Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(21):3858-3866. doi:10.7150/jca.22855 This issue Cite
Research Paper
Runs of homozygosity associate with decreased risks of lung cancer in never-smoking East Asian females
Yi-Xiao Chen1,2, Yan Guo2, Shan-Shan Dong2, Xiao-Feng Chen2, Jia-Bin Chen2, Yu-Jie Zhang2, Shi Yao2, Hlaing Nwe Thynn2, Liqiang Zhi1 ![]() , Tie-Lin Yang1,2
, Tie-Lin Yang1,2 ![]()
1. Department of Joint Surgery, Honghui Hospital; The Key Laboratory of Biomedical Information Engineering of Ministry of Education, Xi'an Jiaotong University, Xi'an, P. R. China
2. Key Laboratory of Biomedical Information Engineering of Ministry of Education, School of Life Science and Technology, Xi'an Jiaotong University, Xi'an, Shaanxi Province, 710049, P. R. China
Received 2017-8-18; Accepted 2018-2-24; Published 2018-10-5
Abstract
Although genome-wide association studies (GWASs) have identified some risk single-nucleotide polymorphisms in East Asian never-smoking females, the unexplained missing heritability is still required to be investigated. Runs of homozygosity (ROHs) are thought to be a type of genetic variation acting on human complex traits and diseases. We detected ROHs in 8,881 East Asian never-smoking women. The summed ROHs were used to fit a logistic regression model which noteworthily revealed a significant association between ROHs and the decreased risk of lung cancer (P < 0.05). We identified 4 common ROHs regions located at 2p22.1, which were significantly associated with decreased risk of lung cancer (P = 2.00 × 10-4 - 1.35 × 10-4). Functional annotation was conducted to investigate the regulatory function of ROHs. The common ROHs were overlapped with potential regulatory elements, such as active epigenome elements and chromatin states in lung-derived cell lines. SOS1 and ARHGEF33 were significantly up-regulated as the putative target genes of the identified ROHs in lung cancer samples according to the analysis of differently expressed genes. Our results suggest that ROHs could act as recessive contributing factors and regulatory elements to influence the risk of lung cancer in never-smoking East Asian females.
Keywords: lung cancer, runs of homozygosity, GWASs, genetic risk factors, regulatory elements
Introduction
Lung cancer is a major public health problem worldwide and constitutes an enormous burden on global society [1]. Epidemiological studies of lung cancer have shown that the highest incidence rates among females occur in North America, Europe, Australia and East Asia [2]. The highest incidence rate among females occurs in Northern America, which is 33.8 cases per 100,000. For the East Asian females, the incidence rate is 19.2 cases per 100,000 in average. Moreover, lung cancer is the most commonly diagnosed cancer among females in China and North Korea [3]. Although most of lung cancers are attributed to tobacco smoking, genetic factors also play a pivotal role in lung cancer development [3]. Heritability of lung cancer has been estimated to be 31% [4]. Moreover, it is now fairly accepted that lung cancer occurring in lifetime never-smokers is distinct from smoking-associated lung cancer [5]. Compared to never-smokers, the risk of developing lung cancer is 20-40 times higher in lifetime smokers [6]. Furthermore, it has been reported that there are different clinical features and outcomes of lung cancer between never-smokers and smokers. Several studies have reported a higher proportion of adenocarcinoma histology in never-smokers with lung cancer compared to smokers [5, 7]. Never-smokers with lung cancer have better response rates to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors than smokers [2, 8]. Identifying genetic factors in never-smoking females could exclude environmental confounding risk factors and offer new insights into the progression of lung cancer.
Previous genome-wide association studies (GWASs) have identified over 70 susceptibility single nucleotide polymorphisms (SNPs) associated with lung cancer (http://www.genome.gov/gwastudies), including some SNPs identified in never-smoking females in Asia. For instance, susceptibility loci at 3q28, 5p15.33, 6p21.1, 6p21.32, 6q22.2, 9p21, 10q25.2, 12q13.13 and 17q24.3 are reported to be associated with the risk of lung cancer in never-smoking Asian females [9-13]. However, the susceptibility loci identified by GWASs only accounted for about 10% of the total heritability in Asian populations, remaining a large part of the heritability to be interpreted [14]. Therefore, studies with innovative methodologies are needed to detect other genetic factors.
Runs of homozygosity (ROHs) are referred to as a consecutively homozygous segment with large numbers of SNPs along chromosomes. ROHs can represent as a new type of genetic variation since ROHs varies among individuals and populations [15-17]. With the advancement of genome-wide SNP array, genome-wide homozygosity can be assessed conveniently using high-density SNPs data [18]. ROHs can reflect the level of inbreeding and reveal non-additive genetic effects, hence complex traits and diseases could be influenced by ROHs and corresponding recessive genetic effects. Several studies have reported the associations between ROHs and complex traits or diseases, including height, bone mineral density, Alzheimer's disease and thyroid cancer [17, 19-21]. In the investigation of the association between ROHs and the risk of lung cancer, Cheng Wang et al. found that the ROHs level was negatively related to the risk of lung cancer, and a ROHs region at 14q23.1 was associated with the risk of lung cancer in Han Chinese population [22]. This study used population comprising both males and females. In East Asia, the incidence rates of lung cancer in males are much higher than in females, suggesting the gender difference in lung cancer risks [23]. Besides, smoking and never-smoking individuals were used in this study at the same time, it was difficult to examine whether the identified genetic variants were associated with lung carcinogenesis or nicotine addiction [24]. Therefore, the investigation of the effects of ROHs in never-smoking females with a larger sample size may enhance the understanding of the relationship between ROHs and lung cancer.
In this study, we screened ROHs on whole-genome regions in 8,881 never-smoking East Asian females and filtered the common ROHs regions by statistical methods. With the purpose of examining underlying function of ROHs, we annotated these common ROHs regions with located genes and investigated their effects on neighbor genes. We also investigated the underlying regulatory function of the common ROHs regions with annotation using epigenetics markers, regulatory elements and long-range interaction data. We identified differentially expressed genes (DEGs) between lung adenocarcinoma samples and control samples to evaluate the expression level of genes possibly regulated by ROHs. Our results reveal that ROHs play an important role as recessive genetic factors and act as regulatory elements in the underlying genetic mechanism of lung cancer.
Materials and Methods
Subjects
A total of 8,881 never-smoking East Asian females were enrolled in this study, including 4,922 (55.42%) lung cancer cases and 3,959 (44.58%) controls from China, South Korea, Japan, Singapore, Taiwan, and Hong Kong. The mean age of the subjects was 58.2 years old. The diagnoses of all cases were confirmed histologically. All subjects had complete phenotypes, including age, sex, and histological type of lung cancer. The basic characteristics of the subjects are shown in Table 1. The phenotype and genotype data were obtained from Database of Genotypes and Phenotypes (dbGaP). The data we used (phs000716.v1.p1) have passed the embargo date [25]. All samples used in this study have been reported in Lan et al [25].
Basic characteristics of the study samples
| Characteristic | Never-smoking East Asian females | GSE40791 |
|---|---|---|
| Sample Size (n) | 8,881 | 194 |
| Age (year) | 58.2(9.29) | 68.01(10.46) |
| Female (%) | 100% | 42.8% |
| Cases (%) | 55.42% | 48.45% |
| Never-smokers (%) | 100.00% | 7.22% |
| Histology | ||
| Adenocarcinoma (%) | 73.04% | 100% |
| Squamous (%) | 13.41% | 0% |
| Other (%) | 13.55% | 0% |
Age values are presented as mean (standard deviation).
Genotyping and quality control
Samples were genotyped using two similar high-density SNP arrays (Illumina 610Q SNP microarray and Illumina 660W SNP microarray). The intersection of SNPs on both arrays exceeded 570,000 SNPs. The genotyping data passed the quality control (QC) with measurements have been provided by the previous GWASs [25]. The individuals with over 2% missing rate were excluded. SNPs with call rate less than 95% or minor allele frequency (MAF) less than 5% were excluded. Only autosomal SNPs were analyzed in this study. After QC, 4,922 cases, 3,959 controls and 420,680 autosomal SNPs were remained for subsequent analyses. We estimated the proportion of phenotypic variance explained by SNPs among all of the 8,881 individuals using a software called genome-wide complex trait analysis (GCTA) [26], with the estimated prevalence of lung cancer in East Asian females as 19.2 cases per 100,000 (Globocan 2012 http://globocan.iarc.fr/Default.aspx).
Identification of ROHs
In this study, ROHs were defined as segments with at least 50 consecutive homozygous SNPs and ROHs calling were performed using PLINK v1.07 (http://pngu.mgh.harvard.edu/purcell/plink/) [27]. We set a range of the minimum length for calling ROHs segments, including 0.50 Mb, 1.00 Mb, 1.25 Mb, 1.50 Mb, 1.75 Mb, 2.00 Mb and 3.00 Mb. At the same time, ROHs detected in this study must have more than one SNP per 50 kb, and the gap between two adjacent ROHs segments was set more than 1.00 Mb. PLINK used a sliding window of 5.00 Mb with at least 50 SNPs to define ROHs. One heterozygous site and five missing calls in a window were allowed.
Association analyses between ROHs and lung cancer
A logistic regression model was used to examine the association between the summed ROHs and lung cancer. Age and three significant principal components (EV1, EV2, and EV4) obtained from the software GCTA [26] were used as covariates. The summed ROHs were defined as the total length of ROHs in an individual. After fitting the logistic regression model, FROHs and βFROHs were used to examine the degree of homozygosity and the effects of ROHs on phenotype. FROH was defined as the ratio of the summed ROHs to the total genome length (approximately 2.5 × 106) and βFROHs was the estimated effect of FROHs on the trait calculated by FROHs divided by the standard deviation of the summed ROHs [28].
The common ROHs regions selection
The “--homozyg-group” option of PLINK was used to produce files consisted of the overlapping ROHs regions which were divided into pools including the ratio of cases and controls containing the same overlapping ROHs regions. Although ROHs regions were defined with parameters described above, some overlapping regions were small because of little common segments in different individuals, e.g. only including one SNP. Such regions were excluded from the subsequent annotation. Overlapping ROHs regions contained in less than 5% individuals were also excluded from subsequent analyses. The chi-squared goodness-of-fit test was then performed to detect the common regions, which were defined as the overlapping ROHs regions with significant different ratios in cases and controls. The Benjamini and Hochberg (BH) procedure was used for multiple-testing corrections.
Inbreeding coefficients calculation and statistical analysis
We used PLINK to get the genomic inbreeding coefficients estimated (F) [27]. The inbreeding coefficients were calculated from all the homozygous SNPs counts on each autosome. The differences between cases and controls were tested using the Student's t-test on whole-genome and a single chromosome separately. We fitted a logistic regression model with inbreeding coefficients and the covariates on the case-control status of lung cancer. The covariates were the same as in the previous regression model, including age and three principal components (EV1, EV2, and EV4).
Testing the effects of natural selection
We used haplotter (http://haplotter.uchicago.edu/) to evaluate the effects of natural selection on the common ROHs [29]. Haplotter is a tool that can detect positive selection in a genomic region using the HapMap Phase II data. We estimated the integrated haplotype score (iHS), Fay and Wu's H and fixation index (Fst) for ROHs separately. The value of iHS was used to detect signals of the recent selection through scanning SNPs data at the whole genome. Voight et al. reported that SNPs with |iHS| > 2 indicated a powerful signal of selection [29]. Fay and Wu's H is powerful to detect positive selection based on the frequencies of the polymorphisms in the region [30]. Fst can be used to measure the level of differentiation at the locus between populations due to selection [31].
Functional annotation of the common ROHs regions
To evaluate the underlying regulatory function of the common regions, we annotated the common ROHs regions in different levels. First, we identified genes located in the common ROHs regions using ANNOVAR [32]. The common ROHs regions were also annotated for histone markers in A549 cell line by using the ChIP-seq data from ENCODE in the UCSC genome database [33]. To assess the chromatin states of ROHs regions, we downloaded 15-state chromatin state segmentation data generated by ChromHMM based on the Roadmap histone modification data [34]. We identified the common ROHs regions within putative enhancer regions, including “Enhancers” and “Genic enhancers”. We annotated the chromatin states of the common ROHs regions in A549 cells, IMR90 cells and normal lung tissue cells, separately. To detect the regulatory function of ROHs regions through long-range interaction, we used the Hi-C data downloaded from 4D genome (http://4dgenome.research.chop.edu/) [35]. We only annotated the Hi-C regions in IMR90 because high-quality Hi-C data from other lung cell lines are still unavailable. We identified long-range interaction regulated genes which overlapped with transcription start sites (including “Active TSS” and “Flanking TSS” according to 15-state chromatin state segmentation annotation) lied within long-range interaction pairs of enhancer regions.
Gene expression profiling of lung tumors
DEGs from lung adenocarcinoma tissues were identified from a publicly available dataset (GEO accession number: GSE40791). The dataset consists of 94 lung adenocarcinoma samples and 100 normal controls. Basic information of GSE40791 samples was summarized in Table 1. The age row of GSE40791 samples refers to the age at surgery. The gene expression data were generated on Affymetrix human genome u133 plus 2.0 arrays. Before statistical analyses, the gene expression data were normalized by using the Robust Multi-array Average (RMA) method [36]. The conditions of differential expression were calculated by using the unpaired t-test. The significant cutoff was set as P-value < 0.05.
Results
Estimates of heritability explained by SNPs
We estimated the heritability with GWAS data in 8,881 unrelated individuals. The SNPs explained on average 18.68% (s.d. 2.57%) of the proportion of the total phenotypic variance, which was approximate to the result of previous published studies. Sampson et al. estimated that the heritability of the lung cancer in Asian population was 12.1% (95 % CI = 6.40% to 17.7%) based on the GWAS dataset [37].
Identification of ROHs on autosomal chromosomes in subjects
We identified total ROHs regions in all 8,881 individuals with different minimum length thresholds. Summary statistics of the total ROHs size and the total ROHs number are shown in Table 2. The total number and the total size of ROHs in individuals decreased with the minimum ROHs size threshold increased. The mean size per ROHs became longer with the minimum length increased except the threshold of ROHs size of 3 Mb. When the threshold of the minimum length for detecting ROHs was set above 1.50 Mb, the lengths of ROHs which carried by some individuals were shorter than the set threshold (Table 2). Using 1.5 Mb as the minimum threshold of ROHs length, 99 % of cases and 99 % of controls were detected with more than 1 ROHs. When the threshold was set as 3 Mb, we detected 4 % of cases and 6 % of controls carrying one or more ROHs.
Summary of ROHs Characteristics identified with different length
| Size | Total ROHs size | Total ROHs number | Mean size per ROHs (kb) | ||||
|---|---|---|---|---|---|---|---|
| Mean(kb) | Range | Mean(n) | Range | ||||
| Min(kb) | Max(kb) | Min(n) | Max(n) | ||||
| > 500 kb | 148,270 | 58,436 | 445,666 | 176.90 | 75 | 272 | 837.8 |
| > 1.00 Mb | 51,936 | 13,854 | 378,231 | 34.79 | 11 | 115 | 1,471 |
| > 1.25 Mb | 32,633 | 2,769 | 358,867 | 17.39 | 2 | 81 | 1,808 |
| > 1.50 Mb | 21,650 | - | 350,432 | 9.32 | - | 58 | 2,153 |
| > 1.75 Mb | 14,884 | - | 343,937 | 5.12 | - | 44 | 2,513 |
| > 2.00 Mb | 10,938 | - | 334,826 | 2.99 | - | 33 | 2,760 |
| > 3.00 Mb | 5,947 | - | 335,623 | 0.84 | - | 25 | 2,185 |
Size: the minimum threshold for calling ROHs segments. The mean and range data were calculated by adding together all the values for the sizes of ROHs in each individual.
Associations between ROHs and lung cancer risk
To examine whether the total length of ROHs per individual between cases and controls could affect case-control status, we performed a logistic regression analysis. The values of βFROHs were all less than zero (ranging from -11.09 to -12.20). Therefore, we found that the increased level of ROHs was strongly associated with the decreased lung cancer risk under all of the different lengths conditions (P < 0.05) (Table 3).
Effects of genome-wide burden of ROHs on subjects
| Size | P | βFROH | βFROH-se |
|---|---|---|---|
| > 500 kb | 0.007 | -11.50 | 4.24 |
| > 1.00 Mb | 0.010 | -11.09 | 4.33 |
| > 1.25 Mb | 0.007 | -11.81 | 4.38 |
| > 1.50 Mb | 0.009 | -11.58 | 4.42 |
| > 1.75 Mb | 0.006 | -12.20 | 4.48 |
| > 2.00 Mb | 0.007 | -12.13 | 4.52 |
| > 3.00 Mb | 0.009 | -12.08 | 4.65 |
P: P-value for association with the logistic regression model; β: the estimated effects of ROHs on whole genome in units of standard deviations; se: standard error.
Identification of common ROHs regions
We identified ROH_14857 (P = 1.43 × 10-4, adjusted P = 0.044), ROH_14715 (P = 1.41 × 10-4, adjusted P = 0.044), ROH_14344 (P = 1.35 × 10-4, adjusted P = 0.044) and ROH_14342 (P = 2.00 × 10-4, adjusted P = 0.045) as the common ROHs regions. These regions were used in the subsequent analyses. Consistent with results of the logistic model (Table 4), the number of controls which had the common ROHs regions were more than the number of cases. The frequencies of the common ROHs regions were over 0.01 within the subjects (range from 0.048 to 0.050). Notably, we found that all of the common ROHs regions were located in chromosome 2. Each region contains one or two protein-coding genes (DHX57, MORN2, ARHGEF33 and SOS1). Genes with exonic regions overlapped with these common ROHs regions are also listed in Table 4.
Basic characteristics of significant ROHs associated with lung cancer
| ROH | Chr | Position | Freq | Cases: controls | Chi2 | P | Pad | Located gene | iHS max | Fay and Wu's H max | Fst max | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Start | End | ||||||||||||
| ROH_14857 | 2 | 39025631 | 39047154 | 0.048 | 198:230 | 14.47 | 1.43×10-4 | 0.044 | DHX57 | 1.284 | -22.097 | 0.360 | |
| ROH_14715 | 2 | 39084756 | 39135927 | 0.049 | 200:232 | 14.49 | 1.41×10-4 | 0.044 | DHX57, MORN2 | 2.623 | -36.364 | 0.433 | |
| ROH_14344 | 2 | 39198965 | 39226271 | 0.049 | 206:238 | 14.57 | 1.35×10-4 | 0.044 | ARHGEF33, SOS1 | 1.798 | -39.925 | 0.290 | |
| ROH_14342 | 2 | 39247288 | 39293530 | 0.050 | 207:237 | 13.85 | 2.00×10-4 | 0.044 | SOS1 | 2.010 | -56.961 | 0.271 | |
Freq: frequency; Chr: chromosome; Chromosomal positions are shown according to NCBI Build 37 (hg19); P: P-value for testing the differences of homozygosity status between cases and controls with two-sided chi-square test; Pad: P-value adjusting with the Benjamini and Hochberg (BH) procedure; Located gene: ROHs located gene; iHS max: the maximal absolute values for iHS; Fay and Wu's H max: the maximal absolute values for Fay and Wu's H; and Fst max: the maximal absolute values for Fst; iHS max, Fay and Wu's H max and Fst max were derived for Asian population from Haplotter (http://haplotter.uchicago.edu/) using Phase II HapMap Project data.
Measurement and association between Inbreeding coefficient and lung cancer
We calculated the inbreeding coefficient (F) using SNPs information of all samples. We found a significant difference of F between cases and controls on whole-genome level (P-value = 8.65 × 10-4). The means and standard deviations (SDs) for F in cases and controls were 0.0004 (0.1277) and 0.0110 (0.1087), separately. Furthermore, we calculated F for each chromosome separately. As shown in Table 5, chromosomes 2, 6, 8 and 16 showed the significant differences in the three inbreeding coefficients (P-value = 0.0026, 0.044, 0.0004 and 0.0369, separately). All of the common ROHs regions were located in chromosome 2. After fitting a logistic regression model with F, F also showed significant P-value of 0.0087 with a negative estimate of effect as -4.774.
P-values for the statistical analyses of inbreeding coefficients between cases and controls
| Chromosome | Total length | FI |
|---|---|---|
| 1 | 249250621 | 0.088905 |
| 2 | 243199373 | 0.002689 |
| 3 | 198022430 | 0.717751 |
| 4 | 191154276 | 0.575738 |
| 5 | 180915260 | 0.171943 |
| 6 | 171115067 | 0.043707 |
| 7 | 159138663 | 0.489741 |
| 8 | 146364022 | 0.000393 |
| 9 | 141213431 | 0.207553 |
| 10 | 135534747 | 0.163951 |
| 11 | 135006516 | 0.223446 |
| 12 | 133851895 | 0.599272 |
| 13 | 115169878 | 0.985238 |
| 14 | 107349540 | 0.543299 |
| 15 | 102531392 | 0.151867 |
| 16 | 90354753 | 0.036879 |
| 17 | 81195210 | 0.75997 |
| 18 | 78077248 | 0.282267 |
| 19 | 59128983 | 0.303959 |
| 20 | 63025520 | 0.561602 |
| 21 | 48129895 | 0.995754 |
| 22 | 51304566 | 0.537312 |
*bold values represent significant differences between cases and controls at P < 0.05
Likewise, FROH was also higher in controls than cases. The means and standard deviations (SDs) for FROH were 0.0204 (0.0094) in cases and 0.0211 (0.0109) in controls. We detected a significant difference for FROH between cases and controls (P-value = 0.0013). Moreover, the inbreeding coefficient and FROH were significantly associated with each other according to Pearson's correlation coefficient (r = 0.7517, P-value = 2.2 × 10-16).
Natural selection on the common ROHs
We used iHS, Fay and Wu's H test and Fst to measure the selective pressure of ROHs. In the East Asian population, ROH_14715 and ROH_14342 showed extremely positive iHS scores (iHS > 2) of 2.623 and 2.010, respectively. The Fay and Wu's H test scores for all four ROHs were extremely negative and less than -10 (from -56.961 to -22.097). According to the Fst test, the Fst values of the ROHs were derived as 0. 360, 0. 433, 0. 290, and 0. 271, respectively. Based on the threshold of Fst > 0.2 used by Thomsen et al [17], we found that all of the ROHs were different between Yoruba and East Asian populations with the Fst values.
Regulatory functional annotation of the common ROHs regions
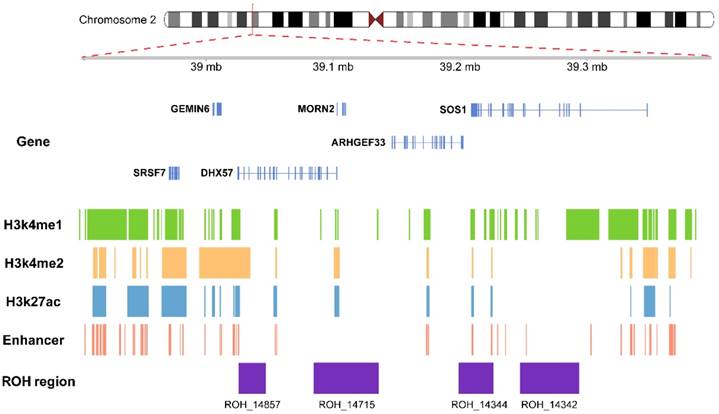
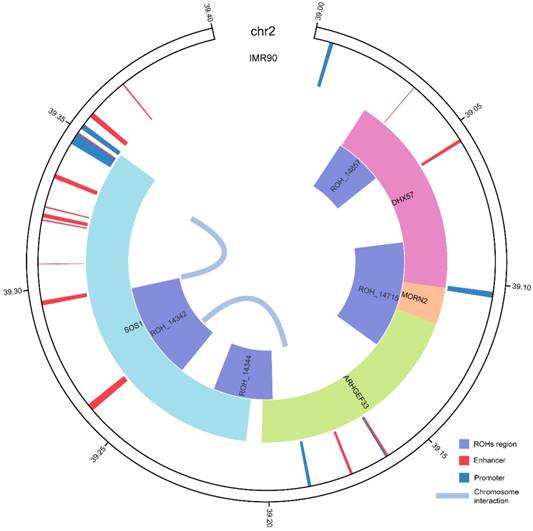
We annotated the common ROHs regions with active histone markers in A549 cell line, including H3K27ac, H3K4me1 and H3K4me2. As shown in Figure 1, ROH_14342, ROH_14344 and ROH_14715 were enriched in a set of activated enhancer histone modifications in A549 cell line, including H3K27ac, H3K4me1 and H3K4me2. Therefore, these 3 ROHs regions were regarded as enhancers in A549 cell line according to the chromatin states annotation (Figure 1). In IMR90 cell line, ROH_14342 and ROH_14857 were also annotated as enhancers. The results of the common ROHs regions annotated with chromatin states and long-range interactions are shown in Figure 2. We also explored the effects of ROHs on genes as distal regulatory elements. In IMR90 cell line, we found that the active promoter regions of SOS1 and ARHGEF33 were the target regions of enhancer within ROH_14342.
Functional annotation for the common ROHs regions. The common ROHs regions overlapped with histone marks which classified as active enhancer in A549 cell line, including H3k4me1, H3k4me2 and H3k27ac, and annotated as enhancers according to 15-state chromatin state segmentation.
Regulatory annotation of the common ROHs regions in IMR90 cell line. ROH_14342 and ROH_14857 were annotated with enhancers and interactions within the chromosome regions in IMR90 cell line. SOS1 and ARHGEF33 with active promoters were the target genes of the enhancer region overlapped with ROH_14342. Status of the enhancer regions were annotated according to 15-state chromatin state segmentation data generated by ChromHMM based on the Roadmap histone modification data. The interactions between regions on the chromosome detected by chromatin capture Hi-C are shown in the center.
DEGs of the common ROHs regions
We evaluated the expression levels for the four genes located in the common ROHs in lung cancer samples. The gene expression data of 100 normal samples and 94 lung adenocarcinoma samples were obtained by using microarray. We found that SOS1 (P = 1.21×10-8) and ARHGEF33 (P = 3.25×10-4) were significantly up-regulated in lung cancer samples.
Discussion
In this study, we screened whole-genome ROHs in 8,881 never-smoking East Asian females. We found that low level of ROHs was associated with the risk of lung cancer. We successfully identified four ROHs significantly associated with lung cancer. Our study reveals that ROHs might be involved in the development of lung cancer through affecting gene expression in never-smoking East Asian females.
In this study, we used two indicators containing the total length and the numbers of ROHs to reveal the level of homozygosity in an individual from different perspectives. According to the statistical summaries, most of the ROHs regions in the subjects were relatively short. These results indicate that homozygosity in the subjects is more likely to be the consequence of selective pressure compared with inbreeding [38]. In other words, ROHs in the samples inherited from common ancestors are improbable. Consistent with the previous analyses, our results support the conclusion that the ROHs might be the consequences of selection. Some studies have reported that consanguineous parents are more likely to generate long ROHs [39, 40]. In contrast, short ROHs are supposed to be a risk factor of complex traits and diseases [41]. Therefore, in this study, ROHs could present as a type of genetics variant in the population, and contribute genetics effects to the risk of lung cancer. The distribution of total length and the numbers of ROHs in this study are consistent with the results of previous studies that detected ROHs in East Asian populations under the same minimum length [28, 42, 43]. For instance, at the minimum length of ROHs >1.50 Mb, we found that the mean size of total ROHs was 21.65 Mb, while Joshi et al. reported that the size of ROHs ranged from 20.00 Mb to 39.00 Mb in diverse East Asian populations [28].
We found that the risk of lung cancer decreased with the increasing homozygosity in East Asian females. Our results are consistent with the results of a previous study [22]. Because we did not detect ROHs in some individuals with the threshold of minimum length at 1.25 Mb, we considered that the results of chi-square test were meaningful when the minimum length is below 1.25 Mb. However, defining ROHs with shorter length may overestimate the true level of homozygosity in individual genome [15]. Several studies have indicated that outbred populations usually carry ROHs with the length of more than 1.00 Mb [39]. Considering most of previous studies defined ROHs with a minimum length of 1.00 Mb to balance those factors, we also chose 1.00 Mb as length of ROHs regions to explore their functions.
We identified four ROHs significantly associated with the risk of lung cancer. The proportion of ROHs in cases and controls indicated that more controls had these four ROHs. For example, we detected ROH_14857 in 198 cases and 230 controls. The chi-square test showed that the distribution of the ROHs was significantly different between cases and controls. Therefore, these consequences and the results of logistic regression pointed out the same conclusion that the increased level of ROHs was strongly associated with the decreased risk of lung cancer. Furthermore, all the identified common ROHs regions located in chromosome 2p22.1. Lui et al. detected the high level of amplification in small cell lung samples at chromosome 2p22 [44]. According to Yan et al., gains of chromosome 2p were associated with advanced clinical stage and metastases of lung squamous cell carcinomas [45]. Pifarre et al. found that chromosome 2p25-p22 was frequent targets for replication errors (RER) in lung cancer, and RER-positive tumors were correlated with worse survival [46]. Therefore, the increased ROHs might suggest the deletion of heterozygosity mutation, and consequently lead to the activation of tumor suppressor genes and/or the inactivation of oncogene located in this region. We found that these four ROHs located at 2p22.1 overlapped with several genes, including SOS1, ARHGEF33, DHX57, and MORN2. The ROHs at 14q23.1 reported by Wang et al.[47] was also detected in our study. However, there was no significant association between it and lung cancer after multiple-testing corrections.
Intriguingly, we found that the common ROHs regions also had potential regulatory function to distal genes. As the results of chromatin state segmentation annotation in IMR90 cell line and A549 cell line, ROH_14342 was annotated as enhancers in these two cell lines. These results suggest that ROHs can be distal regulatory elements.
SOS1 and ARHGEF33 were potentially regulated by ROH_14342 according to the results of long-range interaction annotation and DEGs. SOS1 is a famous guanine nucleotide-exchange factor, acting in directing exchange of RAS-GDP to RAS-GTP and leading to the ERK activation[48]. Sequentially, the activation of RAS-RAF-MEK-ERK-MAP kinase pathway accelerates tumor cell proliferation [49, 50]. Several studies found that the reduction in SOS1 expression and corresponding RAS-MAPK activity depression could be great importance in anticancer activity [51-53]. ARHGEF33 has been identified as the gene for cell differentiation, primary function of cell survival and caspase-dependent cell death pathway. Considering ROHs are associated with lower risk of lung cancer, we suggest that the specific ROH regions could display as inhibitors to down-regulate the cell proliferation related genes. These results provide the new evidence that more homozygous levels inhibit the expression of cancer-associated genes and consequently lower the risk of lung cancer.
There are some potential limitations of our study. Some other genetic factors which could influence the detection of ROHs such as one-copy deletion were not able to be definitely excluded [54]. Therefore, we used five missing calls in one window when calling ROHs. That reduced the absence of one allele in genotype calling affecting in ROHs identification. More explorations of the common ROHs regions require more details of sequencing and chromosome conformation capture techniques.
In conclusion, our study provides evidence that ROHs could act as recessive contributing factors and regulatory elements for the risk of lung cancer, which offers a new insight for discovering missing heritability of lung cancer. For confirming and illuminating the potential mechanism of lung cancer, further molecular studies would be demanded.
Acknowledgements
Funding: This work was supported by the National Natural Science Foundation of China (81573241, 31471188); China Postdoctoral Science Foundation (2016M602797); Natural Science Basic Research Program Shaanxi Province (2016JQ3026); Zhejiang Provincial Natural Science Foundation of China (LGF18C060002); and the Fundamental Research Funds for the Central Universities.
The authors thank the Female Lung Cancer Consortium in Asia (FLCCA), which aims to learn about the etiology of lung cancer among never-smoking women in Asia. During the preparation of this manuscript, we did not collaborate with the researchers of the FLCCA. Therefore, our study does not necessarily reflect the opinions of them. The GWAS dataset we used in this study is available from dbGaP (http://www.ncbi.nlm.nih.gov/gap) under accession number phs000716.v1.p1.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Siegel RL, Ward EM, Jemal A. Global Cancer Incidence and Mortality Rates and Trends-An Update. Cancer Epidemiol Biomarkers Prev. 2016;25:16-27
2. Subramanian J, Govindan R. Lung cancer in never smokers: A review. Journal of Clinical Oncology. 2007;25:561-70
3. Ahmad A, Gadgeel S. Lung Cancer and Personalized Medicine. Springer International Publishing. 2016
4. Munoz M, Pong-Wong R, Canela-Xandri O, Rawlik K, Haley CS, Tenesa A. Evaluating the contribution of genetics and familial shared environment to common disease using the UK Biobank. Nat Genet. 2016;48:980-3
5. Subramanian J, Govindan R. Lung cancer in 'Never-smokers': a unique entity. Oncology (Williston Park, NY). 2010;24:29-35
6. Ozlu T, Bulbul Y. Smoking and lung cancer. Tuberkuloz ve toraks. 2005;53:200-9
7. Quinn AM, Newman WG, Hasleton PS. Risk Factors for Lung Cancer in Never Smokers: A Recent Review Including Genetics. Current Respiratory Medicine Reviews. 2016;12:74-117
8. Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers—a different disease. Nature Reviews Cancer. 2007;7:778-90
9. Hosgood III HD, Wang W-C, Hong Y-C, Wang J-C, Chen K, Chang I-S. et al. Genetic variant in TP63 on locus 3q28 is associated with risk of lung adenocarcinoma among never-smoking females in Asia. Hum Genet. 2012;131:1197-203
10. Hsiung CA, Lan Q, Hong Y-C, Chen C-J, Hosgood III HD, Chang I-S. et al. The 5p15. 33 locus is associated with risk of lung adenocarcinoma in never-smoking females in Asia. PLoS Genet. 2010;6:e1001051
11. Lan Q, Hsiung CA, Matsuo K, Hong Y-C, Seow A, Wang Z. et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nature genetics. 2012;44:1330-5
12. Wang Z, Seow WJ, Shiraishi K, Hsiung CA, Matsuo K, Liu J. et al. Meta-analysis of genome-wide association studies identifies multiple lung cancer susceptibility loci in never-smoking Asian women. Human molecular genetics. 2016;25:620-9
13. Timofeeva MN, Hung RJ, Rafnar T, Christiani DC, Field JK, Bickeböller H. et al. Influence of common genetic variation on lung cancer risk: meta-analysis of 14 900 cases and 29 485 controls. Human molecular genetics. 2012;21:4980-95
14. Sampson JN, Wheeler WA, Yeager M, Panagiotou O, Wang Z, Berndt SI. et al. Analysis of Heritability and Shared Heritability Based on Genome-Wide Association Studies for 13 Cancer Types. JNCI-J Natl Cancer Inst. 2015;107:11
15. Ku CS, Naidoo N, Teo SM, Pawitan Y. Regions of homozygosity and their impact on complex diseases and traits. Hum Genet. 2011;129:1-15
16. Gamsiz ED, Viscidi EW, Frederick AM, Nagpal S, Sanders SJ, Murtha MT. et al. Intellectual Disability Is Associated with Increased Runs of Homozygosity in Simplex Autism. American Journal of Human Genetics. 2013;93:103-9
17. Thomsen H, Chen BW, Figlioli G, Elisei R, Romei C, Cipollini M. et al. Runs of homozygosity and inbreeding in thyroid cancer. Bmc Cancer. 2016;16:11
18. Kijas JW. Detecting regions of homozygosity to map the cause of recessively inherited disease. Methods in molecular biology (Clifton, NJ). 2013;1019:331-45
19. Nalls MA, Guerreiro RJ, Simon-Sanchez J, Bras JT, Traynor BJ, Gibbs JR. et al. Extended tracts of homozygosity identify novel candidate genes associated with late-onset Alzheimer's disease. Neurogenetics. 2009;10:183-90
20. Yang TL, Guo Y, Zhang LS, Tian Q, Yan H, Papasian CJ. et al. Runs of Homozygosity Identify a Recessive Locus 12q21.31 for Human Adult Height. Journal of Clinical Endocrinology & Metabolism. 2010;95:3777-82
21. Yang TL, Guo Y, Zhang JG, Xu C, Tian Q, Deng HW. Genome-wide Survey of Runs of Homozygosity Identifies Recessive Loci for Bone Mineral Density in Caucasian and Chinese Populations. Journal of Bone and Mineral Research. 2015;30:2119-26
22. Wang C, Xu Z, Jin G, Hu Z, Dai J, Ma H. et al. Genome-wide analysis of runs of homozygosity identifies new susceptibility regions of lung cancer in Han Chinese. J Biomed Res. 2013;27:208-14
23. Torre LA, Siegel RL, Jemal A. Lung Cancer Statistics. Advances in experimental medicine and biology. 2016;893:1-19
24. Chanock SJ, Hunter DJ. Genomics: when the smoke clears. Nature. 2008;452:537-8
25. Lan Q, Hsiung CA, Matsuo K, Hong Y-C, Seow A, Wang Z. et al. Genome-wide association analysis identifies new lung cancer susceptibility loci in never-smoking women in Asia. Nat Genet. 2012;44:1330-5
26. Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: A Tool for Genome-wide Complex Trait Analysis. American Journal of Human Genetics. 2011;88:76-82
27. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. American Journal of Human Genetics. 2007;81:559-75
28. Joshi PK, Esko T, Mattsson H, Eklund N, Gandin I, Nutile T. et al. Directional dominance on stature and cognition in diverse human populations. Nature. 2015;523:459-62
29. Voight BF, Kudaravalli S, Wen X, Pritchard JK. A Map of Recent Positive Selection in the Human Genome. PLoS Biology. 2006;4:e72
30. Fay JC, Wu CI. Hitchhiking under positive Darwinian selection. Genetics. 2000;155:1405-13
31. Biswas S, Akey JM. Genomic insights into positive selection. Trends in genetics: TIG. 2006;22:437-46
32. Wang K, Li MY, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research. 2010;38:7
33. Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM. et al. ENCODE Data in the UCSC Genome Browser: year 5 update. Nucleic Acids Research. 2013;41:D56-D63
34. Ernst J, Kellis M. ChromHMM: automating chromatin state discovery and characterization. Nature methods. 2012;9:215-6
35. Teng L, He B, Wang J, Tan K. 4DGenome: a comprehensive database of chromatin interactions. Bioinformatics. 2015;31:2560-4
36. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of affymetrix GeneChip probe level data. Nucleic Acids Research. 2003:31
37. Sampson JN, Wheeler WA, Yeager M, Panagiotou O, Wang Z, Berndt SI. et al. Analysis of Heritability and Shared Heritability Based on Genome-Wide Association Studies for 13 Cancer Types. JNCI-J Natl Cancer Inst. 2015:107
38. Voight BF, Kudaravalli S, Wen XQ, Pritchard JK. A map of recent positive selection in the human genome. Plos Biology. 2006;4:446-58
39. McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L. et al. Runs of homozygosity in European populations. American Journal of Human Genetics. 2008;83:359-72
40. Kirin M, McQuillan R, Franklin CS, Campbell H, McKeigue PM, Wilson JF. Genomic Runs of Homozygosity Record Population History and Consanguinity. Plos One. 2010:5
41. Weiss LA, Pan L, Abney M, Ober C. The sex-specific genetic architecture of quantitative traits in humans. Nature Genetics. 2006;38:218-22
42. Gibson J, Morton NE, Collins A. Extended tracts of homozygosity in outbred human populations. Hum Mol Genet. 2006;15:789-95
43. Li LH, Ho SF, Chen CH, Wei CY, Wong WC, Li LY. et al. Long contiguous stretches of homozygosity in the human genome. Human Mutation. 2006;27:1115-21
44. Lui WO, Tanenbaum DM, Larsson C. High level amplification of 1p32-33 and 2p22-24 in small cell lung carcinomas. International journal of oncology. 2001;19:451-7
45. Yan W, Song L, Liang Q, Fang Y. Progression analysis of lung squamous cell carcinomas by comparative genomic hybridization. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2005;26:158-64
46. Pifarre A, Rosell R, Monzo M, De Anta JM, Moreno I, Sanchez JJ. et al. Prognostic value of replication errors on chromosomes 2p and 3p in non-small-cell lung cancer. British journal of cancer. 1997;75:184-9
47. Wang C, Xu Z, Jin G, Hu Z, Dai J, Ma H. et al. Genome-wide analysis of runs of homozygosity identifies new susceptibility regions of lung cancer in Han Chinese. J Biomed Res. 2013;27:208-14
48. Li N, Batzer A, Daly R, Yajnik V, Skolnik E, Chardin P. et al. GUANINE-NUCLEOTIDE-RELEASING FACTOR HSOS1 BINDS TO GRB2 AND LINKS RECEPTOR TYROSINE KINASES TO RAS SIGNALING. Nature. 1993;363:85-8
49. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S. et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54
50. Wilhelm SM, Carter C, Tang LY, Wilkie D, McNabola A, Rong H. et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Research. 2004;64:7099-109
51. Xiao Z, Li L, Li Y, Zhou W, Cheng J, Liu F. et al. Rasfonin, a novel 2-pyrone derivative, induces ras-mutated Panc-1 pancreatic tumor cell death in nude mice. Cell Death & Disease. 2014;5:9
52. Lv ZH, Yang LZ. miR-124 inhibits the growth of glioblastoma through the downregulation of SOS1. Molecular Medicine Reports. 2013;8:345-9
53. Liu K, Jiang T, Ouyang YB, Shi Y, Zang YJ, Li N. et al. Nuclear EGFR impairs ASPP2-p53 complex-induced apoptosis by inducing SOS1 expression in hepatocellular carcinoma. Oncotarget. 2015;6:16507-16
54. McCarroll SA, Kuruvilla FG, Korn JM, Cawley S, Nemesh J, Wysoker A. et al. Integrated detection and population-genetic analysis of SNPs and copy number variation. Nature Genetics. 2008;40:1166-74
Author contact
![]() Corresponding authors: Tie-Lin Yang, Ph.D., Liqiang Zhi, Ph.D., Department of Joint Surgery, Honghui Hospital; The Key Laboratory of Biomedical Information Engineering of Ministry of Education, Xi'an Jiaotong University, Xi'an, P. R. China. Phone: 86-29-62818386; E-Mail: yangtielinxjtu.edu.cn; zhiliqiang2011com
Corresponding authors: Tie-Lin Yang, Ph.D., Liqiang Zhi, Ph.D., Department of Joint Surgery, Honghui Hospital; The Key Laboratory of Biomedical Information Engineering of Ministry of Education, Xi'an Jiaotong University, Xi'an, P. R. China. Phone: 86-29-62818386; E-Mail: yangtielinxjtu.edu.cn; zhiliqiang2011com