Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(20):3802-3811. doi:10.7150/jca.29233 This issue Cite
Research Paper
Oestrogen Inhibits VEGF Expression And Angiogenesis In Triple-Negative Breast Cancer By Activating GPER-1
Chen Wang, Jiehao Li, Shuang Ye, Yaxing Zhang, Ping Li, Ling Wang, Ting-huai Wang ![]()
Department of Physiology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou, People's Republic of China
Received 2018-8-14; Accepted 2018-8-18; Published 2018-10-1
Abstract
Triple-negative breast cancer (TNBC) is the most malignant type of breast cancer with ample vascularisation and high vascular endothelial growth factor (VEGF) expression. The sex steroid hormone oestrogen is involved in several cellular activities associated with TNBC regulation. However, the role of oestrogen in VEGF expression and angiogenesis in TNBC remains unclear. In this study, we found that treatment with 17β-oestradiol (E2) inhibited VEGF mRNA and protein expression in the TNBC cell lines MDA-MB-468 and MDA-MB-436. To further elaborate on the phenomenon of E2-regulated angiogenesis, we showed that conditioned medium from the TNBC cell line MDA-MB-468 treated with E2 inhibits the tube formation ability of human umbilical vein endothelial cells (HUVECs). Additionally, the G-protein-coupled oestrogen receptor-1 (GPER-1)-specific agonist G-1 has a function similar to that of E2. While G-15, the selective antagonist of GPER-1, notably reversed the inhibitory effects of E2 and G-1 on VEGF expression and tube formation, suggesting that GPER-1 is involved in the E2-induced angiogenesis suppression in TNBC cells. Moreover, E2 inhibited in vivo tumour growth and angiogenesis and reduced the expression levels of VEGF, NF-κB/p65, STAT3, and the endothelial marker CD34 in MDA-MB-468 xenograft tumours. Our findings provide important evidence that E2 can inhibit VEGF expression and angiogenesis in TNBC by activating GPER-1, offering additional insight into tumour angiogenesis and targets for drug intervention in TNBC.
Keywords: oestrogen, TNBC, angiogenesis, VEGF, GPER-1
Introduction
Breast cancer is a hormone-dependent tumour that is closely related to endocrine function, and it is the most common cause of cancer death in women [1]. Based on the expression of oestrogen receptors (ERs), progesterone receptors (PRs) and human epidermal growth factor receptor 2 (HER-2), breast cancer is divided into the following types: hormone receptor-positive breast cancer; HER-2-overexpressing breast cancer and triple-negative breast cancer (TNBC) [2]. TNBC is characterized by a lack of ER, PR and HER-2 expression [3]. Although TNBC comprises only 15% of all the breast cancer molecular subtypes, it is the most malignant, with a higher risk of relapse and metastasis than the other subtypes [4]. Recently, significant progress has been made in the treatment of HER-2-overexpressing and ER-positive breast cancer [5-8]; however, TNBC still lacks effective treatment strategies [4, 9].
Oestrogen, a sex steroid hormone that normally activates the classical ER and induces ER binding to the oestrogen response element located in the promoters of target genes, regulates multiple biological functions [10]. It is reported that prolonged exposure to oestrogen can lead to breast cancer [11]. Since TNBC lacks ER and PR, it is generally accepted that oestrogen is not directly involved in regulating TNBC development. Recently, researchers found that oestrogen treatment significantly inhibits the proliferation and tumour growth of the TNBC cell line MDA-MB-231[12]. In addition, several studies have demonstrated that G-protein-coupled oestrogen receptor-1 (GPER-1), which was established as a new ER [13] that mediates oestrogenic signalling in various normal and malignant cell types [14-18], is closely related to tumourigenesis [19] and recurrence [20] in TNBC patients and has a significant effect on oestrogen-induced proliferation of TNBC cell lines [21-23]. These findings suggest that there might be a direct regulatory relationship between oestrogen and TNBC development. However, the role of oestrogen in TNBC angiogenesis remains elusive.
Tumour angiogenesis is an essential step in tumour development, and previous data support its central role in breast cancer growth and metastasis [24]. Vascular endothelial growth factor-A (VEGF-A, also VEGF) is an important member of the VEGF family and has a strong pro-angiogenic function, and thus, it has been assessed in both clinical and experimental studies [24]. In this regard, oestrogen is reported to induce VEGF expression in endometrial epithelial cells [25], colon cancer [26], the central nervous system [27, 28] and breast cancer [29, 30]. TNBC is characterized by high vascularisation and exhibits significantly higher VEGF expression than other subtypes [31]. In cases where targeted therapy agents failed to substantially improve the survival of TNBC patients [32], a VEGF inhibitor, bevacizumab, can effectively inhibit tumour angiogenesis and has shown clinical efficacy in patients [33, 34]. Therefore, we wondered whether control of VEGF expression and angiogenesis in TNBC may be associated with an improvement in patient prognosis after treatment.
A previous study indicated that GPER-1 mediated the oestrogen-induced expression and function of VEGF in HER-2-overexpressing breast cancer [35]. However, whether this association exists in TNBC has not been evaluated. Consequently, in the present study, we aimed to explore the role of oestrogen in VEGF expression and angiogenesis in TNBC in vitro and in vivo. Furthermore, we investigated the potential role of GPER-1 in the relationship between oestrogen and TNBC angiogenesis. Additionally, we studied the influence of nuclear factor kappa B (NF-κB)/p65 and signal transducer and activator of transcription 3 (STAT3) on TNBC angiogenesis in response to oestrogen in vivo.
Materials and Methods
Reagents
17β-oestradiol (E2) was obtained from Sigma-Aldrich (St. Louis, MO, USA). G-1 and G-15 were obtained from Tocris Bioscience (Ellisville, MO, USA).
Cell culture
MCF-7, SKBR3, MDA-MB-468 and MDA-MB-436 cells were obtained from American Type Culture Collection (Manassas, VA, USA). HUVECs were cultured as previously described [36]. The cell lines were cultured in phenol red-free Dulbecco's modified Eagle's medium (DMEM)/F12 supplemented with 10% foetal bovine serum, 100 U/ml penicillin and 100 µg/ml streptomycin at 5% CO2 and 37°C.
Western blot analysis
Cell lysis buffer containing Tris-HCl (pH 6.8), glycerol, SDS, NaF, Na3VO4 and PMSF was added to the cells to extract proteins, which were then resolved by SDS-PAGE and transferred to PVDF membranes (Millipore, Temecula, CA, USA). The membranes with protein were washed with TBST, blocked with 5% BSA for 30 minutes at room temperature and examined with primary antibodies against GPER-1 (1:1000 dilution, Abcam, Cambridge, MA, USA), ERα (1:1000 dilution, Cell Signalling Technology, Danvers, MA, USA) and β-actin (1:1000 dilution, Santa Cruz Biotechnology Inc., USA) at 4°C overnight. The membranes were then washed three times with TBST for 10 minutes, incubated with peroxidase-conjugated goat anti-mouse or anti-rabbit (1:2000 dilution, Cell Signalling Technology) IgG for 1 h at room temperature and washed three more times with TBST. Labelled proteins were detected with enhanced chemiluminescence.
Quantitative real-time PCR (qRT-PCR)
TRIzol reagent (Invitrogen, Carlsbad, CA) was used to extract total RNA. RNA quality was determined with a NanoDrop 2000. cDNA synthesis was performed in a reaction mix containing 500 ng/µl total RNA, 2 µl of 5*PrimScrip RT Mix (Takara, Japan) and RNase-Free dH2O. qRT-PCR was performed with an iCycler using SYBR Premix Ex Taq II (Takara, Japan), PCR Forward Primer, PCR Reverse Primer, cDNA and RNase-free dH2O. This part of the experimental procedure was performed on ice. Reactions were run on a CFX-96 real-time PCR system (Bio-Rad Laboratories Inc.). Primer Express version 2.0 software (Applied Biosystems Inc.) was used to design the primers. For VEGF and β-Actin, the primers were as follows: VEGF (forward (fwd)) 5'-TGCAGATTATGCGGATCAAACC-3' and (reverse (rev)) 5'-TGCATTCACATTTGTTGTGCTGTAG-3'; β-Actin, (fwd) 5'-AACTTCTCCTCGAAACCCTCCA and (rev) 5'-GTCGTGTACTGTGAGGTGCATACTT-3'. The housekeeping gene β-Actin acted as an internal reference.
Enzyme-linked immunosorbent assay (ELISA)
VEGF levels in serum and the supernatants of cultured TNBC cells were measured using an ELISA kit according to the manufacturer's instructions (R&D Systems, USA).
Immunofluorescence microscopy
Coverslips were transferred into a six-well plate. After trypsin digestion of TNBC cells, 2 ml of cells was added per well. After 18 h of drug treatment, the culture medium was discarded, and the cells were washed twice with precooled (4°C) PBS and fixed with 1 ml of 4% paraformaldehyde for 30 minutes. Triton X-100 (1 ml of 0.1% in PBS) was added for 5 minutes to perforate the cells, which were then incubated in 1% BSA (PBS preparation) for 30 minutes. The cells were then incubated with an antibody against VEGF (Abcam, Cambridge, MA, USA), washed three times with PBST and incubated with the secondary antibody at room temperature for 50 minutes. Nuclei were counterstained with 1:1000 4′-6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific). Cells were observed and imaged with an Olympus BX41 microscope and an Olympus DP70 digital camera.
Conditioned Medium
MDA-MB-468 cells were cultured in the aforementioned medium, washed with PBS and then cultured in serum-free medium for 24 h. Then, the cells were treated for 18 h with E2 and G-1. Subsequently, the cell culture medium was collected and centrifuged at 3500 rpm for 5 minutes, and the supernatant was extracted as the conditioned medium.
Tube Formation Assay
The day before the experiment, growth factor-reduced Matrigel (BD Biosciences, Bedford, MA, USA) was placed at 4°C overnight. On the day of the experiment, 150 μl of Matrigel was added per well to a 48-well plate, avoiding air bubbles. The plate was shaken so that the gel spread around the bottom of the well and then placed at 4°C on a balance for 15 minutes, after which it was transferred to an incubator for 30 minutes for curing. The starved HUVECs were digested, counted, resuspended in conditioned medium from TNBC cells and seeded onto the Matrigel at a density of 2-5 × 104 cells/well. The plate was placed in an incubator for 4-8 h before images were captured. Tube formation was quantified using the NIH software ImageJ.
Animal experiments
Four-week-old female BALB/c (nu/nu) mice were obtained from Sun Yat-sen University. All animals were bred and housed at the Animal Experiment Center of Zhongshan School of Medicine, Sun Yat-sen University. The care and use of all animals followed the guidelines for laboratory animals, and the protocol was approved by Sun Yat-sen University Animal Policy and Welfare Committee. The mice were anaesthetized and underwent a bilateral ovariectomy (OVX) through a 1-cm dorsal incision. Meanwhile, E2 pellets (0.72 mg, 60-day release, Innovative Research) and control pellets (Innovative Research) were implanted subcutaneously. After surgery, the mice were allowed to recover for 1 week, and tumours were established by injecting MDA-MB-468 cells (5 × 106 in 100 ml of Matrigel mixture) into the mammary fat pads of the mice. Callipers were used to measure the size of tumours every 5 days, and tumour volumes were calculated using the formula TV = length × [width] 2 × 0.5. Forty-five days later, the mice were sacrificed, and the tumour specimens were harvested for further study.
Determination of serum oestrogen in vivo
Orbital blood, placed in a frozen centrifuge tube, was allowed to stand at room temperature for 2 h, after which it was centrifuged at 3500 rpm for 15 minutes at 4°C, and the supernatant was collected for measurement. Oestrogen levels in serum were assessed using chemiluminescence techniques (Abbott Laboratories i2000 Automatic Chemiluminescence Immune Analyser).
Immunohistochemistry (IHC)
Tumour samples were fixed in 4% paraformaldehyde and embedded in paraffin. Cross-sections with a thickness of 4 μm were prepared using a microtome. One hour after baking at 60°C, tumour sections were deparaffinised and rehydrated. Antigen retrieval was performed after the samples were washed three times with PBS and cooled for 1 h at room temperature. Triton X-100 (0.1%) was added for 5 minutes to perforate membranes, followed by incubation with goat serum for 15 minutes and incubation with a primary antibody overnight at 4°C. Samples were rewarmed for 45 minutes the next day, followed by incubation with the secondary antibody for 1 h at room temperature. Samples were then washed and immersed in PBS, after which the nuclei were counterstained with haematoxylin. The following primary antibodies were used: anti-CD34 (1:100 dilution, Abcam, Cambridge, MA, USA), anti-VEGF (1:100 dilution, Abcam), anti-NF-κB/p65 (1:100 dilution, Abcam) and anti-STAT3 (1:100 dilution, Abcam) antibodies. To investigate the microvessel density (MVD), we scanned the hot spots at low magnification and counted the blood vessels in a field of view at 400 × magnification. Images were captured using a Nikon microscope (Nikon, Japan).
Statistical analyses
The data, which were attained from experiments repeated at least three times unless otherwise specified, are presented as the means ± SD. Two-tailed unpaired Student's t-tests and one-way ANOVA followed by Bonferroni tests were performed for two groups and multiple comparisons, respectively. Statistical analysis was performed using SPSS 16.0 software. A two-sided p-value < 0.05 was considered statistically significant.
Results
E2 inhibits VEGF expression in TNBC cell lines
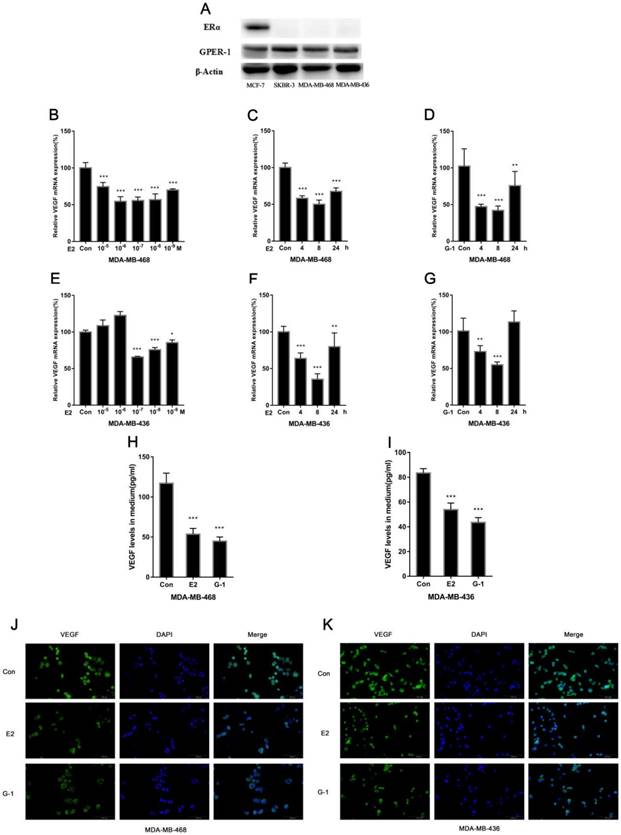
We investigated the effect of E2 on VEGF expression in the TNBC cell lines MDA-MB-468 and MDA-MB-436. Cells were treated with physiological concentrations of E2 (10-5-10-9 M) for 8 h, and we found that E2 treatment significantly decreased VEGF mRNA expression (Fig. 1B, 1E). The effect was relatively obvious at 10-6 M E2 in MDA-MB-468 cells and 10-7 M in MDA-MB-436 cells. Furthermore, E2 decreased VEGF mRNA expression in cells over time (Fig. 1C, 1F). As mentioned earlier, GPER-1 is highly expressed in TNBC; we used the ERα-positive breast cancer cell line MCF-7 and the ERα-negative breast cancer cell line SKBR3 as positive controls to compare the GPER-1 expression in the two TNBC cell lines. We found that GPER-1 was highly expressed and that ERα was not expressed in these cells (Fig. 1A). In addition, the GPER-1-specific agonist G-1 (1 μM) displayed effects similar to those of E2 in both cell lines (Fig. 1D, 1G). We quantitatively detected the secretion of VEGF by ELISA, and the results showed that E2 and G-1 significantly decreased VEGF expression (Fig. 1H, 1I), which was confirmed by immunofluorescence staining (Fig. 1J, 1K). Collectively, our data suggest that E2 can suppress VEGF expression in TNBC cell lines.
E2 inhibits VEGF expression in TNBC cell lines. (A) ERα and GPER-1 expression levels were analysed by western blot. Actin was used for normalization. MDA-MB-468 cells were treated with different concentrations (10-5-10-9 M) of E2 for 8 h (B) or with 10-6 M E2 (C) or 1 μM G-1 for various time periods (4 h, 8 h, 24 h) (D), and VEGF mRNA was measured by qRT-PCR. MDA-MB-436 cells were treated with different concentrations (10-5-10-9 M) of E2 for 8 h (E) or with 10-7 M E2 (F) or 1 μM G-1 for various time periods (4 h, 8 h, 24 h) (G), and VEGF mRNA was measured by qRT-PCR. (H) MDA-MB-468 cells were treated with 10-6 M E2 or 1 μM G-1 for 18 h, and VEGF secretion in the medium was detected via ELISA. (I) MDA-MB-436 cells were treated with 10-7 M E2 or 1 μM G-1 for 18 h, and VEGF secretion in the medium was detected via ELISA. (J) MDA-MB-468 cells were treated with 10-6 M E2 or 1 μM G-1 for 18 h, and (K) MDA-MB-436 cells were treated with 10-7 M E2 or 1 μM G-1 for 18 h; VEGF expression was detected by immunofluorescence staining. The data are presented as the mean ± SD of three independent experiments. *p < 0.05 compared with control, **p < 0.01 compared with control, and ***p < 0.001 compared with control.
E2 inhibits tube formation in TNBC cells
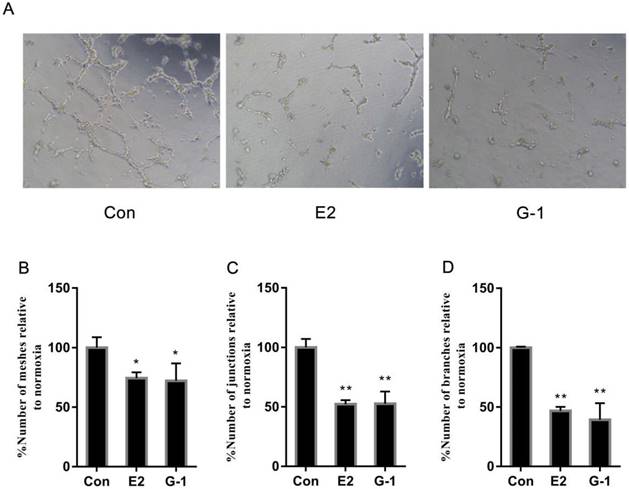
Herein, having established that E2 initiated the downregulation of VEGF, we then further analysed its effects on the angiogenesis of TNBC cells in vitro using tube formation assays. We cultured HUVECs in conditioned medium from MDA-MB-468 cells that were previously treated with E2 (10-6 M) and G-1 (1 μM) to construct an angiogenesis model. Unlike in the control group, we found that a network of tubules could not be generated in HUVECs grown in medium from MDA-MB-468 cells treated with E2 and G-1 (Fig. 2A). Further supporting this phenomenon, the number of meshes, junctions and branches that were recapitulated (Fig. 2B, 2C, 2D) indicated that E2 could suppress new blood vessel formation in TNBC cells.
E2 inhibits tube formation in TNBC cells. (A) Tube formation was evaluated in HUVECs cultured for 6 h in medium that was collected from the TNBC cell line MDA-MB-468 treated with vehicle, 10-6 M E2 or 1 μM G-1 for 18 h. Quantification of the number of meshes (B), the number of branching junctions (C) and the number of branches (D) observed in HUVECs. The data are presented as the mean ± SD of three independent experiments. *p < 0.05 compared with control, and **p < 0.01 compared with control.
GPER-1 is involved in the inhibition of VEGF expression in TNBC cell lines by E2
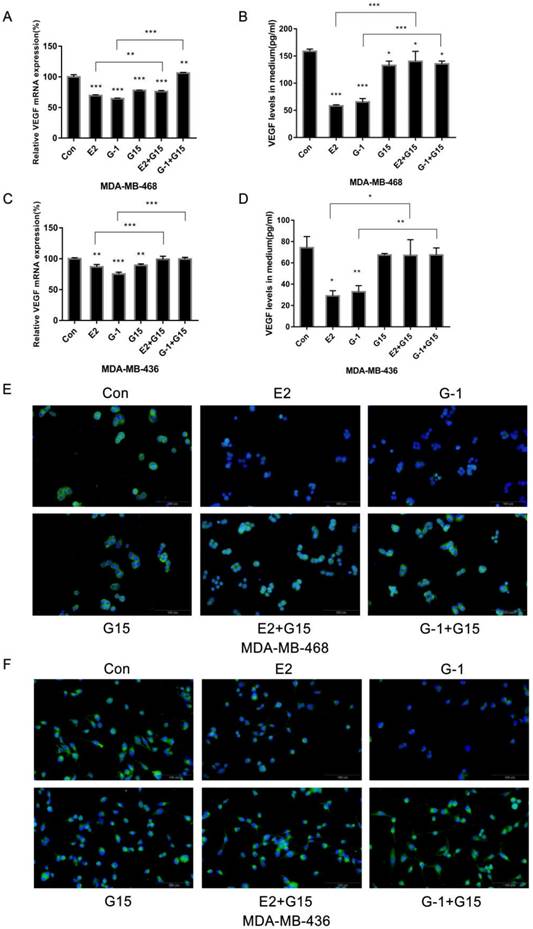
Since TNBC cells highly expressed GPER-1, we explored whether GPER-1 participated in the response to E2. We added the antagonist G-15 (5 μM) to block GPER-1 function. The results revealed that G-15 notably reversed the inhibitory effects of E2 and G-1 on VEGF expression (Fig. 3A, 3C). Quantification of VEGF by ELISA revealed that blocking GPER-1 significantly reversed the inhibition effects of E2 on VEGF secretion in TNBC cell lines (Fig. 3B, 3D). Immunofluorescence in MDA-MB-468 and MDA-MB-436 cells after blocking of GPER-1 further confirmed the role of GPER-1 in the downregulation of VEGF protein expression in TNBC cells induced by E2 (Fig. 3E, 3F).
GPER-1 is involved in the inhibition of VEGF expression in TNBC cell lines induced by E2. (A) MDA-MB-468 cells were treated with 10-6 M E2, 1 μM G-1, 5 μM G-15 or a combination for 8 h, and VEGF mRNA was measured by qRT-PCR. (B) MDA-MB-468 cells were treated with 10-6 M E2, 1 μM G-1, 5 μM G-15 or a combination for 18 h, and VEGF secretion in the medium was detected via ELISA. (C) MDA-MB-436 cells were treated with 10-7 M E2, 1 μM G-1, 5 μM G-15 or a combination for 8 h, and VEGF mRNA was measured by qRT-PCR. (D) MDA-MB-436 cells were treated with 10-7 M E2, 1 μM G-1, 5 μM G-15 or a combination for 18 h, and VEGF secretion in the medium was detected via ELISA. VEGF expression in MDA-MB-468 (E) and MDA-MB-436 (F) cells was detected by immunofluorescence staining. The data are presented as the mean ± SD of three independent experiments. *p < 0.05 compared with control, **p < 0.01 compared with control, and ***p < 0.001 compared with control.
GPER-1 is involved in the E2-induced reduction in tube formation in TNBC cells
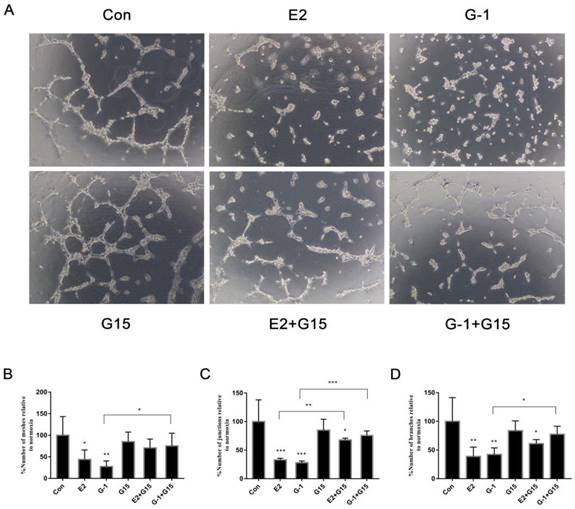
After elucidating the effect of GPER-1 on VEGF expression in TNBC cells, we aimed to investigate the role of GPER-1 in TNBC cell angiogenesis induced by E2 in vitro. Conditioned medium from MDA-MB-468 cells treated with 10-6 M E2 or 1 μM G-1 for 18 h extensively suppressed tube formation, whereas the HUVECs formed a series of reticular structures in the conditioned medium from MDA-MB-468 cells treated with 5 μM G-15 or with the agonist and antagonist together to block GPER-1 (Fig. 4A). Further supporting this phenomenon, the number of meshes, junctions and branches that were spotted (Fig. 4B, 4C, 4D), suggesting that GPER-1 may be a target of E2 participates in the inhibition of tube formation in TNBC cells.
GPER-1 is involved in the E2-induced reduction of tube formation in TNBC cells. (A) Tube formation was evaluated in HUVECs cultured for 6 h in medium collected from MDA-MB-468 cells treated with vehicle, 10-6 M E2, 1 μM G-1, 5 μM G-15 or a combination for 18 h. Quantification of the number of meshes (B), the number of branching junctions (C) and the number of branches (D) in HUVECs. The data are presented as the mean ± SD of three independent experiments. *p < 0.05 compared with control, **p < 0.01 compared with control, and ***p < 0.001 compared with control.
Effect of E2 on TNBC xenograft tumours in vivo
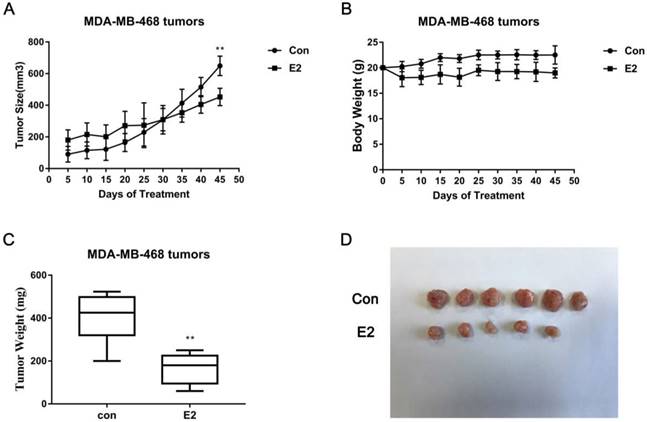
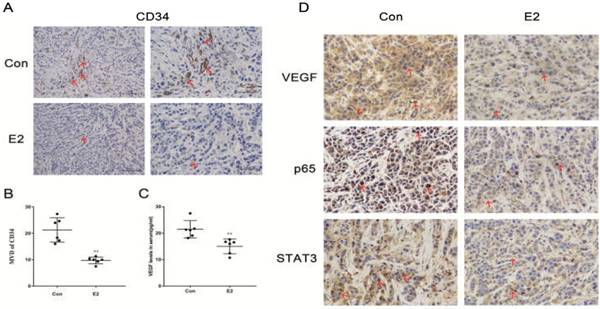
Next, we used an in vivo model system. After female BALB/c (nu/nu) mice were anaesthetized and bilaterally ovariectomized, they were randomly divided into two groups: one group received E2 pellets (0.72 mg, 60-day release), and the other served as the control group. After 45 days of treatment, we harvested the tumours and serum and conducted a series of experiments. Serum oestrogen levels were determined by chemiluminescence analysis. The results exhibited a sharp reduction (p < 0.001) in serum oestrogen levels in control mice compared with the levels in E2 group mice (Table 1), which demonstrated that we successfully established an ovariectomized animal model. Tumour growth and body weight (BW) were recorded every 5 days for each mouse. The tumour growth (Fig. 5A) and weights (Fig. 5C) of the control mice were higher than those of the mice that received E2 treatment, while the initial BW in the two groups showed no differences (Fig. 5B). Representative tumour images are shown in Fig. 5D. We calculated the microvascular density (MVD) by labelling the CD34 in the endothelium to evaluate the degree of angiogenesis. The results showed that the MVD values in the E2 group were significantly lower than those in the control group (Fig. 6A, 6B); this pattern was also observed with respect to the expression of VEGF, p65 and STAT3 in MDA-MB-468 xenografts (Fig. 6C, 6D). All of these data demonstrate that E2 treatment may inhibit TNBC tumour growth and angiogenesis in vivo.
E2 concentration in E2 and control group (pg/ml)
| Mean±SD | P value | |
|---|---|---|
| E2 group | 1327.67±162.17 | <0.001 |
| Control group | 24.33 ± 3.33 |
E2 inhibits MDA-MB-468 xenograft growth. (A) E2 treatment decreased the size of the MDA-MB-468 tumours (n = 6). Mice were administered E2 pellets or control pellets, as described in the Materials and Methods. (B) The BWs of xenograft mice bearing MDA-MB-468 tumours (n = 6). (C) Weights of MDA-MB-468 xenograft tumours treated with vehicle or E2 (n = 6). (D) Representative images scaled to the same ratio as that of the image of excised tumours from the two groups. ** p < 0.01 compared with control.
In vivo angiogenesis effect of E2 on MDA-MB-468 xenografts in nude mice. (A) On the 45th day post-implantation, MDA-MB-468 tumour tissues from control or E2 group mice were dissected and stained for CD34. Scale bar, 100 μm. (B) Determination of angiogenesis in the TNBC xenografts via analysis of the MVD, as described in the Materials and Methods. The bars correspond to the mean ± SD. **p < 0.01 versus the control group. (C) Serum VEGF levels in the two groups of xenograft mice that were bilaterally ovariectomized (n = 6) were detected via ELISA. ** p < 0.01 versus the control group. (D) Tumour tissue sections from the control and E2 groups were subjected to IHC detection of VEGF, p65 and STAT3. Scale bar, 50 μm.
Discussion
In this study, we demonstrated that E2 inhibits VEGF expression and tube formation in TNBC cell lines by activating GPER-1 in vitro and suppresses tumour growth and angiogenesis as well as the expression levels of VEGF, NF-κB/p65, STAT3 and the endothelial marker CD34 in MDA-MB-468 xenograft tumours.
TNBC is a type of breast cancer that lacks ER, PR and HER-2/neu [3]. In recent years, researchers have suggested that GPER-1 is highly expressed in TNBC cells [21, 37] and in the endometrium [38, 39], ovaries [40], thyroid [41] and lungs [42]. In our study, we used the ERα-positive breast cancer cell line MCF-7 and the ERα-negative breast cancer cell line SKBR3 as positive controls to examine the relative GPER-1 expression in the TNBC cell lines MDA-MB-468 and MDA-MB-436 and found that GPER-1 was highly expressed in both TNBC cell lines and that ERα was not expressed, consistent with the above studies.
Oestrogens promote cell migration, invasion and angiogenesis in breast cancer by binding to ERs [43]. Tumour angiogenesis is essential in tumour metastasis [24], and VEGF is an important potential proangiogenic factor that is involved in breast cancer angiogenesis [44]. As previously mentioned [2], breast cancer is divided into hormone receptor-positive breast cancer, HER-2-overexpressing breast cancer and TNBC. Studies have suggested that ERα, which is activated by oestrogen, increases VEGF levels in ER-positive breast cancer through binding to VEGF- related oestrogen response elements [28]. Similarly, E2 promotes VEGF expression and angiogenesis via GPER-1 together with the EGFR/ERK/c-FOS signalling pathway in HER-2-overexpressing breast cancer [35]. However, another study found that E2 treatment inhibited tumour formation in ERβ-expressing TNBC cell lines through inhibition of cyclin-dependent kinases 1 and 7 [12]. Transfection of the androgen receptor-positive TNBC cell lines MDA-MB-231 and Hs5787 with ERβ1 also resulted in fewer pulmonary metastases in vivo compared with empty vector-transfected cells through inhibition of ZEB1 [45]. In addition, treatment with the GPER-1-specific agonist G-1 can suppress cell migration and tumour angiogenesis in vitro and in vivo through NF-kB/IL-6 in TNBC [37]. The above studies suggest that activation of ERβ or GPER-1 by oestrogens may potentially inhibit TNBC progression. Our findings also suggest that E2 inhibits VEGF expression in a concentration- and time- dependent manner and GPER-1 might be involved in the E2-induced suppression of angiogenesis in TNBC cell lines. Additionally, E2 also repressed tumour growth and angiogenesis in MDA-MB-468 xenograft tumours in vivo. Overall, the complexity of the function of E2 and GPER-1 might be due to differences in breast cancer subtypes, cell types, agonist specificity, receptor expression ratios and the activation of signalling molecules.
NF-κB is a typical transcription factor that is involved in the modulation of inflammatory responses and the expression of interleukin (IL)-6, which enhances tumour growth and metastasis in TNBC [46, 47]. IL-6 activates Janus kinase (JAK), which can further induce STAT3 tyrosine phosphorylation. STAT3 participates in the transcription and translation of target genes involved in angiogenesis in several cancers [48, 49]. Our previous study showed that E2 has an anti-inflammatory effect in vascular smooth muscle cells [50]. Here, we confirmed that E2 treatment reduced the expression of NF-κB/p65, STAT3, VEGF and CD34 and decreased the nuclear localization of NF-κB/p65 and STAT3 in primary tumour xenografts. Therefore, the inhibitory effect of E2 on angiogenesis may be related to the suppressed expression and nuclear translocation of NF-κB/p65 and STAT3. Some researchers have suggested that NF-κB and STAT3 can regulate the function of quite a few genes involved in anti-apoptosis and cell cycle control in a cooperative manner [51, 52]. Currently, whether cooperation exists between NF-κB and STAT3 in oestrogen-induced TNBC angiogenesis remains to be elucidated.
In conclusion, the results presented here reveal that oestrogen inhibits VEGF expression and angiogenesis in TNBC by activating GPER-1. The present findings shed further light on E2 affects GPER-1-positive TNBC progression and may provide novel insights to identify prognostic markers and targets for drug intervention.
Abbreviations
E2: 17β-oestradiol; TNBC: triple-negative breast cancer; VEGF: vascular endothelial growth factor; GPER-1: G-protein-coupled oestrogen receptor-1; HUVECs: human umbilical vein endothelial cells; ERs: oestrogen receptors; PRs: progesterone receptors; HER-2: human epidermal growth factor receptor 2; STAT3: signal transducer and activator of transcription 3; NF-κB/p65: nuclear factor kappa B/p65.
Acknowledgements
This study was funded by the National Natural Science Foundation of China (NSFC) (No.81372818 and No. 81572585).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians. 2017;67:7-30
2. Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T. et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:1160-7
3. Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z. et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10:5367-74
4. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:4429-34
5. Baselga J, Campone M, Piccart M, Burris HA, Rugo HS, Sahmoud T. et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. The New England journal of medicine. 2012;366:520-9
6. Johnston SR. New strategies in estrogen receptor-positive breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:1979-87
7. Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nature reviews Clinical oncology. 2011;9:16-32
8. Higgins MJ, Baselga J. Targeted therapies for breast cancer. The Journal of clinical investigation. 2011;121:3797-803
9. Gluz O, Liedtke C, Gottschalk N, Pusztai L, Nitz U, Harbeck N. Triple-negative breast cancer-current status and future directions. Annals of oncology: official journal of the European Society for Medical Oncology. 2009;20:1913-27
10. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G. et al. Mechanisms of estrogen action. Physiological reviews. 2001;81:1535-65
11. Russo J, Russo IH. The role of estrogen in the initiation of breast cancer. The Journal of steroid biochemistry and molecular biology. 2006;102:89-96
12. Reese JM, Bruinsma ES, Monroe DG, Negron V, Suman VJ, Ingle JN. et al. ERbeta inhibits cyclin dependent kinases 1 and 7 in triple negative breast cancer. Oncotarget. 2017;8:96506-21
13. Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625-30
14. Zhou Z, Qiao JX, Shetty A, Wu G, Huang Y, Davidson NE. et al. Regulation of estrogen receptor signaling in breast carcinogenesis and breast cancer therapy. Cellular and molecular life sciences: CMLS. 2014;71:1549
15. Maggiolini M, Picard D. The unfolding stories of GPR30, a new membrane-bound estrogen receptor. The Journal of endocrinology. 2010;204:105-14
16. Mizukami Y. In vivo functions of GPR30/GPER-1, a membrane receptor for estrogen: from discovery to functions in vivo. Endocrine journal. 2010;57:101-7
17. Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A. et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer research. 2007;67:1859-66
18. Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009;28:523-32
19. Chen ZJ, Wei W, Jiang GM, Liu H, Wei WD, Yang X. et al. Activation of GPER suppresses epithelial mesenchymal transition of triple negative breast cancer cells via NF-kappaB signals. Molecular oncology. 2016;10:775-88
20. Steiman J, Peralta EA, Louis S, Kamel O. Biology of the estrogen receptor, GPR30, in triple negative breast cancer. American journal of surgery. 2013;206:698-703
21. Girgert R, Emons G, Grundker C. Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: possible application in targeted therapy. Breast cancer research and treatment. 2012;134:199-205
22. Girgert R, Emons G, Grundker C. 17beta-estradiol-induced growth of triple-negative breast cancer cells is prevented by the reduction of GPER expression after treatment with gefitinib. Oncol Rep. 2017;37:1212-8
23. Girgert R, Emons G, Grundker C. Inhibition of GPR30 by estriol prevents growth stimulation of triple-negative breast cancer cells by 17beta-estradiol. BMC cancer. 2014;14:935
24. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298-307
25. Huang TS, Chen YJ, Chou TY, Chen CY, Li HY, Huang BS. et al. Oestrogen-induced angiogenesis promotes adenomyosis by activating the Slug-VEGF axis in endometrial epithelial cells. J Cell Mol Med. 2014;18:1358-71
26. Yang J, Xiong LJ, Xu F, Zhao X, Liu B, Cai KL. et al. Estrogen inhibits colon polyp formation by reducing angiogenesis in a carcinogen-induced rat model. Int J Endocrinol. 2013;2013:453898
27. Barouk S, Hintz T, Li P, Duffy AM, MacLusky NJ, Scharfman HE. 17beta-estradiol increases astrocytic vascular endothelial growth factor (VEGF) in adult female rat hippocampus. Endocrinology. 2011;152:1745-51
28. Hyder SM, Nawaz Z, Chiappetta C, Stancel GM. Identification of functional estrogen response elements in the gene coding for the potent angiogenic factor vascular endothelial growth factor. Cancer Res. 2000;60:3183-90
29. Sengupta K, Banerjee S, Saxena N, Banerjee SK. Estradiol-induced vascular endothelial growth factor-A expression in breast tumor cells is biphasic and regulated by estrogen receptor-alpha dependent pathway. Int J Oncol. 2003;22:609-14
30. Madeo A, Maggiolini M. Nuclear alternate estrogen receptor GPR30 mediates 17beta-estradiol-induced gene expression and migration in breast cancer-associated fibroblasts. Cancer Res. 2010;70:6036-46
31. Linderholm BK, Hellborg H, Johansson U, Elmberger G, Skoog L, Lehtio J. et al. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Annals of oncology: official journal of the European Society for Medical Oncology. 2009;20:1639-46
32. Oakman C, Viale G, Di Leo A. Management of triple negative breast cancer. Breast. 2010;19:312-21
33. Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD. et al. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:1810-6
34. von Minckwitz G, Eidtmann H, Rezai M, Fasching PA, Tesch H, Eggemann H. et al. Neoadjuvant chemotherapy and bevacizumab for HER2-negative breast cancer. The New England journal of medicine. 2012;366:299-309
35. De Francesco EM, Pellegrino M, Santolla MF, Lappano R, Ricchio E, Abonante S. et al. GPER mediates activation of HIF1alpha/VEGF signaling by estrogens. Cancer Res. 2014;74:4053-64
36. Zheng S, Huang J, Zhou K, Xiang Q, Zhang Y, Tan Z. et al. Progesterone enhances vascular endothelial cell migration via activation of focal adhesion kinase. J Cell Mol Med. 2012;16:296-305
37. Liang S, Chen Z, Jiang G, Zhou Y, Liu Q, Su Q. et al. Activation of GPER suppresses migration and angiogenesis of triple negative breast cancer via inhibition of NF-kappaB/IL-6 signals. Cancer Lett. 2017;386:12-23
38. He YY, Cai B, Yang YX, Liu XL, Wan XP. Estrogenic G protein-coupled receptor 30 signaling is involved in regulation of endometrial carcinoma by promoting proliferation, invasion potential, and interleukin-6 secretion via the MEK/ERK mitogen-activated protein kinase pathway. Cancer Sci. 2009;100:1051-61
39. Leblanc K, Sexton E, Parent S, Belanger G, Dery MC, Boucher V. et al. Effects of 4-hydroxytamoxifen, raloxifene and ICI 182 780 on survival of uterine cancer cell lines in the presence and absence of exogenous estrogens. Int J Oncol. 2007;30:477-87
40. Smith HO, Arias-Pulido H, Kuo DY, Howard T, Qualls CR, Lee SJ. et al. GPR30 predicts poor survival for ovarian cancer. Gynecol Oncol. 2009;114:465-71
41. Vivacqua A, Bonofiglio D, Albanito L, Madeo A, Rago V, Carpino A. et al. 17beta-estradiol, genistein, and 4-hydroxytamoxifen induce the proliferation of thyroid cancer cells through the g protein-coupled receptor GPR30. Mol Pharmacol. 2006;70:1414-23
42. Siegfried JM, Hershberger PA, Stabile LP. Estrogen receptor signaling in lung cancer. Semin Oncol. 2009;36:524-31
43. Zheng S, Huang J, Zhou K, Zhang C, Xiang Q, Tan Z. et al. 17beta-Estradiol enhances breast cancer cell motility and invasion via extra-nuclear activation of actin-binding protein ezrin. PLoS One. 2011;6:e22439
44. Roskoski R Jr. Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncol Hematol. 2007;62:179-213
45. Song W, Tang L, Xu Y, Sun Q, Yang F, Guan X. ERbeta1 inhibits metastasis of androgen receptor-positive triple-negative breast cancer by suppressing ZEB1. Journal of experimental & clinical cancer research: CR. 2017;36:75
46. Sun X, Mao Y, Wang J, Zu L, Hao M, Cheng G. et al. IL-6 secreted by cancer-associated fibroblasts induces tamoxifen resistance in luminal breast cancer. Oncogene. 2014
47. Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol. 2014;26:54-74
48. Dmitrieva OS, Shilovskiy IP, Khaitov MR, Grivennikov SI. Interleukins 1 and 6 as Main Mediators of Inflammation and Cancer. Biochemistry (Mosc). 2016;81:80-90
49. Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A. et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer Cell. 2009;16:487-97
50. Jiang P, Xu J, Zheng S, Huang J, Xiang Q, Fu X. et al. 17beta-estradiol down-regulates lipopolysaccharide-induced MCP-1 production and cell migration in vascular smooth muscle cells. J Mol Endocrinol. 2010;45:87-97
51. Bohlig L, Rother K. One function-multiple mechanisms: the manifold activities of p53 as a transcriptional repressor. J Biomed Biotechnol. 2011;2011:464916
52. Vaughan CA, Singh S, Windle B, Sankala HM, Graves PR, Andrew Yeudall W. et al. p53 mutants induce transcription of NF-kappaB2 in H1299 cells through CBP and STAT binding on the NF-kappaB2 promoter and gain of function activity. Arch Biochem Biophys. 2012;518:79-88
Author contact
![]() Corresponding author: Professor Ting-huai Wang, Department of Physiology, Zhongshan School of Medicine, Sun Yat-sen University, 74 Zhongshan Road II, Guangzhou 510080, CHINA. Tel: 020-87330647; E-mail address: wangthsysu.edu.cn
Corresponding author: Professor Ting-huai Wang, Department of Physiology, Zhongshan School of Medicine, Sun Yat-sen University, 74 Zhongshan Road II, Guangzhou 510080, CHINA. Tel: 020-87330647; E-mail address: wangthsysu.edu.cn