Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(16):2981-2986. doi:10.7150/jca.25640 This issue Cite
Review
A comprehensive look at the role of hyperlipidemia in promoting colorectal cancer liver metastasis
Yimin Shen1#, Caihua Wang2#, Yuezhong Ren1 ![]() , Jun Ye2
, Jun Ye2 ![]()
1. 1 Department of Endocrinology and Metabolism, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang Province, China
2. 2 Department of Gastroenterology, The Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, Zhejiang Province, China
#: These authors contributed equally to this work.
Received 2018-2-21; Accepted 2018-6-9; Published 2018-7-30
Abstract
Colorectal cancer (CRC) is one of the most malignant cancers, and it tends to migrate to the liver and has a high mortality rate. Several mechanisms behind the metastasis of CRC have been identified, including hyperlipidemia. For example, hyperlipidemia can lead to enhanced stemness and neutrophil infiltration, which increases CRC metastasis. There are three primary aspects to the relationship between hyperlipidemia and CRC metastasis: hyperlipidemia (1) promotes the initial metastatic properties of CRC, (2) stimulates CRC cells to leave the vasculature, and (3) facilitates the development of CRC metastasis. In this study, we provide a comprehensive overview of the role that hyperlipidemia played in CRC metastasis to help reduce the mortality associated with CRC metastasis from the standpoint of metabolic. We also review cancer metastasis.
Keywords: Colorectal cancer, hyperlipidemia, liver metastasis, cancer stem cell
Introduction
Colorectal cancer (CRC) is one of the most common neoplasms of the digestive system [1]. It is the third leading cause of cancer-related deaths in the United States [2]. The 5-year survival rate is >90% in patients with early-stage disease, compared with 10-15% in patients with distant metastases [3]. The liver is the most important target site of CRC metastasis [4]. The progression of metastasis mainly involves three steps. First, the primary cancer cells disseminate locally into the vascular system. Second, circulating cancer cells can either be arrested by the innate immune system or extravasate through vascular walls into distant tissues. Finally, the micro-metastatic colonies settle in the parenchyma and proliferate further [5, 6]. There are several mechanisms closely related to CRC distant metastasis. For example, transforming growth factor-β (TGF-β) is a major inducer of epithelial-mesenchymal transition (EMT) program [7]. EMT program is considered as a crucial step in metastatic cascades by making CRC cells acquire migratory abilities [8]. Some factors, such as vascular endothelial growth factor (VEGF), contribute to vascular leakiness and therefore increase the rate at which cancer cells leave the vascular system and metastasize [9]. Recent studies have provided new insights into the profound influence of lipids, via distinct capabilities, on CRC metastasis. Some studies have indicated that hyperlipidemia can exacerbate several severe diseases such as cardiovascular disease, which remains the primary cause of mortality in Western countries [10, 11]. Recent epidemiologic studies have also suggested that lipid metabolism plays important roles in the development of many cancers, including prostate and breast [12, 13]. Furthermore, decreased concentrations of high-density lipoprotein cholesterol (HDL) have been related to increased proinflammatory cytokines such as interleukin 6 (IL-6) and tumor necrosis factor-a receptors, which promote CRC cells growth and proliferation [14]. In the clinic, many epidemiological studies have demonstrated that several lifestyle choices that lead to high levels of lipids, such as high-fat diet (HFD) status, stimulate the CRC progression [15]. Clinical evidence suggests that statins can reduce the incidence of CRC-related mortality [16]. Therefore, it is worth understanding the mechanisms by which hyperlipidemia affects CRC metastasis. For example, a HFD lifestyle is responsible for increased CRC cell stemness, and enhanced stemness may stimulate the initial metastatic capability [17]. Similarly, a HFD status enhances macrophage infiltration, which promotes the migration of CRC cells to distant organs [18, 19]. IL-8 acts as the obesity-induced inflammation, and a higher level of it is also associated with CRC metastasis [20]. In addition, enhanced level of insulin-like growth factor-1 (IGF-1) found in obese individuals accelerates formation of the liver metastasis microenvironment [21]. CRC patients who consume a HFD accumulate more reactive oxygen species (ROS), which eventually stimulate the progression of metastasis by degrading molecules such as PKC and protein tyrosine phosphatases [6].
Several mechanisms involving hyperlipidemia and CRC metastasis have been identified in clinical experiments. In addition to these well-known molecular changes that promote malignant behavior, this review highlights the most recent developments in our understanding of how hyperlipidemia affects CRC metastasis to the liver.
Hyperlipidemia stimulates the initial migratory properties of CRC
Cancer stem cells (CSCs) are an important driving force behind CRC metastasis [22, 23]. CSCs have the ability to self-renew and undergo multi-lineage differentiation [24, 25]; they also play a key role in both original tumorigenicity and the ability of tumors to migrate [26]. Moreover, the dissemination of CSCs may help metastatic colonies to form in distant organs and descendant cells to adjust to the new microenvironment [27]. CRC recurrence and metastasis after chemotherapy are sometimes the result of present CSCs [28].
Serum lipid levels play an essential role in the progression of CSCs. In the context of a HFD, CRC cell stemness can be enhanced by modifying signaling pathways and genetic expression. A HFD induces tumor progression by acting on the RBP4-STRA6 pathway, which stimulates stemness by activating signal transducer and activator of transcription 3 (STAT3) [17]. Low-density lipoprotein cholesterol (LDL) also enhances the expression of stemness genes including Sox2, Oct4, Nanog, and Bmi 1 [6]. Enhanced CRC cell stemness is required for the later stages of migration.
It is generally accepted that the acquisition of pro-invasive capacities is associated with EMT [29]. The EMT program allows epithelial cells to acquire a mesenchymal phenotype [30], which involves loss of cell-cell adhesion, alteration of cytoskeletal organization, and loss of apical-basal polarity [31, 32].
EMT can be activated in a variety of cancer cells [33], and enables cells to acquire stem cell-like features and the ability to migrate [34-36]. After activation of EMT, CRC cells can differentiate and become resistant to apoptosis [37, 38]. Moreover, cancer cells within the CSC-enriched subpopulation exhibit aspects of EMT program activation [32]. LDL has the ability to activate the MAPK pathway, which is an indispensable signaling cascade during EMT that is regulated by PKC and TGF-β/Smad signaling [6]. The MAPK pathway is also involved in a wide range of cellular responses, including cell proliferation, differentiation, and survival [39]. Consistent with these findings, lipids specifically affect the early metastatic seeding of CRC in distant organs.
Hyperlipidemia promotes the departure of CRC cells from the vasculature
Many cells within the bloodstream, including endothelial cells, platelets, lymphocytes, and neutrophils, promote metastasis [40, 41]. For example, after degranulation, mast cells (MCs) could secrete tryptase to degrade extracellular matrix (ECM) components to help the invasion and metastasis of CRC [42]. All of these cell types may protect cancer cells from immune attack, especially that involving natural killer (NK) cells and γδ T cells [43-45]. NK cells and γδ T cells are crucial for the innate immune response that initiates a series of immunological effects on cancer cells [46, 47]. Hypercholesterolemia-induced oxidative stress downregulates the expression of Tet1 in hemopoietic stem cells, resulting in increased DNA hypermethylation and histone modifications. This ultimately leads to dysfunction and reduced differentiation of NK cells and γδT cells, thereby causing an inadequate immune surveillance [45].
Hyperlipidemia is significantly correlated with a higher proportion of blood neutrophil [48, 49]. The accumulation of neutrophils is mainly the result of enhanced granulopoiesis and mobilization from the bone marrow, as well as increasing levels of CCR1, CCR2, CCR5, and CXCR2 [50]. Neutrophils are critical for cancer metastasis [51]. They can induce a leaky vasculature and increase the attachment of cancer cells to endothelial monolayers via matrix metalloproteinase MMP9 and BV8, thereby facilitating the adhesion and extravasation of CRC cells via the vascular system [52, 53]. Neutrophils also capture circulating cancer cells, either by the cell surface molecule CD11b or by releasing neutrophil extracellular traps, which contributes to increased metastasis formation [41, 44, 54]. Neutrophils are also effectors of angiogenesis, since they provide CRC cells with more selective escape routes [55]. Therefore, neutrophils promote metastasis around the vascular system.
Among immune cells, dendritic cells (DCs) are critically involved in CRC metastasis; they have the capacity of immune activation and the ability to recognize and absorb cancer cells [56]. Under the conditions of obesity and CRC, the adipocyte microenvironment delivers immunosuppressive signals to induce DC differentiation and shift DC immune surveillance toward T lymphocyte antigen presentation; these effects are mediated by increased expression of inhibitory molecules and decreased IL-12/IL-10 levels [57].
CRC patients with thrombocytosis (high platelet count) have a poor prognosis [58]. Platelets protect cancer cells from immune elimination and allow better adaption to the blood environment, adherence to endothelial cells, and migration of cancer cells [59, 60]. In addition, central obesity status helps boost platelet activity in the circulation [61]. It activates platelets both locally and systemically via three main approaches [62]. First, obesity stimulates the production of ROS, result in increased isoprostane levels. Second, obesity increases the levels of inflammatory adipokines. Third, obesity leads to an increased platelet activity via the NO/cGMP/PKG and PGI2/cAMP/PKA pathways [62]. Activated platelets increase their adhesion to cancer cells, forming hetero-aggregates that promote metastasis; heterodimer formation occurs between platelet integrin αIIbβ3 and cancer cell receptors such as integrin αvβ3 [63].
Understanding the influence of the immune cells and platelets discussed here will provide more insight into how hyperlipidemia affects circulating tumor cells (CTCs) in CRC. CRC cells become CTCs once they enter the vasculature [64]. CTCs often exhibit both epithelial and mesenchymal traits that are a prerequisite for successful formation of a metastatic colony [5]. CTCs can potentially undergo necrosis in the bloodstream in response to high hemodynamic shear stress, which inhibits metastasis. In a hyperlipidemic state, the elevated levels of lipids would slow down the hemodynamics, and the resulting reduced flow shear stress would stimulate CTC migration [65].
Hyperlipidemia facilitates the formation of CRC metastases
After exiting the vasculature, CRC cells are most likely to metastasize to the liver [66]. The formation of distant metastasis is correlated with several modifications, especially MMP9.
At sites of metastasis, increased numbers of neutrophils release granules containing neutrophil elastase and MMP9, which remodel the ECM in the tumor microenvironment. This remodeling allows CRC cells to overcome the constraints of cell-cell and cell-matrix interactions and to migrate more freely [67]. Obesity also contributes to the recruitment and transformation of M1-macrophages into M2-macrophages [68]; this transformation is a metastasis-promoting phenotype that occurs in response to the immunosuppressive cytokines secreted by tumor tissues [69]. Taken together, these studies suggest that tumor-associated neutrophils and the macrophages involved in hyperlipidemia further accelerate CRC metastasis.
In a metastatic microenvironment, high levels of plasma triglycerides, total serum cholesterol, and saturated fatty acids contribute to ROS-induced oxidative stress [70]. ROS are produced via intracellular metabolism and include superoxide, the hydroxyl radical, and H2O2 [71, 72]. ROS degrade molecules such as PKC and protein tyrosine phosphatases and regulate downstream molecules of MAPK and PAK, which are both involved in cancer migration [6].
Obese subjects have an increased risk of non-alcoholic fatty liver disease (NAFLD) [73]. NAFLD includes steatosis and its progression to non-alcoholic steatohepatitis [74]. The NAFLD provides a microenvironment that is more susceptible to CRC metastasis by secreting increased levels of various signaling molecules, including MMPs [75]. In addition, NAFLD is associated with liver cell injury and inflammation [76]. Inflammatory cells such as neutrophils might be recruited in the NAFLD and contribute to CRC progression [77].
The relationship between obesity and liver metastasis also involves circulating levels of IGF-1 [21]. Obesity is accompanied by elevated circulating IGF-1 levels and insulin resistance [78-80]. IGF-1 is a procarcinogen produced mainly by the liver that regulates the host innate immune system [21]. It also reduces cancer cell death, enhances cell proliferation, influences the host microenvironment, and modulates the pro-inflammatory response to promote liver metastasis [81, 82].
The metastatic microenvironment in the liver also contains stromal cells and endothelial cells [83], which exert oxidative stress under conditions of hyperlipidemia. The interaction between oxidative stress and inflammation can stimulate metastasis. Obese subjects are apt to get increased circulating VEGF level as well [84]. The elevated level of VEGF is a predictor of liver metastasis. And a higher serum VEGF level represents a worse prognostic for patients with liver metastases [85]. In addition, PIGF/VEGFR-1 signaling in obese subjects contributes to a pro-cancer immune microenvironment [86].
Collectively, high lipid levels further influence CRC metastasis by excreting increased levels of granules and stimulating the accumulation of ROS and IGF-1, which confer a special advantage to tumor cells and promote metastasis progression (Table 1).
The risk factors of the formation of CRC metastases in the presence of high lipid levels
| subjects | Factors | Results | Risk of CRC metastasis |
|---|---|---|---|
| high level of lipid status | neutrophils↑ | MMP9, NE, CD11b, NETs… | ↑ |
| M1->M2 (macrophages) | metastasis-promoting phenotype | ↑ | |
| ROS↑ | oxidative stress | ↑ | |
| NAFLD | Permissive microenvironment | ↑ | |
| IGF-1↑ | Permissive microenvironment | ↑ |
Conclusion: principles and outlook
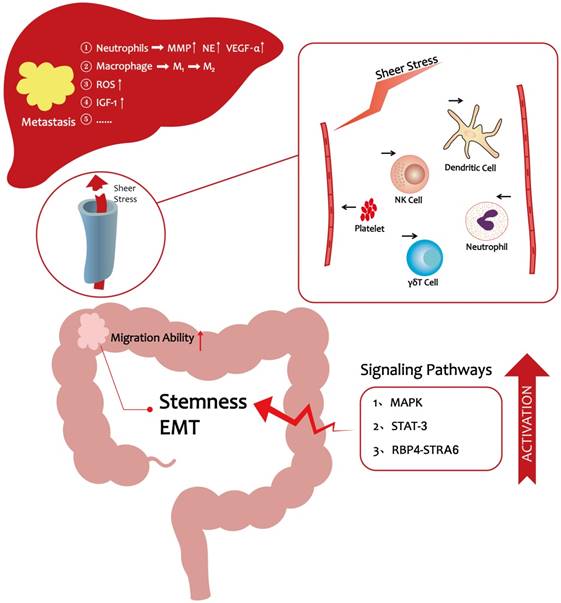
As the preceding discussions indicated, significant progress has been made over the past decade in elucidating the cellular, molecular, and signaling pathway programs that drive CRC liver metastasis. It is now evident that hyperlipidemia promotes the entry of CRC cells into the vasculature and their subsequent migration to the distant liver. A HFD lifestyle could enhance CRC cells stemness to promote their migration ability [17]. In vivo, lipids enhance platelet activation and the numbers of neutrophils to protect CRC cells from immune attack [48-50, 59, 60], thereby making it easier for cancer cells to escape from the vascular system. In a pre-metastatic microenvironment, hyperlipidemia facilitates liver metastasis via a variety of mechanisms including accumulation of tumor-associated neutrophils and ROS production (Figure 1) [6, 51]. Moreover, obesity, which is directly related to hyperlipidemia, is believed to be a risk factor for liver metastasis in CRC [21]. These observations provide novel insights into how lifestyle changes could help reduce the likelihood of CRC metastasis. For example, a low-fat diet and exercise are recommended to reduce the risk of metastasis. Clinical evidence showed that statins can reduce the mortality of CRC [16]. Statins are known to be the inhibition of Ras signaling and may activate the bone morphogenetic protein (BMP) signaling pathway in CRC cells [87]. Therefore, we wonder whether we can target Ras or BMP signaling pathway to reduce CRC metastatic properties still need to be further explored. In conclusion, CRC metastasis is highly related to hyperlipidemia, and restoration of lipid levels may reduce CRC metastasis-associated mortality.
The progression of CRC liver metastasis under hyperlipidemic conditions
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81773065), Zhejiang Provincial Natural Science Foundation of China (No. LY16H160019).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Angeli JP, Garcia CC, Sena F. et al. Lipid hydroperoxide-induced and hemoglobin-enhanced oxidative damage to colon cancer cells. Free Radic Biol Med. 2011;51(2):503-15
2. Siegel R, DeSantis C, Jemal A. Colorectal cancer statistics, 2014. Ca-Cancer J Clin. 2014;64(2):104-17
3. Geng L, Chaudhuri A, Talmon G. et al. MicroRNA-192 suppresses liver metastasis of colon cancer. Oncogene. 2014;33(46):5332-40
4. Fernandez MC, Rayes R, Ham B. et al. The type I insulin-like growth factor regulates the liver stromal response to metastatic colon carcinoma cells. Oncotarget. 2017;8(32):52281-93
5. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell. 2017;168(4):670-91
6. Wang C, Li P, Xuan J. et al. Cholesterol Enhances Colorectal Cancer Progression via ROS Elevation and MAPK Signaling Pathway Activation. Cell Physiol Biochem. 2017;42(2):729-42
7. Hirakawa M, Takimoto R, Tamura F. et al. Fucosylated TGF-beta receptors transduces a signal for epithelial-mesenchymal transition in colorectal cancer cells. British journal of cancer. 2014;110(1):156-63
8. Su L, Luo Y, Yang Z. et al. MEF2D Transduces Microenvironment Stimuli to ZEB1 to Promote Epithelial-Mesenchymal Transition and Metastasis in Colorectal Cancer. Cancer research. 2016;76(17):5054-67
9. Anderberg C, Cunha SI, Zhai Z. et al. Deficiency for endoglin in tumor vasculature weakens the endothelial barrier to metastatic dissemination. J Exp Med. 2013;210(3):563-79
10. Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. The Lancet. 2014;384(9943):626-35
11. Rosamond W, Flegal K, Furie K. et al. Heart disease and stroke statistics-2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117(4):e25-146
12. Nelson ER, Wardell SE, Jasper JS. et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342(6162):1094-8
13. His M, Zelek L, Deschasaux M. et al. Prospective associations between serum biomarkers of lipid metabolism and overall, breast and prostate cancer risk. European journal of epidemiology. 2014;29(2):119-32
14. van Duijnhoven FJ, Bueno-De-Mesquita HB, Calligaro M. et al. Blood lipid and lipoprotein concentrations and colorectal cancer risk in the European Prospective Investigation into Cancer and Nutrition. Gut. 2011;60(8):1094-102
15. Day SD, Enos RT, McClellan JL. et al. Linking inflammation to tumorigenesis in a mouse model of high-fat-diet-enhanced colon cancer. Cytokine. 2013;64(1):454-62
16. Nielsen SF, Nordestgaard BG, Bojesen SE. Statin use and reduced cancer-related mortality. N Engl J Med. 2013;368(6):576-7
17. Karunanithi S, Levi L, DeVecchio J. et al. RBP4-STRA6 Pathway Drives Cancer Stem Cell Maintenance and Mediates High-Fat Diet-Induced Colon Carcinogenesis. Stem cell reports. 2017;9(2):438-50
18. Liu Z, Brooks RS, Ciappio ED. et al. Diet-induced obesity elevates colonic TNF-alpha in mice and is accompanied by an activation of Wnt signaling: a mechanism for obesity-associated colorectal cancer. The Journal of nutritional biochemistry. 2012;23(10):1207-13
19. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39-51
20. Pietrzyk L, Torres A, Maciejewski R. et al. Obesity and Obese-related Chronic Low-grade Inflammation in Promotion of Colorectal Cancer Development. Asian Pacific journal of cancer prevention: APJCP. 2015;16(10):4161-8
21. Wu Y, Brodt P, Sun H. et al. Insulin-like growth factor-I regulates the liver microenvironment in obese mice and promotes liver metastasis. Cancer Res. 2010;70(1):57-67
22. Sampieri K, Fodde R. Cancer stem cells and metastasis. Semin Cancer Biol. 2012;22(3):187-93
23. de Sousa e Melo F, Kurtova AV, Harnoss JM. et al. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature. 2017;543(7647):676-80
24. Ji Y, Yang C, Tang Z. et al. Adenylate kinase hCINAP determines self-renewal of colorectal cancer stem cells by facilitating LDHA phosphorylation. Nat Commun. 2017;8:15308
25. Zeuner A, Todaro M, Stassi G. et al. Colorectal cancer stem cells: from the crypt to the clinic. Cell stem cell. 2014;15(6):692-705
26. Manic G, Signore M, Sistigu A. et al. CHK1-targeted therapy to deplete DNA replication-stressed, p53-deficient, hyperdiploid colorectal cancer stem cells. Gut. 2018;67(5):903-17
27. Gerger A, Zhang W, Yang D. et al. Common cancer stem cell gene variants predict colon cancer recurrence. Clin Cancer Res. 2011;17(21):6934-43
28. Todaro M, Francipane MG, Medema JP. et al. Colon cancer stem cells: promise of targeted therapy. Gastroenterology. 2010;138(6):2151-62
29. Gulhati P, Bowen KA, Liu J. et al. mTORC1 and mTORC2 regulate EMT, motility, and metastasis of colorectal cancer via RhoA and Rac1 signaling pathways. Cancer research. 2011;71(9):3246-56
30. Meng F, Wu G. The rejuvenated scenario of epithelial-mesenchymal transition (EMT) and cancer metastasis. Cancer Metastasis Rev. 2012;31(3-4):455-67
31. Vincent CT, Fuxe J. EMT, inflammation and metastasis. Seminars in cancer biology. 2017;47:168-9
32. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nature reviews Clinical oncology. 2017;14(10):611-29
33. Beck B, Lapouge G, Rorive S. et al. Different levels of Twist1 regulate skin tumor initiation, stemness, and progression. Cell stem cell. 2015;16(1):67-79
34. Liu W, Wang S, Sun Q. et al. DCLK1 promotes epithelial-mesenchymal transition via the PI3K/Akt/NF-kappaB pathway in colorectal cancer. International journal of cancer. 2018;142(10):2068-79
35. Tsoumas D, Nikou S, Giannopoulou E. et al. ILK Expression in Colorectal Cancer Is Associated with EMT, Cancer Stem Cell Markers and Chemoresistance. Cancer genomics & proteomics. 2018;15(2):127-41
36. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29(34):4741-51
37. Nieto MA. Epithelial plasticity: a common theme in embryonic and cancer cells. Science. 2013;342(6159):1234850
38. Puisieux A, Brabletz T, Caramel J. Oncogenic roles of EMT-inducing transcription factors. Nature cell biology. 2014;16(6):488-94
39. Lundberg IV, Wikberg ML, Ljuslinder I. et al. MicroRNA Expression in KRAS- and BRAF-mutated Colorectal Cancers. Anticancer research. 2018;38(2):677-83
40. Labelle M, Begum S, Hynes RO. Platelets guide the formation of early metastatic niches. Proc Natl Acad Sci U S A. 2014;111(30):E3053-61
41. Spicer JD, McDonald B, Cools-Lartigue JJ. et al. Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells. Cancer research. 2012;72(16):3919-27
42. Marech I, Ammendola M, Gadaleta C. et al. Possible biological and translational significance of mast cells density in colorectal cancer. World journal of gastroenterology. 2014;20(27):8910-20
43. Paolino M, Choidas A, Wallner S. et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. 2014;507(7493):508-12
44. Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nature reviews Immunology. 2015;15(2):73-86
45. Tie G, Yan J, Khair L. et al. Hypercholesterolemia Increases Colorectal Cancer Incidence by Reducing Production of NKT and gammadelta T Cells from Hematopoietic Stem Cells. Cancer Res. 2017;77(9):2351-62
46. Tallerico R, Todaro M, Di Franco S. et al. Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J Immunol. 2013;190(5):2381-90
47. Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nat Rev Immunol. 2013;13(2):88-100
48. Tolani S, Pagler TA, Murphy AJ. et al. Hypercholesterolemia and reduced HDL-C promote hematopoietic stem cell proliferation and monocytosis: studies in mice and FH children. Atherosclerosis. 2013;229(1):79-85
49. Herz J, Sabellek P, Lane TE. et al. Role of Neutrophils in Exacerbation of Brain Injury After Focal Cerebral Ischemia in Hyperlipidemic Mice. Stroke. 2015;46(10):2916-25
50. Drechsler M, Megens RT, van Zandvoort M. et al. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. 2010;122(18):1837-45
51. Liang W, Ferrara N. The Complex Role of Neutrophils in Tumor Angiogenesis and Metastasis. Cancer immunology research. 2016;4(2):83-91
52. Shojaei F, Singh M, Thompson JD. et al. Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(7):2640-5
53. Swierczak A, Mouchemore KA, Hamilton JA. et al. Neutrophils: important contributors to tumor progression and metastasis. Cancer metastasis reviews. 2015;34(4):735-51
54. Cools-Lartigue J, Spicer J, McDonald B. et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. The Journal of clinical investigation. 2013;123(8):3446-58
55. Tazzyman S, Niaz H, Murdoch C. Neutrophil-mediated tumour angiogenesis: subversion of immune responses to promote tumour growth. Seminars in cancer biology. 2013;23(3):149-58
56. Pardoll D. Cancer and the Immune System: Basic Concepts and Targets for Intervention. Semin Oncol. 2015;42(4):523-38
57. Del Corno M, D'Archivio M, Conti L. et al. Visceral fat adipocytes from obese and colorectal cancer subjects exhibit distinct secretory and omega6 polyunsaturated fatty acid profiles and deliver immunosuppressive signals to innate immunity cells. Oncotarget. 2016;7(39):63093-105
58. Erpenbeck L, Schon MP. Deadly allies: the fatal interplay between platelets and metastasizing cancer cells. Blood. 2010;115(17):3427-36
59. Haemmerle M, Taylor ML, Gutschner T. et al. Platelets reduce anoikis and promote metastasis by activating YAP1 signaling. Nat Commun. 2017;8(1):310
60. Liang H, Yang C, Zhang B. et al. Hydroxyethyl starch 200/0.5 decreases circulating tumor cells of colorectal cancer patients and reduces metastatic potential of colon cancer cell line through inhibiting platelets activation. Med Oncol. 2015;32(5):151
61. Li Q, Chen H. Epigenetic modifications of metastasis suppressor genes in colon cancer metastasis. Epigenetics. 2011;6(7):849-52
62. Anfossi G, Russo I, Trovati M. Platelet dysfunction in central obesity. Nutr Metab Cardiovasc Dis. 2009;19(6):440-9
63. Gay LJ, Felding-Habermann B. Contribution of platelets to tumour metastasis. Nat Rev Cancer. 2011;11(2):123-34
64. Kang Y, Pantel K. Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer cell. 2013;23(5):573-81
65. Regmi S, Fu A, Luo KQ. High Shear Stresses under Exercise Condition Destroy Circulating Tumor Cells in a Microfluidic System. Sci Rep. 2017;7:39975
66. Pugh SA, Harrison RJ, Primrose JN. et al. T cells but not NK cells are associated with a favourable outcome for resected colorectal liver metastases. BMC cancer. 2014;14:180
67. Dumitru CA, Lang S, Brandau S. Modulation of neutrophil granulocytes in the tumor microenvironment: mechanisms and consequences for tumor progression. Seminars in cancer biology. 2013;23(3):141-8
68. Huh JH, Kim HM, Lee ES. et al. Dual CCR2/5 Antagonist Attenuates Obesity-Induced Insulin Resistance by Regulating Macrophage Recruitment and M1/M2 Status. Obesity. 2018;26(2):378-86
69. Smith HA, Kang Y. The metastasis-promoting roles of tumor-associated immune cells. Journal of molecular medicine. 2013;91(4):411-29
70. Ding C, Zhao Y, Shi X. et al. New insights into salvianolic acid A action: Regulation of the TXNIP/NLRP3 and TXNIP/ChREBP pathways ameliorates HFD-induced NAFLD in rats. Scientific reports. 2016;6:28734
71. Poillet-Perez L, Despouy G, Delage-Mourroux R. et al. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox biology. 2015;4:184-92
72. Gough DR, Cotter TG. Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell death & disease. 2011;2:e213
73. Hassan K, Bhalla V, El Regal ME. et al. Nonalcoholic fatty liver disease: a comprehensive review of a growing epidemic. World journal of gastroenterology. 2014;20(34):12082-101
74. Bale G, Steffie AU, Ravi Kanth VV. et al. Regional differences in genetic susceptibility to non-alcoholic liver disease in two distinct Indian ethnicities. World J Hepatol. 2017;9(26):1101-7
75. Mendonsa AM, VanSaun MN, Ustione A. et al. Host and tumor derived MMP13 regulate extravasation and establishment of colorectal metastases in the liver. Molecular cancer. 2015;14:49
76. Mohlke KL. Nonsynonymous variants and fatty liver disease. Nat Genet. 2008;40(12):1394-5
77. VanSaun MN, Lee IK, Washington MK. et al. High fat diet induced hepatic steatosis establishes a permissive microenvironment for colorectal metastases and promotes primary dysplasia in a murine model. Am J Pathol. 2009;175(1):355-64
78. Verdile G, Keane KN, Cruzat VF. et al. Inflammation and Oxidative Stress: The Molecular Connectivity between Insulin Resistance, Obesity, and Alzheimer's Disease. Mediators of inflammation. 2015;2015:105828
79. Pendyala S, Neff LM, Suarez-Farinas M. et al. Diet-induced weight loss reduces colorectal inflammation: implications for colorectal carcinogenesis. The American journal of clinical nutrition. 2011;93(2):234-42
80. Jung SY, Ho G, Rohan T. et al. Interaction of insulin-like growth factor-I and insulin resistance-related genetic variants with lifestyle factors on postmenopausal breast cancer risk. Breast cancer research and treatment. 2017;164(2):475-95
81. Janakiram NB, Mohammed A, Rao CV. Role of lipoxins, resolvins, and other bioactive lipids in colon and pancreatic cancer. Cancer Metastasis Rev. 2011;30(3-4):507-23
82. Sinicrope FA, Foster NR, Sargent DJ. et al. Obesity is an independent prognostic variable in colon cancer survivors. Clin Cancer Res. 2010;16(6):1884-93
83. Psaila B, Lyden D. The metastatic niche: adapting the foreign soil. Nature reviews Cancer. 2009;9(4):285-93
84. Hursting SD, Hursting MJ. Growth signals, inflammation, and vascular perturbations: mechanistic links between obesity, metabolic syndrome, and cancer. Arteriosclerosis, thrombosis, and vascular biology. 2012;32(8):1766-70
85. Divella R, Daniele A, R DEL. et al. Circulating Levels of VEGF and CXCL1 Are Predictive of Metastatic Organotropismin in Patients with Colorectal Cancer. Anticancer research. 2017;37(9):4867-71
86. Incio J, Tam J, Rahbari NN. et al. PlGF/VEGFR-1 Signaling Promotes Macrophage Polarization and Accelerated Tumor Progression in Obesity. Clin Cancer Res. 2016;22(12):2993-3004
87. Voorneveld PW, Reimers MS, Bastiaannet E. et al. Statin Use After Diagnosis of Colon Cancer and Patient Survival. Gastroenterology. 2017;153(2):470-9 e4
Author contact
![]() Corresponding authors: Dr. Jun Ye, Department of Gastroenterology, The Second Affiliated Hospital, Zhejiang University School of Medicine, 88 Jiefang Road, Hangzhou, Zhejiang 310009, China. Tel: 86-571-87784642; Fax: 86-571-87022776; E-mail: wzmcyejunedu.cn and Dr. Yuezhong Ren, Department of Endocrinology and Metabolism, The Second Affiliated Hospital, Zhejiang University School of Medicine, 88 Jiefang Road, Hangzhou, Zhejiang 310009, China. Tel: 86-571-87784642; Fax: 86-571-87022776; E-mail: renyuezedu.cn
Corresponding authors: Dr. Jun Ye, Department of Gastroenterology, The Second Affiliated Hospital, Zhejiang University School of Medicine, 88 Jiefang Road, Hangzhou, Zhejiang 310009, China. Tel: 86-571-87784642; Fax: 86-571-87022776; E-mail: wzmcyejunedu.cn and Dr. Yuezhong Ren, Department of Endocrinology and Metabolism, The Second Affiliated Hospital, Zhejiang University School of Medicine, 88 Jiefang Road, Hangzhou, Zhejiang 310009, China. Tel: 86-571-87784642; Fax: 86-571-87022776; E-mail: renyuezedu.cn