Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(19):3435-3446. doi:10.7150/jca.26101 This issue Cite
Research Paper
A miRNA Combination as Promising Biomarker for Hepatocellular Carcinoma Diagnosis: A Study Based on Bioinformatics Analysis
Jia Ji1#, Hao Chen2#, Xiao-Ping Liu3, Yu-Hui Wang1, Chang-Liang Luo1, Wu-Wen Zhang1, Wen Xie1 ![]() , Fu-Bing Wang1
, Fu-Bing Wang1 ![]()
1. Department of Laboratory Medicine, Zhongnan Hospital of Wuhan University, Wuhan 430071, P.R. China
2. Department of Pathology, Zhongnan Hospital of Wuhan University, Wuhan 430071, P.R. China
3. Center for Evidence-Based and Translational Medicine, Zhongnan Hospital of Wuhan University, Wuhan 430071, P.R. China
# These authors contributed equally to this work.
Received 2018-3-16; Accepted 2018-6-9; Published 2018-9-8
Abstract
Background: miRNAs dysregulate in hepatocellular carcinoma (HCC), showing promise for diagnostic biomarkers which may be found through exploration of differentially expressed miRNAs when comparing HCC and normal liver tissues.
Materials and Methods: In the present research, candidate miRNAs were selected and verified using screening dataset GSE12717 and training dataset GSE10694, respectively. A miRNA combination was constructed using stepwise logistic regression analysis and validated using two datasets GSE74618 and TCGA. Target genes of miRNAs in the combination were obtained using a miRNA target gene prediction database. Functional analysis was conducted using an online tool DAVID. We also analyzed the mRNA-Seq data of project LIHC from TCGA to identify the hub target genes of the miRNAs.
Results: A miRNA combination, which is composed of hsa-miR-221 and hsa-miR-29c was defined in this study. The miRNA combination is more effective in discriminating HCC patients from normal individuals than individual miRNAs. Additionally, the combined miRNAs showed a lower misdiagnosis rate than AFP in HCC diagnosis. In terms of the functional analysis, a total of 27 target genes of hsa-miR-221 and 96 target genes of hsa-miR-29c were obtained. Among which, INSIG1 was the common target of the two miRNAs. It was also found that both previously mentioned miRNAs played important roles in the regulation of transcription, cell proliferation, and involvement in cancer-related pathways. Lastly, 2 hub target genes of hsa-miR-221 and 16 hub target genes of hsa-miR-29c were obtained.
Conclusion: We established a miRNA combination as a promising tool for HCC diagnosis, and the target genes we predicted provide possible points of penetration for researching these two miRNAs in HCC.
Keywords: Hepatocellular carcinoma, MicroRNA combination, Biomarker, Diagnose, Bioinformatics analysis, Public gene database
Introduction
Liver cancer is one of the most common digestive system tumors, and the second leading cause of cancer related death worldwide. In less developed areas, new cancer cases are expected to reach up to 21.6 million people annually by 2030[1]. Among the primary liver cancer cases, HCC accounts for approximately 80% of them [2]. Despite the advancement of detection methods and treatment, the overall prognosis of HCC patients is still sobering.
AFP as a traditional serologic marker has been used for HCC diagnosis for decades [3, 4]. And 20ng/ml was acut-off value of AFP commonly used in clinical HCC diagnosis [5]. However, AFP is not significant in all HCC cases. It is reported that, AFP levels are elevated in only 60-70% of overall HCC patients, and nonspecific increase was also observed in non-HCC diseases such as chronic hepatitis or cirrhosis [6-8]. What's more, AFP level is frequently normal in early stage HCC [9]. Thus, novel diagnostic targets with higher sensitivity and specificity are urgently needed.
MicroRNAs (miRNAs) represent RNA molecules that act in a combinatorial manner via binding to the 3' untranslated regions (UTRs) of mRNA transcripts to negatively regulate target genes. Accumulating evidence has demonstrated the significant role of miRNAs in many cancers [10-15]. With the deepening of miRNA research, miRNAs are becoming a promising biomolecule for tumor diagnosis, prognosis and therapy [16-19]. As one of the leading causes of cancer related death worldwide, HCC has been intensively studied for exploring its pathophysiology. So far, the roles of miRNA in the biological behavior of HCC, such as proliferation, metastasis, invasion, recurrence and drug resistance, have been widely explored [20-23]. Some miRNAs have been reported to act as potential biomarkers for HCC diagnosis [24-26]. However, most of previous studies focused on individual miRNAs which were less sensitive and specific for diagnosis than miRNA profiling [27]. Since miRNA profiling is increasingly used in tumor detection, it might be a promising tool for HCC detection as well.
In recent years, microarray and high-throughput sequencing technology has undergone rapid development and has showed superiority over traditional techniques [28-30]. The ability of simultaneously examining the expression of thousands of genes makes these tools useful for studying disease [29]. Nowadays, they have been increasingly applied by numerous researchers towards finding new biomarkers for diagnosis and prognosis of various diseases [31, 32], and their application in HCC research is also increasing [33-35].
Owing to information globalization, public gene databases like Gene Expression Omnibus (GEO) [36] Database, The Cancer Genome Atlas (TCGA) and ArrayExpress are more accessible and easy to use for researchers to conduct their investigation. The data in these databases sheds new light on the novel methods for the diagnosis, prognosis and treatment of HCC.
In this work, a miRNA combination consisting of hsa-miR-221 and hsa-miR-29c was established using logistic regression analysis based on gene expression data from public gene databases. The miRNA combination is superior to AFP in distinguishing HCC patients from healthy individuals. More importantly, it is able to detect AFP-negative HCC and early-stage HCC. We also predicted the target genes of hsa-miR-221 and hsa-miR-29c and found INSIG1 was the common target of two miRNAs. Target genes which have not been experimentally verified may be a breakthrough in the exploration of these two miRNAs.
Materials and Methods
Study Design
HCC is one of the leading killers among cancer patients. It has been reported that miRNAs become dysregulated as part of the pathology of HCC. This study aims to find suitable biomarkers for HCC diagnosis through exploring dysregulated miRNAs between HCC tissues and normal liver tissues. In total, 4 independent miRNA expression profile datasets and 1 mRNA expression profile dataset were downloaded from GEO and TCGA. Datasets with smaller sample size were applied in screening and identifying the biomarker, while datasets with larger sample size were used for validation. The mRNA expression profile dataset was used to identify the hub target genes. The detailed information of 5 datasets is shown in Table S1. Figure 1 shows an overview of the strategy.
Overview of the strategy
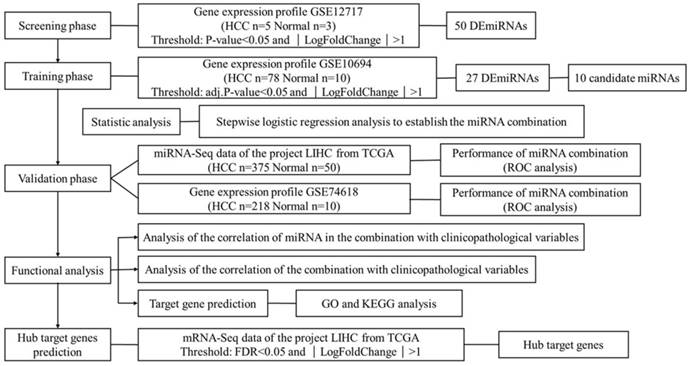
Dataset collection and miRNA screening
An original microarray dataset, GSE12717 was downloaded from NCBI-Gene Expression Omnibus database (NCBI-GEO) (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE12717), which is based on the platform of GPL7274 CapitalBio human/mouse/rat non-coding RNA microarray. Expression of 347 miRNAs in 5 human HCC tumor samples and 3 healthy liver samples were available, of which 313 human miRNAs were selected for further analysis. There were two replicates for each sample and their average was determined to conduct the differential analysis. Significantly different miRNAs were identified using R package “limma” [37] with a p-value <0.05 and |LogFoldChange|>1 were identified as the threshold values to judge differentially expressed miRNAs(DEmiRNAs).
Verification of DEmiRNAs and establishment of miRNA combination
In order to verify the DEmiRNAs of the GSE12717, we downloaded a new dataset GSE10694 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE10694), showing the expression profile of 111 miRNAs in 78 HCC tumor tissues, 78 corresponding noncancerous liver tissues and 10 normal liver tissues generated the training dataset. The miRNA expression profile of 110 human miRNAs in 78 HCC tumor tissues and 10 normal liver tissues was selected for analysis. Significantly different miRNAs were identified using R package “limma”, and adjusted p-value<0.05 and |LogFoldChange|>1 were identified as the threshold values to judge differentially expressed miRNAs. After differential analysis of the training dataset, we verified the DEmiRNAs of the screening dataset within the training dataset. Only the differentially expressed miRNAs with the same expressional trend in both datasets were selected as candidate miRNAs.
To establish an ideal diagnostic miRNA combination, a stepwise logistic regression model was constructed based on the expression profile of candidate miRNAs in the training dataset. The diagnostic performance of the miRNA combination was evaluated by the ROC (receiver operating characteristic) analysis.
Validation of the miRNA combination
To validate the diagnostic efficiency of the training combination, two datasets with different testing methods were downloaded. One was the project LIHC from The Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/) with high-throughput miRNA data of 375 HCC tissues and 50 normal liver tissues (The data was downloaded at 3rd July, 2017. Several important parameter options are listed below: the category is “Transcriptome Profiling”, the data type is “miRNA Expression Quantification”, the experimental strategy is “miRNA-Seq” and the workflow type is “BCGSC miRNA Profiling”). The data was downloaded using the GDC Data Transfer Tool from the official TCGA website. The other one was the microarray dataset GSE74618 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74618) which contains 1073 human miRNA data from 218 human HCC samples, 10 tumor-adjacent cirrhotic tissue samples, 10 healthy liver samples and 12 HCC cell lines. Before validating using the miRNA expression data of two validation datasets, a normalization phase was first conducted. Function NormalizeBetweenArrays() of R package “Limma” was used to normalize the log2-transformed expression value of GSE74618 while the miRNA high throughput data of TCGA was normalized using Trimmed Mean of M-values(TMM) method of R package “edgeR”. Because the miRNA high throughput data of TCGA was BCGSC, huge differences exist between the expressions of different miRNAs, and single genes may have a large impact on the value of the miRNA combination. Meanwhile, the miRNA expression value in the 3 datasets downloaded from GEO database was log2-transformed. Thus, we took the log of the normalized expression values before validation using miRNA high throughput data of TCGA. ROC analysis was carried out to validate the diagnostic efficiency of the miRNA combination, and the area under the ROC curve (AUC) was used as an accuracy index for evaluating the diagnostic performance.
Analysis of the correlation of miRNAs and miRNA combination with clinicopathological variables
To investigate the connection between miRNAs and miRNA combinations with important clinicopathological variables of HCC, clinical characteristics including AFP level, pathological grade, TNM stage, tumor stage, lymph-node metastasis, distant metastasis, child-pugh and liver fibrosis of 375 HCC samples was downloaded from TCGA database. Spearman's rank correlation was employed to analyze the relationship between miRNAs and miRNA combinations with clinicopathological variables. P<0.05 was considered statistically significant.
Target gene prediction and functional analysis
Target genes of the miRNAs in the combination were predicted using 3 prediction databases including Pictar (target predictions for all human microRNAs based on conservation in mammals (human, chimp, mouse, rat, dog)) [38] (http://pictar.mdc-berlin.de/), miRDB [39] (http://www.mirdb.org/), miRanda (Good mirSVR score, Conserved miRNA) [40] (http://www.microrna.org/microrna/home.do), and were further validated in the DIANA-TarBase v7.0 [41] database with strong evidence including luciferase reporter assay, WB and RT-PCR (Available online: http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/index). Only genes that were confirmed by all four databases were considered target genes of miRNAs.
Targeting to have a comprehensive investigation of the miRNAs in the diagnostic combination, GO [42] enrichment analysis and KEGG [43] analysis of their target genes were conducted by an online tool DAVID (https://david.ncifcrf.gov/, version 6.8) and P<0.05 was set as the cut-off criterion for significance.
Hub target genes prediction
After finding target genes of miRNAs in the diagnostic combination, in order to identify the chief mRNAs underlying the regulation mechanism of these miRNAs, high-throughput mRNA sequencing data from the LIHC project of TCGA database was introduced (data downloaded at 6th June, 2017). The project contains 374 human HCC samples and 50 normal liver samples, in which the expression of 60244 genes were detected. The gene annotation file was downloaded from Ensembl (GRCh38) [44] database (http://www.ensembl.org/index.html) to distinguish gene variations, and only protein-coding genes were selected for further analysis. The significantly differentially expressed mRNAs were identified using R package “edgeR” [45]. Benjamini-Hochberg method was used to control the false discovery rate (FDR). Genes with FDR<0.05 and |LogFoldChange|>1 were considered the threshold value to judge differentially expressed genes (DEGs).
Considering the negative regulation relationship between miRNA and its target genes, we compared the down-regulated DEGs with genes identified as targets of the up-regulated miRNAs, while the up-regulated DEGs were compared with the target genes of the down-regulated miRNAs. The mutual ones were considered hub target genes.
Statistical analysis
Significantly differentially expressed genes of microarray data were analyzed using R package “limma”, and significantly differentially expressed genes of high-throughput data were analyzed using R package “edgeR” of R language (version 3.2.5).
Spearman's rank correlation was employed to analyze the relationship between miRNAs and miRNA combination with clinicopathological variables. Spearman's rank correlation analysis was performed using SPSS (Version 19, SPSS Inc., Chicago, USA) and P<0.05 was considered statistically significant.
Stepwise logistic regression analysis was used to establish the miRNA combination. The independent variables are the expression of ten candidate miRNAs and the dependent variable is the sample status, that is, whether the sample is tumorous or not. The analysis method is Forward: Conditional. The stepwise logistic regression analysis was performed using SPSS (Version 19, SPSS Inc., Chicago, USA). Receiver operating characteristic (ROC) curves and area under the ROC curve (AUC) were used to evaluate the diagnostic performance of miRNA biomarkers in HCC. GraphPad Prism (version 6.0; GraphPad software) was used to generate the ROC curve.
Results
DEmiRNA analysis and screening of candidate miRNAs
Following the screening criteria, 50 DEmiRNAs (Figure 2A, Table S2) and 27 DEmiRNAs (Figure 2B, Table S3) were obtained from the screening set and training set, respectively. Among these miRNAs, 5 up-regulated miRNAs (hsa-miR-106b, hsa-miR-25, hsa-miR-93, hsa-miR-222, hsa-miR-221) and 5 down-regulated miRNAs (hsa-miR-424, hsa-miR-520f, hsa-miR-29c, hsa-miR-101, hsa-miR-422a) which were differentially expressed in HCC tissues in both screening and training datasets were selected as candidate miRNAs (Table 1).
Microarray assay of miRNAs differentially expressed in the tissues of HCC and normal liver samples. Notes: Heatmap exhibiting the differentially expressed miRNAs in HCC tissues compared to normal liver samples in dataset GSE12717(A) and GSE10694(B). Each column represents an individual sample and each row represents a single miRNA. Expression level of each miRNA in a single sample is depicted according to the color scale. Yellow represents high expression, whereas blue represents low expression. This heatmap was conducted by the “pheatmap” package of R language. p-value<0.05 and |LogFoldChange| >1 were identified as threshold value to judge differentially expressed miRNAs(DEmiRNAs) of GSE12717. adjusted p-value<0.05 and |LogFoldChange| >1 were identified as threshold value to judge differentially expressed miRNAs of GSE10694.
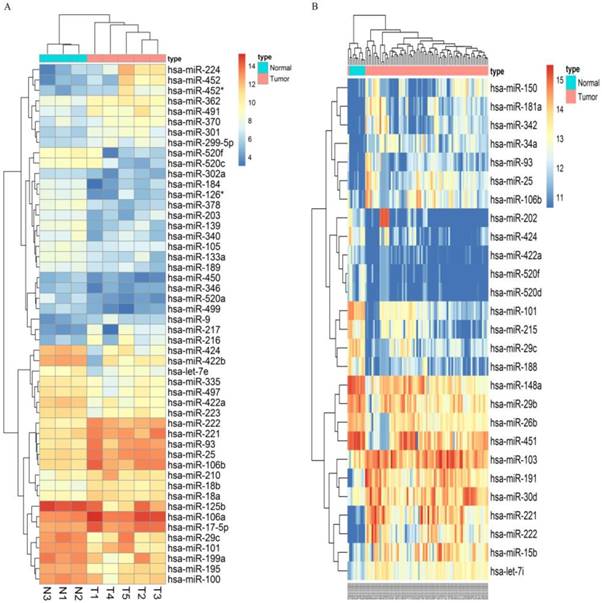
Candidate miRNAs to establish the diagnostic combination
| miRNA | Screening dataset | Training dataset | |||||
|---|---|---|---|---|---|---|---|
| Log FC | P value | type | Log FC | adj.P value | type | ||
| Hsa-miR-424 | -3.49307 | 0.005137 | Down | -1.63196 | 3.03E-07 | Down | |
| Hsa-miR-520f | -3.36844 | 0.013892 | Down | -1.19342 | 8.42E-10 | Down | |
| Hsa-miR-29c | -2.27737 | 0.03304 | Down | -1.84006 | 2.17E-08 | Down | |
| Hsa-miR-101 | -1.82946 | 0.041984 | Down | -2.10304 | 3.77E-09 | Down | |
| Hsa-miR-422a | -2.61368 | 0.001059 | Down | -1.03471 | 1.36E-09 | Down | |
| Hsa-miR-106b | 1.929812 | 0.007399 | Up | 1.270324 | 0.000267 | Up | |
| Hsa-miR-25 | 1.949887 | 0.001366 | Up | 1.690017 | 2.23E-07 | Up | |
| Hsa-miR-93 | 2.217326 | 0.003224 | Up | 1.128434 | 0.000155 | Up | |
| Hsa-miR-222 | 2.491835 | 0.000312 | Up | 2.385102 | 1.36E-09 | Up | |
| Hsa-miR-221 | 1.887786 | 0.004491 | Up | 2.665307 | 5.11E-11 | Up | |
FC: fold change
Establishing a miRNA combination for HCC diagnosis
A stepwise logistic regression model to estimate the risk of HCC was applied on the training dataset. Two miRNAs were selected to form the final combination and the established regression model was Logit P=3.76 × exp(hsa-miR-221) -3.192 × exp(hsa-miR-29c). Even though the diagnostic accuracy of single miRNA was quite satisfactory, the combination is a better indicator for HCC diagnosis due to its higher AUC and sensitivity as compared with single miRNA (AUC=0.9974, 95%CI=0.9907 to 1.000; sensitivity=98.72%, specificity=100.0%, Figure 3A). We also determined the diagnostic accuracy of two selected miRNAs in the combination, which was not as robust as the miRNA combination (Figure 3A).
ROC curves to compare the diagnostic accuracy. Notes: ROC plots for the (A) Training set; (B) Validation set GSE74618; (C) Validation set TCGA; (D) microRNA combination in discriminating HCC patients of TNM stage Ⅰ/Ⅱ from the healthy individuals.
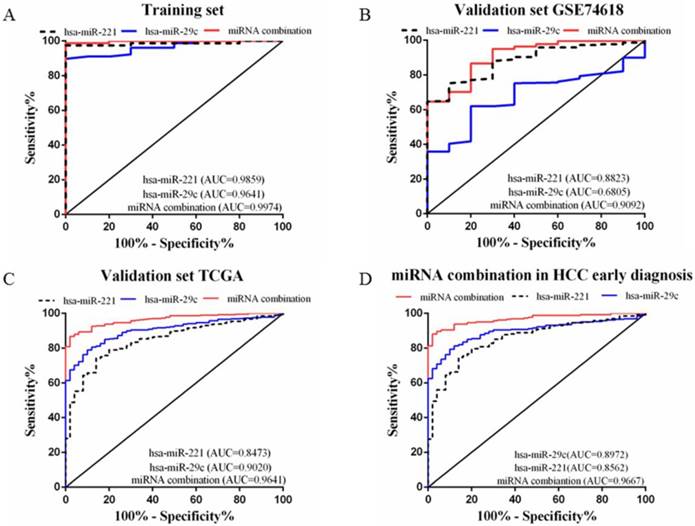
Validating the miRNA combination using two different datasets
In order to validate the diagnostic accuracy of the miRNA combination, we downloaded two datasets with miRNA expression profiles using different testing methods. In the validation set GSE74618, hsa-miR-221 showed mediocre diagnostic accuracy with the AUC=0.88 (95%CI=0.8078 to 0.9569; sensitivity=75.23%, specificity=90.0%) (Figure 3B). However, hsa-miR-29c had no value in distinguishing HCC patients from normal individuals with a P value >0.05(Figure 3B). But the miRNA combination still held a high diagnostic accuracy with the AUC>0.9 (AUC=0.91, 95%CI=0.8294 to 0.9890; sensitivity=86.7%, specificity=80.0%, Figure 3B). In the validation set TCGA, the AUC of hsa-miR-221 was 0.85 (95%CI=0.8002 to 0.8944; sensitivity=74.13%, specificity=86.0%) (Figure 3C) while the AUC of hsa-miR-29c was 0.90 (95%CI=0.8698 to 0.9341; sensitivity=78.93%, specificity=90.0%) (Figure 3C). The diagnostic accuracy of our miRNA combination was very stable with AUC=0.96 (95%CI=0.9474 to 0.9807; sensitivity=86.67%, specificity=98%, Figure 3C).
In the validation phase, the diagnostic accuracy of hsa-miR-221 ranged from 0.85 to 0.88 while the AUC of hsa-miR-29c ranged from 0.68 to 0.90, so the diagnostic accuracy of single miRNAs was much lower and less stable than the miRNA combination (AUC: 0.9020~0.9641). The sensitivity and specificity of hsa-miR-221 ranged from 74.13% to 75.23% and 86% to 90%, respectively. hsa-miR-29c had a specificity of 61.93% to 78.93% and specificity of 80% to 90%. The miRNA combination showed a much higher sensitivity (86.67% to 86.7%), suggesting a lower false-negative rate. All the diagnostic data were summarized in Table 2.
Diagnostic accuracy of miRNAs and the miRNA combination
| Group | miRNA | AUC | P Value | Sensitivity% | Specificity% | 95% CI |
|---|---|---|---|---|---|---|
| Training dataset | hsa-miR-221 | 0.9859 | <0.0001 | 97.44 | 100 | 0.9634-1.000 |
| hsa-miR-29c | 0.9641 | <0.0001 | 89.74 | 100 | 0.9274-1.000 | |
| miRNA combination | 0.9974 | <0.0001 | 98.72 | 100 | 0.9907-1.000 | |
| Validation dataset GSE74618 | hsa-miR-221 | 0.8823 | <0.0001 | 75.23 | 90 | 0.8078-0.9569 |
| hsa-miR-29c | 0.6805 | 0.05376 | 61.93 | 80 | 0.5655-0.7955 | |
| miRNA combination | 0.9092 | <0.0001 | 86.7 | 80 | 0.8294-0.9890 | |
| Validation dataset TCGA | hsa-miR-221 | 0.8473 | <0.0001 | 74.13 | 86 | 0.8002-0.8944 |
| hsa-miR-29c | 0.9020 | <0.0001 | 78.93 | 90 | 0.8698-0.9341 | |
| miRNA combination | 0.9641 | <0.0001 | 86.67 | 98 | 0.9474-0.9807 |
AUC: area under the curve; CI: confidence interval
The correlation of miRNAs and the miRNA combination with HCC clinicopathological variables
To clarify the relationship between HCC and the combination, we analyzed the correlation of miRNAs and the miRNA combination with HCC clinicopathological variables.
Our analysis showed that expression of hsa-miR-221 was positively associated with AFP level, pathological grade, and liver fibrosis, while expression of hsa-miR-29c was negatively associated with high AFP level, pathological grade and tumor stage. The high expression of miRNA combination was closely associated with high AFP level and pathological grade (Table 3).
Correlation of miRNAs and miRNA combination with clinicopathological variables in HCC of TCGA datasets
| Group | Case | hsa-miR-221 | hsa-miR-29c | miRNA combination | ||||
|---|---|---|---|---|---|---|---|---|
| r | p | r | p | r | p | |||
| AFP | ≥20 | 131 | 0.189 | 0.001 | -0.345 | 0.00 | 0.349 | 0.00 |
| <20 | 151 | |||||||
| Grade | 1-2 | 232 | 0.176 | 0.001 | -0.194 | 0.00 | 0.238 | 0.00 |
| 3-4 | 137 | |||||||
| TNM stage | Ⅰ-Ⅱ | 282 | -0.036 | 0.504 | -0.096 | 0.073 | 0.059 | 0.275 |
| Ⅲ-Ⅳ | 91 | |||||||
| Tumor stage | T0-T1 | 184 | -0.056 | 0.210 | -0.13 | 0.02 | 0.03 | 0.562 |
| T2-T4 | 188 | |||||||
| Lymph-node metastasis | N0 | 255 | -0.039 | 0.533 | -0.0102 | 0.101 | 0.256 | 0.071 |
| N1 | 4 | |||||||
| Distant metastasis | M0 | 270 | -0.012 | 0.839 | -0.003 | 0.965 | -0.017 | 0.785 |
| M1 | 4 | |||||||
| Child-pugh | A | 221 | 0.047 | 0.471 | 0.027 | 0.677 | 0.001 | 0.993 |
| B-C | 22 | |||||||
| Liver fibrosis | Yes | 140 | 0.156 | 0.022 | -0.007 | 0.915 | 0.12 | 0.077 |
| No | 76 | |||||||
AFP: alpha-fetoprotein
Even though hsa-miR-221 showed a good diagnostic accuracy, the correlation between its expression and liver cirrhosis may cause misdiagnosis when it is used as a diagnostic biomarker. As for hsa-miR-29c, the close connection with HCC staging clinicopathological variables suggested its potential usage for early HCC diagnosis. However, the volatile diagnostic accuracy impaired reliability in HCC diagnosis. In this study, we found that the expression of miRNA combination showed no significant difference between cirrhosis and non-cirrhosis groups, indicating that the diagnostic accuracy of this combination was independent of cirrhosis status. To explore its potential value in distinguishing patients with different AFP levels and pathological grades, we carried out the following analysis.
The miRNA combination outperformed AFP in HCC diagnosis
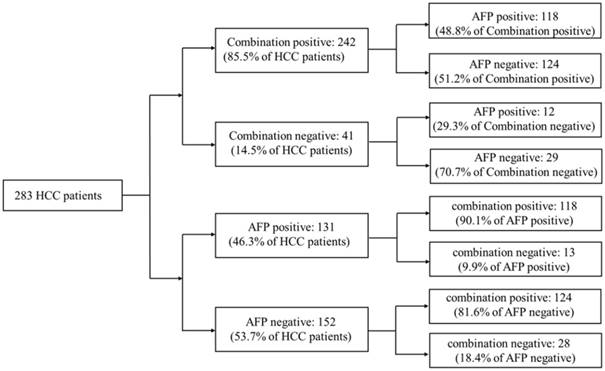
20ng/ml was a cut-off value of AFP commonly used in clinical HCC diagnosis. Aiming to compare the diagnostic accuracy of miRNA combination with AFP, AFP expression data of 283 HCC patients were downloaded from TCGA database. Compared with AFP20 (using 20ng/ml as the cut-off value of AFP in HCC diagnosis), the miRNA combination picked out 85.5% of HCC patients correctly while AFP20 could only distinguish 46.3% of the patient population. We then tested the miRNA combination and AFP levels using a tiered testing strategy, through subjecting the AFP20 negative-diagnosed results to a second review with the miRNA combination. 81.6% of the false-negative diagnosed patients could be picked out correctly. Alternatively, initial screening with the combination followed by AFP, resulted in more than 70% false-negative diagnoses. (Figure 4).
Performance of a two-tiered algorithm for HCC diagnosis incorporation the miRNA combination and an AFP cut-off of 20ng/ml.
The miRNA combination for early HCC diagnosis
Early HCC diagnosis is urgently needed for adequate treatment and study of HCC. In order to explore whether the miRNA combination was able to diagnose early stage HCC, we explored the accuracy of the miRNA combination in distinguishing HCC at TNM stage Ⅰ to Ⅱ. It showed that the miRNA combination more accurately distinguished TNM stage Ⅰ to Ⅱ HCC from normal samples (AUC=0.97), as compared with hsa-miR-221 (AUC=0.86) and hsa-miR29c (AUC=0.90) (Figure 3D).
Target genes of the miRNA combination and functional and pathway enrichment
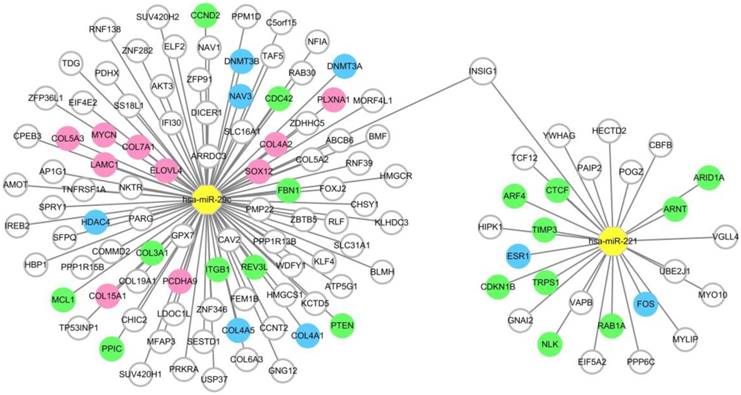
To provide precise and deep insight into the roles of the two miRNAs in the miRNA combination in HCC onset and development, target genes of hsa-miR-221 and hsa-miR-29c were predicted by the prediction database. As a result, a total of 27 target genes of hsa-miR-221 and 96 target genes of hsa-miR-29c were obtained. Among these genes, mRNA INSIG1 was found to be a common target gene for both miRNAs and was breakthrough for the study of the two miRNAs. (Figure 5, Table S4)
MiRNA-target gene network. Notes: The circle represents target gene (mRNA), the yellow octagon represents miRNA. Green circles represent the non-hub target genes which have been validated by experimental study. Blue ones represent the hub target genes which have been validated by experimental study. Pink ones represent the hub target genes which haven't been validated by experimental study. The relationship between the miRNA and gene is represented by a grey line. The network was conducted using Cytoscape_3.5.1.
When performing functional and pathway enrichment analysis of the target genes using the online website DAVID. The result of GO analysis of the hsa-miR-221 target genes was summarized in Figure S1, and the significantly enriched items for each domain were exhibited according to their p values. The hsa-miR-221 target genes were mainly enriched in the biological processes including positive regulation of transcription from the RNA polymerase II promoter, and positive regulation of cell proliferation. The functions of the products of these target genes involve protein binding, transcription factor activity, sequence-specific DNA binding and transcription factor binding. Moreover, their subcellular localization includes nucleus, cytosol, endoplasmic reticulum membrane and transcription factor complex. Their signal pathways were significantly enriched in the estrogen and cancer-related pathways.
The hsa-miR-29c target genes were mainly enriched in the biological processes including negative regulation of transcription from RNA polymerase II promoter, negative regulation of cell proliferation, collagen catabolic process, collagen catabolic process and ability to respond to drugs. The functions of the products of these target genes were protein binding, extracellular matrix organization, protein kinase binding and chromatin binding. Their subcellular localization includes nucleus, cytoplasm, endoplasmic reticulum lumen and extracellular region. Their signal pathways were enriched in focal adhesion, PI3K-Akt signaling pathway, and other cancer-related pathways. Above results were exhibited in Figure S2.
In the functional analysis, we found that both miRNAs played important roles in transcriptional regulation and cell proliferation, and both were involved in cancer-related pathways.
Hub target genes of the two miRNAs in the combination
In order to identify the chief mRNAs that participate in the regulatory mechanism of the two miRNAs in combination, the high-throughput mRNA sequencing data from TCGA was introduced. After differential expression analysis, 4895 differentially expressed genes were shown. Among them 3847 genes were up-regulated while 1048 genes were down-regulated (Figure S3).
Intersection between the differentially expressed genes and the predicted target genes of the two miRNAs was defined according to the negative regulation relationship between miRNAs and their target genes. hsa-miR-221, which was up-regulated in HCC, had two target genes FOS and ESR1 which were significantly down-regulated in HCC tissues. As for hsa-miR-29c, COL15A1, COL4A1, DNMT3A, LAMC1, COL4A2, DNMT3B, HDAC4, SOX12, PLXNA1, COL7A1, NAV3, COL5A3, COL4A5, ELOVL4, PCDHA9 and MYCN were found to be the hub target genes (Figure 5, Table S5).
Discussion
miRNAs have been found in a variety of body fluids, making it a promising non-invasive biomarker in tumor diagnosis[19, 25, 46, 47]. Indeed, many miRNAs can be potentially used in HCC diagnosis [25, 35, 48-51]. Initially HCC research focused on individual miRNAs. However, it has been proposed that miRNA panels were better for tumor diagnosis owing to their higher sensitivity and specificity [35, 52-55]. For example, Lin et al constructed a 7-miRNA classifier for HCC diagnosis [52]. The researchers compared the diagnostic accuracy between their miRNA panel and AFP, and they also indicated the usage of miRNA panels in early HCC diagnosis. Zhang et al constructed a miRNA panel consisting of 3 miRNAs using microarray technology [55]. This panel showed high diagnostic accuracy in distinguishing HCC patients from normal individuals. It also indicated the ability of the miRNA panel to detect early stage HCC and HCC with low AFP.
In this study, we discovered a miRNA combination consisting of hsa-miR-221 and hsa-miR-29c. The combination possessed a high diagnostic accuracy with the AUC>0.9 in both training set and validation set. Its sensitivity ranged from 86.67%~98.72% and the specificity ranged from 80%~100%. Compared with hsa-miR-221 or hsa-miR-29c alone, the combination is undoubtedly more suitable for the HCC diagnosis. In order to compare our miRNA combination with the miRNA combinations of Lin and Zhang, we validated the diagnostic accuracy of their miRNA panels using the two validation datasets (Figure S4). When validated using dataset GSE74618, Lin's 7-miRNA classifier had an AUC<0.9 (AUC=0.88, 95%CI=0.8177 to 0.9401; sensitivity=74.31%, specificity=100.0%), which was inferior to our miRNA combination analyzed by the same dataset. We did not validate the 7-miRNA classifier using TCGA dataset because one of its components, hsa-miR-133a, was not detected in this dataset. As for Zhang's 3-miRNA panel, miR-92-3p and miR-3126-5p were not found in two validation datasets, so validation was unsuccessful in the databases we selected. In our study, we not only screened and verified the expression of candidate miRNAs but also used two validation datasets to validate the diagnostic accuracy of the miRNA combination with different methods.
AFP has been a traditional tumor marker for HCC diagnosis for decades. However, AFP expression can be affected by other non-malignant diseases, which impairs the specificity of AFP for HCC diagnosis. A previous study showed that given a cut-off of 20 ng/ml, AFP has a sensitivity of 41%~65% and a specificity of 80%~90% in HCC detection [56]. We found that when using 20ng/ml as the cut-off, AFP's sensitivity and specificity were both lower than those of our miRNA combination. Meanwhile, the combination had the ability to pick out 90.1% of HCC patients false-negatively diagnosed by AFP which also indicate the advantage over AFP in HCC diagnosis.
HCC is one of the most common cancers worldwide. Despite the development in detection methods and treatment, overall prognosis of HCC patients still shows a sobering picture owing to late diagnosis. Hence, early diagnosis is crucial for current liver cancer research. In our study, we found that the miRNA combination was significantly associated with pathological grade. The miRNA combination showed an ideal diagnostic efficiency with the AUC=0.97in distinguishing TNM stage Ⅰ /Ⅱ HCC. Thus, our miRNA combination may be a more suitable tool for early HCC diagnosis.
We also explored the biological function of the two miRNAs in the combination. Hsa-miR-221 is one of the well-studied oncogenic miRNAs up-regulated in various cancers.[57-59] Numerous reports have indicated that hsa-miR-221 was significantly associated with tumor metastasis, invasion[60-62] and drug resistance[63, 64]. In particular, hsa-miR-221 is linked to the HCC progression from liver cirrhosis.[65] In our research, hsa-miR-221 was significantly associated with pathological grade and liver fibrosis as previously reported. In the GO analysis, the target genes of hsa-miR-221 were related to the transcriptional regulation of tumor-associated genes. The target genes of hsa-miR-221 were related to estrogen signaling and cancer-related signaling. Our data was consistent with other studies.[66]
As a tumor suppressor, hsa-miR-29c suppresses tumor metastasis and invasion,[67-70] and is down-regulated in numerous tumors. It inhibits HCC cell proliferation and induces HCC cell apoptosis.[70, 71] Consistently, our research demonstrated that low hsa-miR-29c level was significantly associated with high pathological grade and tumor stage. In the functional analysis, hsa-miR-29 target genes mainly participated in the negative regulation of transcription, and the pathways involved were related to focal adhesion, PI3K-Akt signaling, cancer signaling and cancer-associated miRNAs. In conclusion, hsa-miR-29c may play an important role in HCC formation and development.
In the functional analysis, 27 target genes of hsa-miR-221 and 96 target genes of hsa-miR-29c were identified using target gene prediction databases. Among them, 11 target genes of hsa-miR-221 and 15 target genes of hsa-miR-29c have been proved by experimental studies. Among the target genes we predicted, INSIG1 was the common target of hsa-miR-221 and hsa-miR-29c. So far as we know, we are the first to report this result. INSIG1 might be a new breakthrough for the study of hsa-miR-221 and hsa-miR-29c in HCC. To predict the hub target genes of hsa-miR-221 and hsa-miR-29c, mRNA-Seq data from TCGA was downloaded and analyzed. According to the negative regulation relationship between miRNAs and their target genes, we found the hub target genes of hsa-miR-221 were FOS and ESR1, while the hub target genes of hsa-miR-29c were COL15A1, COL4A1, DNMT3A, LAMC1, COL4A2, DNMT3B, HDAC4, SOX12, PLXNA1, COL7A1, NAV3, COL5A3, COL4A5, ELOVL4, PCDHA9 and MYCN. These genes may play significant roles in the regulatory mechanism of hsa-miR-221 and hsa-miR-29c in HCC. Some of them have been studied by other researchers, and the ones that have not been studied might lead to new clues for exploring miRNA functions in HCC.
Limitations
Limitations may exist in our study. For example, miRNA expression in different datasets may be greatly affected by detection methods, researcher skills and specimen status. In order to reduce the impacts of these factors on the results, we used multiple datasets with different detection methods to verify the diagnostic accuracy of the combination. However, more clinical samples are still needed to further substantiate the generalizability of the miRNA combination. Further investigations are necessary to confirm the correlations between the miRNAs and predicted target genes.
Conclusion
In conclusion, we constructed a miRNA combination consisting of hsa-miR-221 and hsa-miR-29c. This miRNA combination possessed high diagnostic accuracy in HCC diagnosis. It outperformed AFP in HCC diagnosis and had the ability to identify AFP-negative hepatocellular carcinoma and early-stage HCC patients. Two members of the combination have been proved to have a strong connection with the occurrence and development of HCC. And target genes obtained in the functional analysis phase may provide a new research direction for the exploration of the miRNA in HCC development and tumorigenesis. Thus, our study indicates that the miRNA combination could be a promising biomarker for HCC diagnosis.
Abbreviations
HCC: Hepatocellular Carcinoma; TCGA: The Cancer Genome Atlas; GEO: Gene Expression Omnibus; AFP: alpha-fetoprotein; DEmiRNAs: differentially expressed miRNAs; DEGs: differentially expressed genes; ROC: receiver operating characteristic; AUC: area under the ROC curve; FDR: false discovery rate; GO: Gene ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by Applied Basic Research Program of Science and Technology Bureau Foundation of Wuhan (No. 2016060101010054,) and National Natural Science Foundation of China (grant No. 81672114). This work was also funded by Science and Technology Innovation Fostering Foundation of Zhongnan Hospital of Wuhan University (cxpy20160025) and Wuhan City health and family planning medical talented youth development project. The authors report no conflicts of interest in this work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Soerjomataram I. The Changing Global Burden of Cancer: Transitions in Human Development and Implications for Cancer Prevention and Control. In: (ed.) Gelband H, Jha P, Sankaranarayanan R, Horton S. Cancer: Disease Control Priorities, Third Edition (Volume 3). Washington (DC): The International Bank for Reconstruction and Development / The World Bank (c) 2015 International Bank for Reconstruction and Development / The World Bank. 2015
2. Bosch FX, Ribes J, Diaz M, Cleries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5-s16
3. Wong GL, Chan HL, Tse YK, Chan HY, Tse CH, Lo AO. et al. On-treatment alpha-fetoprotein is a specific tumor marker for hepatocellular carcinoma in patients with chronic hepatitis B receiving entecavir. Hepatology. 2014;59:986-95
4. Leandro G, Zizzari S, Fabris C, Basso D, Elba S, Del Favero G. et al. Do CA 19-9 and TPA play a minor role as compared to AFP in diagnosing primary hepatocellular carcinoma? Oncology. 1989;46:381-5
5. Durazo FA, Blatt LM, Corey WG, Lin JH, Han S, Saab S. et al. Des-gamma-carboxyprothrombin, alpha-fetoprotein and AFP-L3 in patients with chronic hepatitis, cirrhosis and hepatocellular carcinoma. Journal of gastroenterology and hepatology. 2008;23:1541-8
6. Akeyama T, Koyama T, Kamada T. Alpha-fetoprotein in acute viral hepatitis. The New England journal of medicine. 1972;287:989
7. Di Bisceglie AM, Hoofnagle JH. Elevations in serum alpha-fetoprotein levels in patients with chronic hepatitis B. Cancer. 1989;64:2117-20
8. Taketa K. Alpha-fetoprotein: reevaluation in hepatology. Hepatology. 1990;12:1420-32
9. Chen DS, Sung JL, Sheu JC, Lai MY, How SW, Hsu HC. et al. Serum alpha-fetoprotein in the early stage of human hepatocellular carcinoma. Gastroenterology. 1984;86:1404-9
10. Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787-98
11. Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321-33
12. Monzon F. MicroRNA Profiling Reveals Distinct Signatures in B Cell Chronic Lymphocytic LeukemiasCalin GA, Liu CG, Sevignani C, et al (Thomas Jefferson Univ, Philadelphia; Univ of Ferrara, Italy; Istituto Superiore Sanita, Rome; et al) Proc Natl Acad Sci U S A 101:11. Yearbook of Pathology & Laboratory Medicine. 2006:235-6
13. Carleton M, Cleary MA, Linsley PS. MicroRNAs and cell cycle regulation. Cell Cycle. 2007;6:2127-32
14. Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S. et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proceedings of the National Academy of Sciences. 2004;101:2999-3004
15. Farazi TA, Spitzer JI, Morozov P, Tuschl T. miRNAs in human cancer. Journal of Pathology. 2011;223:102-15
16. Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends in Molecular Medicine. 2014;20:460-9
17. Li Z, Rana TM. Therapeutic targeting of microRNAs: current status and future challenges. Nature Reviews Drug Discovery. 2014;13:622
18. Schmidt Marco F. Drug target miRNAs: chances and challenges. Trends in Biotechnology. 2014;32:578-85
19. Vecchione A, Belletti B, Lovat F, Volinia S, Chiappetta G, Giglio S. et al. A microRNA signature defines chemoresistance in ovarian cancer through modulation of angiogenesis. Pnas. 2013;110:9845
20. Chang RM, Yang H, Fang F, Xu JF, Yang LY. MicroRNA-331-3p promotes proliferation and metastasis of hepatocellular carcinoma by targeting PH domain and leucine-rich repeat protein phosphatase. Hepatology. 2014;60:1251-63
21. Fang F, Chang RM, Yu L, Lei X, Xiao S, Yang H. et al. MicroRNA-188-5p suppresses tumor cell proliferation and metastasis by directly targeting FGF5 in hepatocellular carcinoma. Journal of hepatology. 2015;63:874-85
22. Wong CM, Wong CC, Lee JM, Fan DN, Au SL, Ng IO. Sequential alterations of microRNA expression in hepatocellular carcinoma development and venous metastasis. Hepatology. 2012;55:1453-61
23. Xia H, Ooi LL, Hui KM. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology. 2013;58:629-41
24. Giordano S, Columbano A. MicroRNAs: new tools for diagnosis, prognosis, and therapy in hepatocellular carcinoma? Hepatology. 2013;57:840-7
25. Wen Y, Han J, Chen J, Dong J, Xia Y, Liu J. et al. Plasma miRNAs as early biomarkers for detecting hepatocellular carcinoma. Int J Cancer. 2015;137:1679-90
26. Sohn W, Kim J, Kang SH, Yang SR, Cho JY, Cho HC. et al. Serum exosomal microRNAs as novel biomarkers for hepatocellular carcinoma. Experimental & molecular medicine. 2015;47:e184
27. Luo J, Zhao Q, Zhang W, Zhang Z, Gao J, Zhang C. et al. A novel panel of microRNAs provides a sensitive and specific tool for the diagnosis of breast cancer. Molecular medicine reports. 2014;10:785-91
28. Derisi J. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nature Genetics. 1996;14:457-60
29. Raghavachari N, Barb J, Yang Y, Liu P, Woodhouse K, Levy D. et al. A systematic comparison and evaluation of high density exon arrays and RNA-seq technology used to unravel the peripheral blood transcriptome of sickle cell disease. BMC medical genomics. 2012;5:28
30. Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome research. 2008;18:1509-17
31. Russo G, Zegar C, Giordano A. Advantages and limitations of microarray technology in human cancer. Oncogene. 2003;22:6497-507
32. Allantaz F, Chaussabel D, Stichweh D, Bennett L, Allman W, Mejias A. et al. Blood leukocyte microarrays to diagnose systemic onset juvenile idiopathic arthritis and follow the response to IL-1 blockade. The Journal of experimental medicine. 2007;204:2131-44
33. Wen Y, Han J, Chen J, Dong J, Xia Y, Liu J. et al. Plasma miRNAs as early biomarkers for detecting hepatocellular carcinoma. International Journal of Cancer. 2015;137:1679
34. Zhou J, Yu L, Gao X, Hu J, Wang J, Dai Z. et al. Plasma MicroRNA Panel to Diagnose Hepatitis B Virus–Related Hepatocellular Carcinoma. Journal of Clinical Oncology Official Journal of the American Society of Clinical Oncology. 2011;29:4781
35. Zhou J, Yu L, Gao X, Hu J, Wang J, Dai Z. et al. Plasma microRNA panel to diagnose hepatitis B virus-related hepatocellular carcinoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2011;29:4781-8
36. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M. et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic acids research. 2013;41:D991-5
37. Gentleman R, Carey VJ, Huber W. Bioinformatics and computational biology solutions using R and Bioconductor. Springer Science+Business Media. 2005
38. Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ. et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495-500
39. Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic acids research. 2015;43:D146-52
40. Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biology. 2010;11:R90
41. Vlachos IS, Paraskevopoulou MD, Karagkouni D, Georgakilas G, Vergoulis T, Kanellos I. et al. DIANA-TarBase v7.0: indexing more than half a million experimentally supported miRNA:mRNA interactions. Nucleic acids research. 2015;43:D153-9
42. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25-9
43. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic acids research. 1999;27:29-34
44. Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S. et al. Ensembl 2015. Nucleic acids research. 2015;43:D662-9
45. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139-40
46. Cheng G. Circulating miRNAs: roles in cancer diagnosis, prognosis and therapy. Advanced drug delivery reviews. 2015;81:75-93
47. Zhang Y, Zhang D, Wang F, Xu D, Guo Y, Cui W. Serum miRNAs panel (miR-16-2*, miR-195, miR-2861, miR-497) as novel non-invasive biomarkers for detection of cervical cancer. Scientific reports. 2015;5:17942
48. Liao Q, Han P, Huang Y, Wu Z, Chen Q, Li S. et al. Potential Role of Circulating microRNA-21 for Hepatocellular Carcinoma Diagnosis: A Meta-Analysis. PLoS One. 2015;10:e0130677
49. Chen L, Chu F, Cao Y, Shao J, Wang F. Serum miR-182 and miR-331-3p as diagnostic and prognostic markers in patients with hepatocellular carcinoma. Tumour Biol. 2015;36:7439-47
50. Bhattacharya S, Steele R, Shrivastava S, Chakraborty S, Di Bisceglie AM, Ray RB. Serum miR-30e and miR-223 as Novel Noninvasive Biomarkers for Hepatocellular Carcinoma. The American journal of pathology. 2016;186:242-7
51. Hung CH, Hu TH, Lu SN, Kuo FY, Chen CH, Wang JH. et al. Circulating microRNAs as biomarkers for diagnosis of early hepatocellular carcinoma associated with hepatitis B virus. Int J Cancer. 2016;138:714-20
52. Lin XJ, Chong Y, Guo ZW, Xie C, Yang XJ, Zhang Q. et al. A serum microRNA classifier for early detection of hepatocellular carcinoma: a multicentre, retrospective, longitudinal biomarker identification study with a nested case-control study. The Lancet Oncology. 2015;16:804-15
53. Ali HEA, Abdel Hameed R, Effat H, Ahmed EK, Atef AA, Sharawi SK. et al. Circulating microRNAs panel as a diagnostic tool for discrimination of HCV-associated hepatocellular carcinoma. Clin Res Hepatol Gastroenterol. 2017;41:e51-e62
54. Wang C, Ding M, Xia M, Chen S, Van Le A, Soto-Gil R. et al. A Five-miRNA Panel Identified From a Multicentric Case-control Study Serves as a Novel Diagnostic Tool for Ethnically Diverse Non-small-cell Lung Cancer Patients. EBioMedicine. 2015;2:1377-85
55. Zhang Y, Li T, Qiu Y, Zhang T, Guo P, Ma X. et al. Serum microRNA panel for early diagnosis of the onset of hepatocellular carcinoma. Medicine (Baltimore). 2017;96:e5642
56. Daniele B, Bencivenga A, Megna AS, Tinessa V. Alpha-fetoprotein and ultrasonography screening for hepatocellular carcinoma. Gastroenterology. 2004;127:S108-12
57. Kneitz B, Krebs M, Kalogirou C, Schubert M, Joniau S, van Poppel H. et al. Survival in patients with high-risk prostate cancer is predicted by miR-221, which regulates proliferation, apoptosis, and invasion of prostate cancer cells by inhibiting IRF2 and SOCS3. Cancer research. 2014;74:2591-603
58. Falkenberg N, Anastasov N, Rappl K, Braselmann H, Auer G, Walch A. et al. MiR-221/-222 differentiate prognostic groups in advanced breast cancers and influence cell invasion. British journal of cancer. 2013;109:2714-23
59. Quintavalle C, Garofalo M, Zanca C, Romano G, Iaboni M, del Basso De Caro M. et al. miR-221/222 overexpession in human glioblastoma increases invasiveness by targeting the protein phosphate PTPmu. Oncogene. 2012;31:858-68
60. Stinson S, Lackner MR, Adai AT, Yu N, Kim HJ, O'Brien C. et al. TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Science signaling. 2011;4:ra41
61. Qin J, Luo M. MicroRNA-221 promotes colorectal cancer cell invasion and metastasis by targeting RECK. FEBS letters. 2014;588:99-104
62. Rong M, Chen G, Dang Y. Increased miR-221 expression in hepatocellular carcinoma tissues and its role in enhancing cell growth and inhibiting apoptosis in vitro. BMC cancer. 2013;13:21
63. Wei Y, Lai X, Yu S, Chen S, Ma Y, Zhang Y. et al. Exosomal miR-221/222 enhances tamoxifen resistance in recipient ER-positive breast cancer cells. Breast cancer research and treatment. 2014;147:423-31
64. Wang Y, Zhao Y, Herbst A, Kalinski T, Qin J, Wang X. et al. miR-221 Mediates Chemoresistance of Esophageal Adenocarcinoma by Direct Targeting of DKK2 Expression. Annals of surgery. 2016;264:804-14
65. Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B. et al. miR-221 overexpression contributes to liver tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:264-9
66. Rao X, Di Leva G, Li M, Fang F, Devlin C, Hartman-Frey C. et al. MicroRNA-221/222 confers breast cancer fulvestrant resistance by regulating multiple signaling pathways. Oncogene. 2011;30:1082-97
67. Zhang JX, Mai SJ, Huang XX, Wang FW, Liao YJ, Lin MC. et al. MiR-29c mediates epithelial-to-mesenchymal transition in human colorectal carcinoma metastasis via PTP4A and GNA13 regulation of beta-catenin signaling. Annals of oncology: official journal of the European Society for Medical Oncology. 2014;25:2196-204
68. Zou Y, Li J, Chen Z, Li X, Zheng S, Yi D. et al. miR-29c suppresses pancreatic cancer liver metastasis in an orthotopic implantation model in nude mice and affects survival in pancreatic cancer patients. Carcinogenesis. 2015;36:676-84
69. Liu N, Tang LL, Sun Y, Cui RX, Wang HY, Huang BJ. et al. MiR-29c suppresses invasion and metastasis by targeting TIAM1 in nasopharyngeal carcinoma. Cancer Lett. 2013;329:181-8
70. Bae HJ, Noh JH, Kim JK, Eun JW, Jung KH, Kim MG. et al. MicroRNA-29c functions as a tumor suppressor by direct targeting oncogenic SIRT1 in hepatocellular carcinoma. Oncogene. 2014;33:2557-67
71. Wang CM, Wang Y, Fan CG, Xu FF, Sun WS, Liu YG. et al. miR-29c targets TNFAIP3, inhibits cell proliferation and induces apoptosis in hepatitis B virus-related hepatocellular carcinoma. Biochemical and biophysical research communications. 2011;411:586-92
Author contact
![]() Corresponding authors: Fu-Bing Wang, Department of Laboratory Medicine, Zhongnan Hospital of Wuhan University, No 169 Donghu Road, Wuchang District, Wuhan 430071, P.R. China. Email address: wfb20042002com; Tel: +86-27-67813517; Fax: +86-27-67813128; Wen Xie, whxw007com.
Corresponding authors: Fu-Bing Wang, Department of Laboratory Medicine, Zhongnan Hospital of Wuhan University, No 169 Donghu Road, Wuchang District, Wuhan 430071, P.R. China. Email address: wfb20042002com; Tel: +86-27-67813517; Fax: +86-27-67813128; Wen Xie, whxw007com.