (highlighted by the dotted circles). At the same time, to our surprise, some MSCs in the stromal area, which were indicated with dotted circle, showed strong signal of AREG production (Fig. 7B). By co-immunofluorescence staining, we found that AREG and EGFR were co-localized in the PDAC cells (Fig. 7C). Taken together, these results strongly suggest the unique role of AREG in PDAC local invasion, i.e., MSCs in PDAC stroma potently affects the upregulation of AREG in the PDAC cells, which in turn stimulates local invasion of the PDAC cells through the surface receptor EGFR in an autocrine manner. In addition, from the staining pattern, AREG produced from MSCs may also function in part to stimulate the neighbor PDAC cells in a paracrine manner.
Immunohistological analysis of AREG and EGFR expression in clinical PDAC specimens. A and B. AREG was immunostained in the patient-derived PDAC tissues. Left and right images show HE staining and AREG immunostaining, respectively. The dotted circles are indicating PDAC nests at the invasive front (A), and fibroblastic mesenchymal cells (B). Magnifications from top to bottom were x20, x20 and x40 in A and magnification in B was x40. C. Double immunofluorescence staining of AREG and EGFR in the clinical PDAC specimen. AREG was detected by red fluorescence color, while EGFR was detected by green fluorescence color.
Discussion
PDACs are one of the most intractable cancers to be cured even with current therapeutics because of its dense stroma with poor vascularity [13]. Owing to the cancer-surrounding dimension, i.e., strong barrier and scant drug supplying path, the permeability of anti-cancer drugs is largely hampered, leading to their lowered effects [14]. In addition, the PDACs have been shown to be highly invasive even at early stage [1-3]. This is also considered not only due to the cell character of PDAC itself but also by the enriched stroma appending to PDACs. Thus, the well understanding of biological role of PDAC stroma at molecular levels is worthwhile to be able to overcome therapeutic resistance of PDACs. Since PDAC stroma is roughly composed of CAFs and matrix in most part, many scientists have been investigated CAFs' function in tumor progression. However, CAFs source seems to be MSCs and so far multiple roles of MSC on PDAC still remain to be surprisingly unknown. Facing this problem, Kabashima-Niibe et al. first reported that MSCs are involved in EMT of PDACs in the cancer-stroma environment [12]. In this report, we also clarified another unique role of MSCs on PDAC progression. MSCs existed in the PDAC dense stroma sharing typical markers with common MSCs, which are positive for CD105, CD73, CD29, αSMA and CD34-, CD45-. As was mentioned above, the single cellular marker restricted only to bone marrow-derived MSC have been not identified yet, these combined expression are important. Especially, MSC strongly and more frequently expressed CD73, which discriminate it from normal fibroblast, NHDF. Coculture of BxPC3 cells and MSCs demonstrated that MSCs significantly facilitated tumor cell invasion. Our mechanistic analysis through secretory phenotype profiling using protein arrays and subsequent functional assays revealed AREG and MMP-3 as a key factor through their cellular interaction. With coexistence of BxPC3 cells and MSCs, AREG were specifically upregulated in BxPC3 cells and MSCs, which was corroborated as a specific phenomenon by additional coculture assay using other PDAC cell lines such as AsPC1, MiaPaca-2 and Panc-1. In a simple cellular assay using the Boyden chamber transwell, we found that AREG but not MMP-3 enabled to provide cancer cells with strong driving force of invasion, but this might be due to the difference in condition between in vitro culture system and in vivo tumor microenvironment. In the PDAC stroma in vivo, MMP3-mediated matrix collapse adjacent to cancer lesion might contribute to AREG-triggered cancer local invasion as an early event of tumor progression. Actually, Mehner et al. recently reported that MMP-3 expression is a reliable sign of poor survival in pancreatic cancers [15]. Our study revealed that AREG and its receptor EGFR were both upregulated at the interface of the cancer and stroma where cancer invasion actively take place, implying a physiologically significant role of AREG-EGFR binding event with an autocrine manner in PDAC invasiveness.
The present study demonstrated specific upregulation of AREG via cellular interaction between PDAC cells and MSCs which seems to accelerate tumor cell invasion, and it was supported by examination of clinical tissues from PDAC patients. However, detailed mechanism of how AREG in PDAC is significantly upregulated still remains unclear. One of the clues may come from our own and other previous works. Researchers studying the relationship between cancer and inflammation have compiled growing mass of evidence that some extracellular S100 proteins that are induced by cancer inflammatory microenvironments play a critical role in cancer invasion and metastasis. Notably, among the S100 family proteins, S100A11 is overexpressed and actively secreted from pancreatic cancer cells. Our previous study showed that secreted S100A11 was able to function as a ligand of a cell surface receptor RAGE (receptor for advanced glycation endoproducts), resulting in induction of EGF [16] in keratinocytes and IL-1β [17] in endothelial cells. Liu FL [18] reported an interesting finding that IL-1β increases AREG mRNA as well as AREG protein release via TACE-dependent membrane cleavage in fibroblast-like synoviocytes in inflammatory legion. Since RAGE is known to express in both pancreatic cancer cells and MSCs, we speculate that one candidate pathway (S100A11-RAGE) may be workable in both cancer cells with an autocrine manner and MSCs with a paracrine manner for AREG induction. To uncover this complex mechanism in PDAC settings, further advanced studies are required.
EGFR is a member of the ERBB family of receptor tyrosine kinases. In response to stimulation by ligands, the receptor homo or heterodimerizes with other ERBB family members and undergoes autophosphorylation, leading to activation of downstream signaling pathways involved in not only cellular invasion but also survival and growth [19-21]. Since AREG binds either EGFR homodimer (EGFR/EGFR) or EGFR/ERBB2 heterodimer, EGFR specific inhibitors may be effective for PDACs' treatment if escaping the stroma barrier in vivo. In this subject, Wang L et al. [22] demonstrated that Erlotinib, an EGFR specific chemical inhibitor, showed marked anti-cancer functions in BxPC3 cells, which possess no mutation of EGFR. In addition, Larbouret C et al. [23] found that targeting approach to EGFR/HER2 heterodimer on PDACs using the combination of Cetuximab (therapeutic anti-EGFR antibody) with Trastuzumab (therapeutic anti-HER2 antibody) produced remarkable tumor suppressing effects. Taken together, these results indicate uncommon role of the AREG-mediated pathways in PDAC progression and high usefulness of the targeting approach to that pathway to treat PDACs.
In conclusion, our results disclosed another important role of MSCs in abundant stroma of PDAC. When PDAC cells and MSCs coexist, both cells affect each other to switch AREG production especially in cancer side and MMP-3 in MSC side. The identified secretory phenotype may orchestrate to promote cancer invasion, which might be an early event for PDAC progression in vivo. Since EGFR overexpression was observed in the cancer cells especially located at interface between cancer and stroma areas, the secreted AREG readily stimulate the cancer cells at the interface for triggering local invasion. Thus, targeting AREG and its relevant conducting pathways will help with establishing an effective strategy to conquer the therapeutic difficulty of PDAC in the future.
Supplementary Material
Supplementary figures.
Acknowledgements
This research was supported by Grant-in-Aid for Scientific Research on Innovative Areas, No. 25112716 (E.K.) from the Japan Society for the Promotion of Science (JSPS).
Ethics Approval and Consent to Participate
In this study, usage of clinical specimens for immunohistochemistry was approved by the research ethics committees of Niigata University Medical and Dental Hospital. Written informed consent was obtained from each patient for use of the materials.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Liang C, Qin Y, Zhang B. et al. Oncogenic KRAS targets MUC16/CA125 in Pancreatic Ductal Adenocarcinoma. Mol Cancer Res. 2016;15(2):201-12
2. Kang R, Xie Y, Zhang Q, Hou W. et al. Intracellular HMGB1 as a novel tumor suppressor of pancreatic cancer. Cell Research. 2017;27:916-32
3. Keleg S, Buchler P, Ludwig R, Buchler MW, Friess H. Invasion and metastasi in pancreatic cancer. Molecular Cancer. 2003;2(14):1-7
4. Shan T, Chen S, Chen X. et al. Cancer-associated fibroblast enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncology Reports. 2017;37:1971-79
5. von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A. The role of stromal cancer-associated fibroblast in pancreatic cancer. Journal of Hematology & Oncology. 2017;10(76):DOI 10.1186/s13045-017-0448-5
6. Zhang A, Qian Y, Ye Z. et al. Cancer-associated fibroblast promote M2 polarization of macrophages in pancreatic ducatal adenocarcinoma. Cancer Medicine. 2017;6(2):403-70
7. Bolm L, Cigolla S, Wittel UA. et al. The role of fibroblast in pancreatic cancer: extracellular matrix versus paracrine factors. Translational Oncology. 2017;10(4):578-88
8. Hwang RF, Moore T, Arumugam T. et al. Cancer-associated stromal fibroblast promote pancreatic tumor progression. Cancer Res. 2008;68(3):918-26
9. Shinagawa K, Kitadai Y, Tanaka M. et al. Mesenchymal stem cells enhance growth and metastasis of colon cancer. Int. J. Cancer. 2010:127 (10):2323-33
10. Karnoub AE, Dash AB, Weinberg R. et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557-63
11. Beckermann BM, Kallifatidis G, Groth A. et al. VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. British J. Cancer. 2008;99:622-31
12. Kabashima-Niibe A, Higuchi H, Takaishi H. et al. Mesenchymal stem cells regulate epithelial-mesenchymal transition and tumor progression of pancreatic cancer cells. Cancer Sci. 2013;104:157-64
13. Xu Z, Pothula PP, Wilson JS, Apte MV. Pancreatic cancer and its stroma: A conspiracy theory. Worl J Gastroenterol. 2014;20(32):11216-29
14. Lin HJ, Lin J. Seed-in-Soil: pancreatic cancer influenced by tumor microenvironment. Cancer. 2017;9(93):DOI 10.1186/s13045-017-0448-5
15. Mehner C, Miller E, Nassar A, Bamlet WR, Radisky ES, Radisky DC. Tumor cell expression of MMP3 as a prognostic factor for poor survival in pancreatic, pulmonary, and mammary carcinoma. Gene & Cancer. 2015;6(11-12):480-9
16. Sakaguchi M, Sonegawa H, Murata H, Kitazoe M, Futami J-I, Kataoka K, Yamada H, Huh N-h. S100A11, a dual mediator for growth regulation of human keratinocytes. Molecular Biology of the Cell. 2008;19:78-85
17. Sakaguchi M, Murata H, Yamamoto K-I, Ono T, Sakaguchi Y, Motoyama A, Hibino T, Kataoka K, Huh N-h. TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE phosphorylated upon ligand binding. PLoS ONE. 2011;6(8):e23132. doi:10.1371/journal.pone.0023132
18. Liu F-L, Wu C-C, Chang D-M. TACE-dependent amphiregulin release is induced by IL-1β and promotes cell invasion in fibroblast-like synoviocytes in rheumatoid arthritis. Rheumatology. 2014;53:260-8
19. Ranson M. Epidermal growth factor receptor tyrosine kinase inhibitors. British Journal of Cancer. 2004;90:2250-5
20. Wieduwiit MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65(10):1566-84
21. Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti A, Batra SK. Targeting the EGFR signalling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16(1):15-31
22. Wang L, Zhu Z-X, Zhang W-M. Schedule-dependent cytotoxic synergism of pemetrexed and erlotinib in BxPC-3 and PANC-1 human pancreatic cancer cells. Experimental and Therapeutic Medicine. 2011;2:969-75
23. Larbouret C, Gaborit N, Chardes T, Coelho M, Campigna E, Bascoul-Mollevi C, Mach J-P, Azria D, Robert B, Pelegrin A. In pancreatic carcinoma, dual EGFR/HER2 targeting with Cetuximab/Trastuzumab is more effective than treatment with Trastuzumab/Erlotinib or Lapatinib alone: implication of receptors'down-regulation and dimers' disruption. Neoplasia. 2012;14(2):121-30
Author contact
![]() Corresponding author: Eisaku Kondo, M.D., Ph.D. Division of Molecular and Cellular Pathology, Niigata University Graduate School of Medical and Dental Sciences, 1-757 Asahimachi-dori, Chuo-ku, Niigata 951-8510, Japan. Tel: +81-25-227-2102; Fax: +81-25-227-0761; E-mail: ekondoniigata-u.ac.jp
Corresponding author: Eisaku Kondo, M.D., Ph.D. Division of Molecular and Cellular Pathology, Niigata University Graduate School of Medical and Dental Sciences, 1-757 Asahimachi-dori, Chuo-ku, Niigata 951-8510, Japan. Tel: +81-25-227-2102; Fax: +81-25-227-0761; E-mail: ekondoniigata-u.ac.jp