Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(24):4665-4676. doi:10.7150/jca.27295 This issue Cite
Research Paper
Utilization of Reactive Oxygen Species Targeted Therapy to Prolong the Efficacy of BRAF Inhibitors in Melanoma
Long Yuan1, Rosalin Mishra1, Hima Patel1, Safnas Abdulsalam2, Kenneth D. Greis3, Ana Luisa Kadekaro4, Edward J. Merino2, Joan T. Garrett1 ![]()
1. James L. Winkle College of Pharmacy,
2. Department of Chemistry,
3. Department of Cancer Biology,
4. Department of Dermatology, University of Cincinnati, Cincinnati, Ohio
Received 2018-5-16; Accepted 2018-10-13; Published 2018-11-24
Abstract
BRAF mutations occur in about 50% of melanoma patients. FDA approved BRAF and MEK inhibitors have improved the prognosis of patients with BRAF mutations. However, all responders develop resistance typically within one year of treatment. Recent observations demonstrate that BRAF inhibitors induce reactive oxygen species (ROS) in melanoma cells. A100, identified from a library screen, is a ROS-activated prodrug that self-cyclizes into a stable bicyclic ring and causes DNA double strand breaks. We proposed to examine if ROS activated therapy will inhibit tumor growth and evade resistance to BRAF inhibitors. In this study, the BRAF inhibitor dabrafenib was used to generate resistant cell lines (A375DR, SK-MEL-24DR and WM-115DR). Flow cytometry experiments showed that ROS levels are increased in these dabrafenib-resistant cells as compared to parental cells, assessed by both the H2DCFDA and MitoSOX assays. Furthermore, we observed that resistant cells had increased levels of the mitochondrial enzymes SOD2 and PRDX1, which function to reduce ROS levels in the mitochondria. We found that A100 sensitized the resistant melanoma cells to dabrafenib and induced DNA damage. Co-treatment of both A100 and dabrafenib significantly suppressed in vitro cell proliferation and three- dimensional (3D) matrigel growth. This study suggests that the combination of A100 with a BRAF inhibitor could be a potential strategy to treat melanoma patients with BRAF mutations.
Keywords: BRAF, melanoma, dabrafenib, A100, targeted therapy
Introduction
The identification of BRAF mutants in cancer piloted a new era in the treatment of advanced melanomas. Approximately one-half of melanomas contain a BRAFT1799A transversion, encoding the constitutively active BRAF‑V600E oncoprotein. Prior to the approval of BRAF inhibitors, patients with BRAF-mutant melanoma had a worse prognosis than those who expressed wild-type (WT) BRAF [1]. However, the natural history of metastatic BRAF-mutant melanoma has been changed by the recent approval of selective ATP-competitive RAF kinase inhibitors, including vemurafenib and dabrafenib, targeting the mutant BRAF protein. Treatment of melanoma patients with mutant BrafV600E with either BRAF inhibitor resulted in a response rate of about 50% and median progression-free survival of about 5 months [2, 3]. Furthermore, the combination of BRAF inhibitors and MEK inhibitors has been extensively explored with successes in clinical trials [4, 5]. Despite these successes, most treated patients will eventually exhibit disease progression. The acquired resistance to these inhibitors has limited their long-term efficacy, and has stirred interest in understanding the mechanisms underlying resistance.
Reactive oxygen species (ROS) are oxygen containing heterogeneous group with chemically reactive ions and molecules [6]. ROS include hydrogen peroxide (H2O2), superoxide and hydroxyl free radicals. Peroxisomes, the endoplasmic reticulum, and the mitochondria are major compartments for ROS metabolic reactions [7]. ROS are known to modify protein activity and signaling in events critical for cell growth and survival [8]. At the biochemical level, ROS catalyze the formation of disulfides and sulfenic acids leading to reversible changes in protein structure and function. It has recently been shown that BRAF signaling results in transcriptional upregulation of the oxidase NOX4, which promotes ROS generation [9]. It has been demonstrated that BRAF inhibitors induce ROS in melanoma cells through PGC1α-induced mitochondria biogenesis [10]. This BRAF inhibitor-induced ROS has been implicated in cellular adaptation including activation of PDKs (pyruvate dehydrogenase kinases) [11]. Oxidative stress induced by ROS has been demonstrated to activate the MAPK pathway, and p38MAPK may be an apoptosis factor mediated by oxidative stress [12, 13]. Alterations in ROS homeostasis are now recognized as critical events in cancer etiology and resistance. We seek to convert BRAF inhibitor-induced ROS into a lethal phenotype by designing prodrugs selectively activated in the presence of high ROS.
Recently we have characterized a ROS activated pro-drug, A100 (referred to as RAC1 in reference [14]). Biochemically, A100 is an amine-containing compound which oxidatively cyclizes into a stable bicyclic DNA ring after ROS quenching and causes DNA double strand breaks [14, 15]. When reduced A100 is not toxic and it is difficult to be oxidized into its active (cytotoxic) form. A100 is only modestly activated in most cells and is not overly cytotoxic. Conditions with high ROS will lead to greater activation of A100 within cancer cells and thus a selectivity. Our hypothesis is that BRAF inhibition will induce the high ROS conditions as the target cells begin to compensate for the blockade. In the current work, we found that A100 sensitizes BRAF mutant melanoma cells to the BRAF inhibitor dabrafenib as assessed by cells grown in two-dimensional culture and a basement membrane of matrigel. Furthermore, the combination of BRAF inhibitor and ROS activated pro-drug increases ɣ-H2AX, a sensor of DNA damage [16]. We next generated BRAF inhibitor resistant cells and found BRAF inhibitor resistant cells have increased ROS compared to cells sensitive to BRAF inhibition. We found that proteins with antioxidant functions are upregulated in the mitochondria of BRAF inhibitor resistant cells including SOD2 and PRDX1. We observed A100 restores sensitivity to dabrafenib in cells rendered resistant to BRAF inhibition as assessed by cells grown in two-dimensional culture and a basement membrane of matrigel. Analogous to BRAF inhibitor sensitive cells, the combination of BRAF inhibitor and ROS activated pro-drug increases ɣ-H2AX in BRAF inhibitor resistant cells. These findings suggest that A100 and related ROS activated pro-drug compounds could be useful therapeutic agents where a BRAF inhibitor has failed as the first line of treatment in melanoma patients harboring BRAFV600E mutation.
Materials and Methods
Cell culture and inhibitors
Human melanoma cells (A375, SK-MEL-24, WM-115) were obtained from American Type Culture Collection (Manassas, VA). Donor melanocytes were generously provided by Dr. Zalfa Abdel-Malek. Dabrafenib was obtained from LC Laboratories. A375 cells were maintained in DMEM with 10% fetal bovine serum and 1% penicillin-streptomycin at 37°C in 5% CO2. WM-115 and SK-MEL-24 cells were maintained in MEM with 10% and 15% fetal bovine serum respectively, 1% penicillin-streptomycin at 37°C in 5% CO2 in a humidified incubator.
Generation of dabrafenib resistant (DR) cell lines
Briefly, melanoma cell lines (A375, SK-MEL-24, WM-115) were initially treated with 0.01 µM dabrafenib and subjected to gradual dose escalation of dabrafenib (0-2.4 µM) over a span of 2-3 months and finally maintained in 2.4 µM dabrafenib. Media and dabrafenib were replenished every alternate day.
Immunoblotting
Melanoma cells were treated ± 3.2 µM A100 and/or ± 0.8 µM dabrafenib and lysed in RIPA buffer containing protease and phosphatase inhibitors. Mitochondrial extracts were prepared following manufacturer's procedures using the Mitochondria Isolation Kit for Cultured Cells (Thermo Fisher). Nuclear extracts were prepared using the NE-PER Nuclear and Cytoplasmic Extraction Reagents Kit (Thermo Fisher) as per manufacturer's guidelines. Immunoblotting was performed using specific primary antibodies for γ-H2AX, H2AX, HSP60, p-ERK1/2, ERK1/2, PARP, PRDX1, SOD2 (Cell Signaling Technology) and actin (Santa Cruz biotechnology). Anti-rabbit IgG-horse radish peroxidase (HRP) was used as the secondary antibody. Actin, PARP, and HSP60 served as loading controls.
MTT cell proliferation assay
Cells (126c, 1729c, 136C+ melanocytes and A375, WM115, SK-MEL-24 melanoma cells) were plated into a 96-well plate at a density of 3.0 x 104 cells per well in triplicate and allowed to grow for 24 hours. After 24 hours, cells were treated with increasing concentrations of A100 (10 nM-100 µM) and DMSO as control. After 72 hours, the media containing the drug was replaced with 5 mg/mL MTT (3-(4,5-dimethylthiazol- 2-yl)-2,5-diphenyltetrazolium bromide) dissolved in cell line specific media and incubated for 4 hours. After 4 hours, the media was aspirated and crystals were dissolved with isopropanol (Molecular grade, Fisher BioReagents). The absorbance was read at 570 nm in a microplate reader (SPECTRAmax PLUS Microplate Spectrophotometer Plate Reader, Molecular Devices Corporation). The IC50 was computed using Graphpad Prism 7. Data were represented as the mean of at least two independent experiments ± SEM.
Cell viability assay
50,000 cells/well (A375, SK-MEL-24 and WM115 sensitive or dabrafenib resistant) were plated in triplicate in a 6 well plate and allowed to grow in the absence or presence of 0.8 µM dabrafenib (dab) and/ or 3.2 µM A100 as indicated. Medium and inhibitors were replenished every 2 days. After 4-6 days cells were stained with crystal violet in 10% methanol.
Matrigel colony formation assay
A375, SK-MEL-24 and WM115 cells (10,000 cells/well) were seeded on matrigel in 96 well plate and allowed to grow in the absence or presence of 0.8 µM dabrafenib (dab) and/or 3.2 µM A100 as indicated. Medium and inhibitors were replenished every 3 days. Images were captured from 5 random fields of duplicate treatment groups after 9 days of cell seeding. Acini area was determined using ImageJ software where all acini from 10 images per treatment group were measured and averaged.
Measurement of intracellular ROS
The production of intracellular ROS was measured using the oxidation sensitive DCFH-DA by flow cytometry. WM-115, SK-MEL-24 and A375 sensitive and DR cells (4×105) were treated in DMSO and 0.8 μM of dabrafenib for 24 hours. The cells were resuspended in HBSS and incubated with 5 μM of 7-Aminoactinomycin D (7-AAD) or Dichloro-dihydro-fluorescein diacetate (DCFH-DA) for 30 minutes at 37°C. The cells were washed, resuspended in HBSS on ice and centrifuged at 4°C. The green fluorescence (DCFH-DA) and red fluorescence (7-AAD) were measured using the FL-1 and FL-2 channels of the FACS Diva respectively, and analyzed using the CellQuest software. Mitochondrial O2.- generation was measured using the mitochondria-targeting probe MitoSOX Red. Briefly, the cell lines were treated with DMSO or 0.8 μM of dabrafenib for 24 hours. The cells were suspended in HBSS buffer and incubated with 5 μM MitoSOX Red and 7-AAD for 15 minutes at 37°C. The cells were washed, resuspended in HBSS buffer on ice, and centrifuged at 4°C. The red fluorescence was measured using the FL-2 channels of the FACS Diva and analyzed by CellQuest software.
Proteomics analysis
Mitochondrial extracts (20 μg) from three WM-115 samples (control) and three experimental samples (WM-115 DR) were obtained using Mitochondria Isolation Kit for Mammalian Cells (Thermo Fisher Scientific). The mitochondrial proteins were solubilized in Laemmli gel buffer, run for about 2 cm into a preparative mini 1D gel, and digested with trypsin. The extracted peptides were labeled with iTRAQ reagents with a control pool (labeled with the 114 reporter iTRAQ tag) generated from an equal mix of proteins from all samples as the common denominator for relative quantification. Three biological replicates for the two samples groups WM-115 (labeled with the 115 reporter) and WM-115 DR (labeled with the 116 reporter) were compared to the control pools of mitochondrial samples all as described in detail previously [17].
The 20 µg of peptides from each of individual iTRAQ-labeled samples were mixed in equal amounts and further separated by high pH reverse phase HPLC. Briefly, the mixed samples were reconstituted in 1 mL of high-pHRPLC solvent (10% Ammonium Formate in water pH 10) and fractionated by high-pHRPLC chromatography on a XBridge C18, 5 µm, 250 x 4.6 mm column (Waters Corporation) by employing an increasing gradient of solvent B (10 % Ammonium Formate in 90 % Acetonitrile pH 10) on an Ultimate Plus HPLC (LC Packings) with a flow rate of 250 µL/min. 1.5 min fractions (375 µL) over 90 minutes were collected for each separation for a total of 60 fractions. The high pH fractions were then concatenated by pooling every 6th fraction (1, 7, 13, 19, 25, 31, 37, 43, 49, 55; then 2, 8, 14, 20, 26, 32, 38, 44, 50, 56;…6, 12, 18, 24, 30, 36, 42, 48, 54, 60) to generate 6 pooled fractions [18].
NanoLC-MS/MS for protein identification and quantification for each of the six concatenated fractions was performed on a Sciex 5600+ Triple-Tof mass spectrometry system followed by databases searching and relative quantification based on the iTRAQ reporter ion ratios using ProteinPilot software (Sciex) as previously described [17] except that the output files from each of the 6 concatenated fractions were analyzed as a merged search with the ProteinPilot software (version 4.5, revision 1656) against a SWISS-PROT database of human. The following search specification were also included in the search, all biological modifications as variable modifications with methylthiocysteine as a fixed modification. For protein identification and quantification, a minimum of 2 peptides at 99% or greater confidence was required. Relative ratios for the triplicate control and triplicate drug resistant mitochondrial sample were captured as log ratios.
Statistical analysis
Data are shown as the mean ± standard error of mean (SEM) and representative of at least three independent experiments unless indicated otherwise. Statistical analysis was performed by two sample 't'-test using ANOVA (Graph Pad Prism 7). The data was considered statistically significant if p<0.05.
Results
A100 has increased specificity in melanoma cells over melanocytes and induces a sensor of DNA damage in melanoma cells
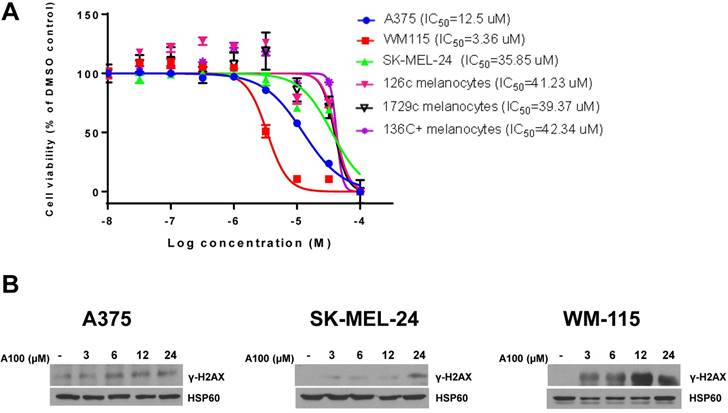
We evaluated the toxicity effect of A100 (10 nM - 100 µM) on 126c, 1729c, 136c+ donor melanocytes and A375, SK-MEL-24 and WM-115 melanoma cell lines using MTT cell proliferation assays. There was variable sensitivity of melanoma cells to A100 with WM115 and A375 cells being more sensitive to A100 versus melanocytes whereas SK-MEL-24 cells had a similar IC50 to melanocytes (Figure 1A). Next, we speculated that increasing the concentration of A100 in A375, SK-MEL-24 and WM-115 melanoma cell lines may induce DNA damage. Double-strand break repair is a complex pathway involving mainly homologous recombination (HR) and non-homologous end joining (NHEJ) [19]. Following the detection of a double-strand break, histone protein H2AX is phosphorylated at Ser 139. The phosphorylated H2AX is termed γ-H2AX. This phosphorylation is one of the most common signals for double-strand break repair and signals to initiate the repair. The melanoma cell lines were treated with increasing concentration of A100 from 3 to 24 µM for 24 hours and nuclear extracts were processed for immunoblotting. The data indicate that increasing concentration of A100 induces γ-H2AX in all three cell lines (Figure 1B). Notably, a higher concentration of A100 (24 µM) was necessary to induce γ-H2AX in SK-MEL-24 cells, correlating with a higher IC50 value in SK-MEL-24 cells. These data indicate that A100 shows preferential toxicity in 2 of 3 melanoma cell lines over melanocytes and that A100 induces DNA damage in melanoma cell lines.
Characterization of A100 in melanoma cells. (A) Melanoma cell lines (A375, WM-115, SK-MEL-24) and melanocytes (126c melanocytes, 1729c melanocytes, 136C+ melanocytes) were plated in triplicate in 96 well plates and treated for 72 hours with A100 from 10 nM, 32 nM, 100 nM, 320 nM, 1µM, 3.2 µM, 10 µM, 32 µM, 100 µM. Cells were treated with MTT at 5 mg/mL for 4 hours and absorbance read at 570 nm in a microtiter plate reader. IC50 values were determined using GraphPad Prism 7 (n=2 independent experiments performed in triplicate ± SEM). (B) A375, SK-MEL-24 and WM-115 cells were treated with 3, 6, 12, 24 µM A100. 24 hours after treatment nuclear extracts were prepared and separated in a 7.5% SDS gel followed by immunoblot analysis with indicated antibodies. Anti-HSP60 antibody is used as a loading control.
A100 sensitizes BRAF mutant melanoma cells to BRAF inhibition
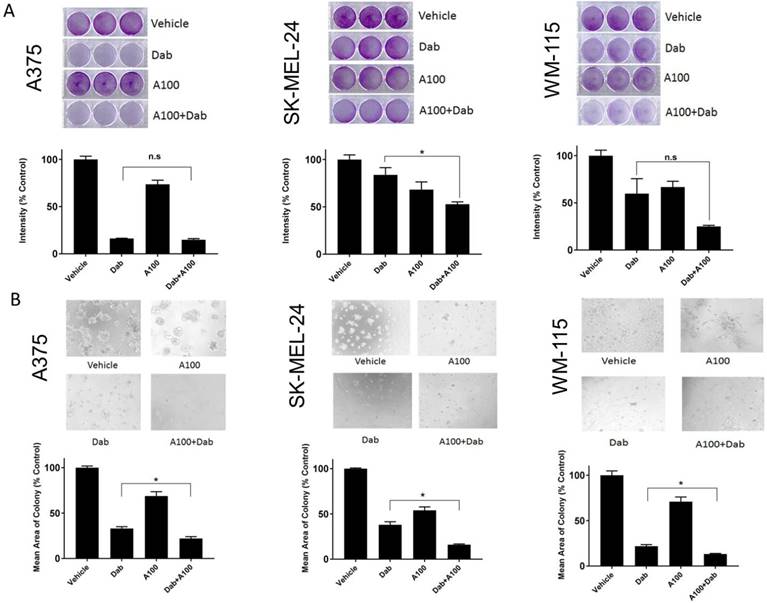
We speculated that the combination of A100 and dabrafenib could be an effective method of inhibiting proliferation in melanoma cell lines harboring endogenous BRAFV600E mutation (A375, SK-MEL-24, and WM-115) [20]. The effect of 0.8 µM dabrafenib (Dab) in the presence or absence of 3.2 µM A100 was assessed on the cell proliferation of melanoma cells using 2D-crystal violet and 3D-matrigel assays. We observed a decrease in cell growth of SK-MEL-24 and WM115 cells when subjected to the combination of A100 and dabrafenib compared to single agent dabrafenib that was statistically significant only for SK-MEL-24 cells (Figure 2A, middle panel). In A375 cells, single agent dabrafenib had almost similar growth inhibitory effect as that of combined treatment of dabrafenib and A100 as indicated by crystal violet (Figure 2A). Examination of cells grown in a basement membrane of matrigel revealed that the addition of A100 to dabrafenib resulted in a statistically significant reduction in colony area compared to dabrafenib alone in the three cell lines (Figure 2B).
A100 sensitizes BRAF mutant melanoma cells to BRAF inhibitor. (A) 50,000 cells/well (A375, SK-MEL-24 and WM115) were plated in triplicate and allowed to grow in the absence or presence of 0.8 µM dabrafenib (dab) and/or 3.2 µM A100 as indicated. Medium and inhibitors were replenished every 2 days. After growth of 4 to 6 days when vehicle treated cells were greater than 80% confluent, cells were stained with crystal violet. Intensities analyzed by odyssey infrared imaging system and represented using bar graph. Statistical analysis by one-way ANOVA (*p < 0.05) SEM (n=3 independent experiments performed in triplicate). (B) A375, SK-MEL-24 and WM115 cells (10,000 cells/well) were seeded in matrigel and allowed to grow in the absence or presence of 0.8 µM dabrafenib (dab) and/or 3.2 µM A100 as indicated. Medium and inhibitors were replenished every 3 days. Images were recorded 9 days after cell seeding. Acini size was determined using ImageJ software. Each bar graph represents the mean acini size + SEM of 5 random fields (*p < 0.05). Data are representative of three independent experiments.
The combination of ROS activated pro-drug A100 and BRAF inhibitor induces γ-H2AX levels, a sensor of DNA damage
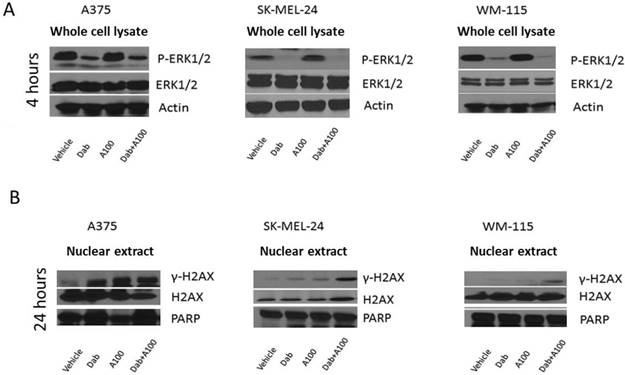
Melanoma patients harboring BRAFV600E mutation have hyperactivation of MAPK pathway which activates ERK1/2 leading to uncontrolled tumor growth [21]. Hence, we examined alteration in MAPK (p-ERK1/2) and γ-H2AX levels in response to combined treatment of dabrafenib and A100. A375, SK-MEL-24 and WM-115 cell lines were treated with dabrafenib in the presence or absence of A100 for 4 hours and processed for immunoblotting. The data indicated that dabrafenib as a single agent had similar effect in reducing phosphorylated ERK1/2 as compared to co-treatment with A100 in A375, SK-MEL-24 and WM-115 cell lines (Figure 3A). To assess γ-H2AX levels, nuclear extracts were prepared post 24 hour treatment with vehicle (DMSO), dabrafenib (0.8 µM), A100 (3.2 µM), or the combination of both. The data indicated that there was a significant upregulation in γ-H2AX levels in A375, SK-MEL-24 and WM-115 cell lines subjected to co-treatment of dabrafenib and A100 as compared to vehicle or individual agents (Figure 3B). These data suggest that A100 in combination with dabrafinib could induce DNA damage in BRAF mutant melanoma cells.
A100 induces DNA damage response in BRAF mutant melanoma cell lines. (A) A375, SK-MEL-24 and WM-115 cells were treated with 0.8 µM dabrafenib (dab) and/or 3.2 µM A100 for 4 hours. Whole cell lysates were prepared and separated in a 7.5% SDS gel followed by immunoblot analysis with indicated antibodies. (B) Nuclear extracts were prepared following 24 h treatment as indicated and separated in a 7.5% SDS gel followed by immunoblot analysis using indicated antibodies.
Dabrafenib resistant (DR) cells grow and have persistent activation of ERK-1/2 in the presence of dabrafenib
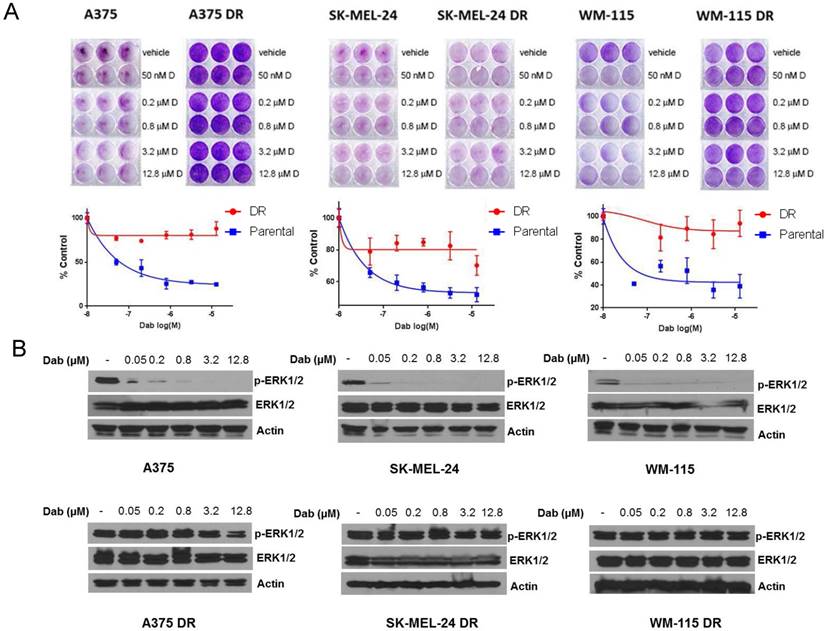
Next, we generated dabrafenib resistant cell lines. We treated three melanoma cell lines (A375, SK-MEL-24 and WM-115) with increasing concentration of dabrafenib (0-2.4 µM) until the cells tolerated and grew in the presence of 2.4 µM dabrafenib. Cell proliferation assays were performed in melanoma cells sensitive to dabrafenib and those rendered dabrafenib resistant (DR). The results indicated that the parental cell lines (A375, SK-MEL-24 and WM-115) treated with increasing concentrations of dabrafenib (0-12.8 µM) exhibited variable sensitivity to dabrafenib with A375 cells being the most sensitive and SK-MEL-24 cells showing less sensitivity to dabrafenib. All DR cell lines showed decreased sensitivity compared to parental cell lines with maintenance of at least 80% proliferation in the presence 12.8 µM dabrafenib (Figure 4A). We examined p-ERK1/2 levels in the presence of increasing concentrations of dabrafenib from 0.05 µM to 12.8 µM. Dabrafenib treatment resulted in a reduction in p-ERK1/2 levels in all three sensitive cell lines even at a concentration of 0.05 µM, while resistant DR cells showed persistent activation of ERK even at 12.8 µM dabrafenib (Figure 4B).
Generation of dabrafenib resistant cell lines. (A) A375, SK-MEL-24 and WM-115 cells were treated with dabrafenib or DMSO with increasing concentrations for two to three months until cells proliferated in the presence of 2.4 µM dabrafenib. The parental and dabrafenib-resistant cells were allowed to grow in the absence or presence of various concentrations of dabrafenib (0, 0.05, 0.2, 0.8, 3.2 and 12.8 µM) as indicated. Cells were stained with crystal violet and quantified using odyssey infrared imaging. Cell proliferation was normalized to DMSO treated cells. Error bars represent ± SEM (n=3 independent experiments performed in triplicate). (B) A375, A375 DR, SK-MEL-24, SK-MEL-24DR, WM-115 and WM-115DR cells were treated with 0.05, 0.2, 0.8, 3.2, 12.8 µM dabrafenib. 4 hours after treatment whole cell lysates were prepared and separated in a 7.5% SDS gel followed by immunoblot analysis with indicated antibodies.
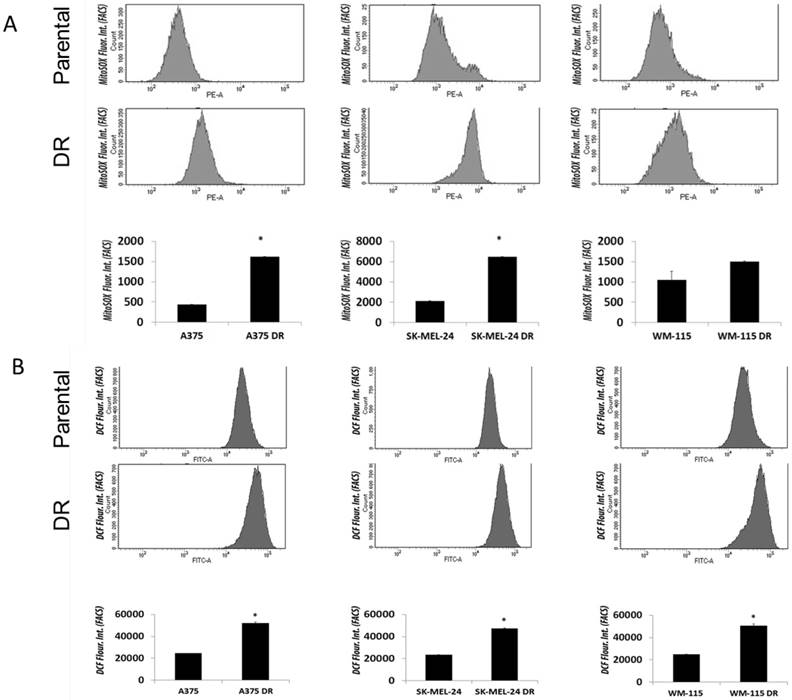
Dabrafenib resistant (DR) melanoma cells exhibit elevated ROS levels compared to respective dabrafenib sensitive parental cell lines
The role of BRAF inhibitors in inducing ROS in melanoma cells through PGC1α-dependent mitochondria biogenesis has previously been explored [10]. We analyzed ROS levels in parental versus dabrafenib resistant cell lines. Superoxide levels were significantly increased in both A375 and SK-MEL-24 DR cell lines compared with parental cell lines as measured using the MitoSOX assay (Figure 5A). Levels of mitochondrial hydrogen peroxide measured using CDCFH2-DA (5,6-carboxy-2′, 7′-dichlorodihydrofluorescein diacetate) probe was significantly increased in all three dabrafenib resistant melanoma cell lines (A375DR, WM-115DR and SK-MEL-24DR) compared to their respective parental cell lines (Figure 5B). Overall, the data indicated that ROS levels are significantly augmented in dabrafenib resistant cell lines in comparison to dabrafenib sensitive cells suggesting a possible involvement of ROS- mediated resistance.
Dabrafenib resistant melanoma cells exhibited elevated ROS levels compared to parental cell lines. (A) Basal superoxide levels in A375/A375DR, SK-MEL-24/SK-MEL-24DR and WM115/ WM115DR cells were measured by MitoSox assay. Results are presented as mean ± SEM from biological triplicates and represented using bar graph. Data are representative of three independent experiments. (B) ROS levels were also measured by DCF assay under above conditions. Results are presented as means ± SEM from biological triplicates and represented graphically. Data are representative of three independent experiments.
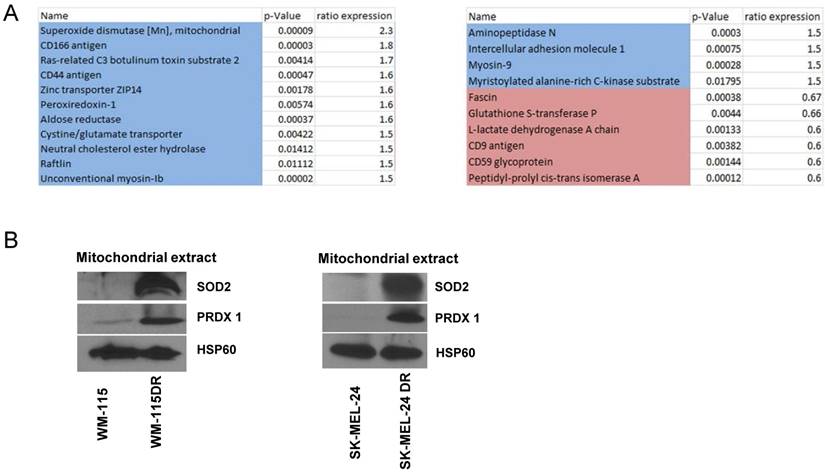
Dabrafenib resistant cell lines exhibit increased ROS-associated mitochondrial enzymes expression compared to parental cell lines
We next aimed to determine changes to mitochondrial proteins in response to dabrafenib resistance. Samples of mitochondrial fractions from WM-115 and WM-115 DR were collected (n=3). Protein changes were identified by comparing a common control pool of all samples by isobaric tags for relative quantitation (iTRAQ) labeling and nano liquid chromatography-tandem mass spectrometry (LC-MS/MS). Protein fold change greater than 1.5 times with significant reproducibility among the biological replicates are shown in Figure 6A. We found that proteins with anti-oxidant functions are increased in DR cells including SOD2 (superoxide dismutase [Mn]) [22] and Peroxiredoxin-1 (PRDX1) [23]. We postulate that these enzymes are increased to compensate for the increased ROS observed in dabrafenib-resistant cells (Figure 5). Immunoblotting data indicated that mitochondrial extracts isolated from dabrafenib resistant WM-115DR cells had elevated levels of SOD2 and PRDX1 compared to parental WM-115. Based on the proteomics and immunoblot data obtained from WM-115 and WM-115DR cells, we further examined whether a similar trend was observed in the upregulation of SOD2 and PRDX1 in SK-MEL24DR and A375DR cell lines versus the respective parental cells. The data indicated a similar increase in the levels of SOD2 and PRDX1 in SK-MEL-24DR compared to SK-MEL-24 parental cells (Figure 6B). We did not observe increased levels of SOD2 or PRDX1 in A375DR cells versus A375 parental cells (data not shown).
Proteomics and immunoblotting analyzing antioxidant enzyme levels in dabrafenib resistant and sensitive cell lines. (A) Six samples of mitochondrial fractions from human melanoma cell lines from WM-115, WM-115DR were collected. Protein changes by comparing a common control pool of all samples by iTRAQ labelling and nano LC-MS/MS were identified. Protein fold change greater than 1.5x were shown in the panel. Blue highlight means more in resistant cells, red means less in resistant cells. Samples were conducted in triplicates. (B) Mitochondrial extracts were collected from WM-115, WM-115DR, SK-MEL-24, SK-MEL-24DR, and SOD2 level was analyzed. HSP60 served as loading control.
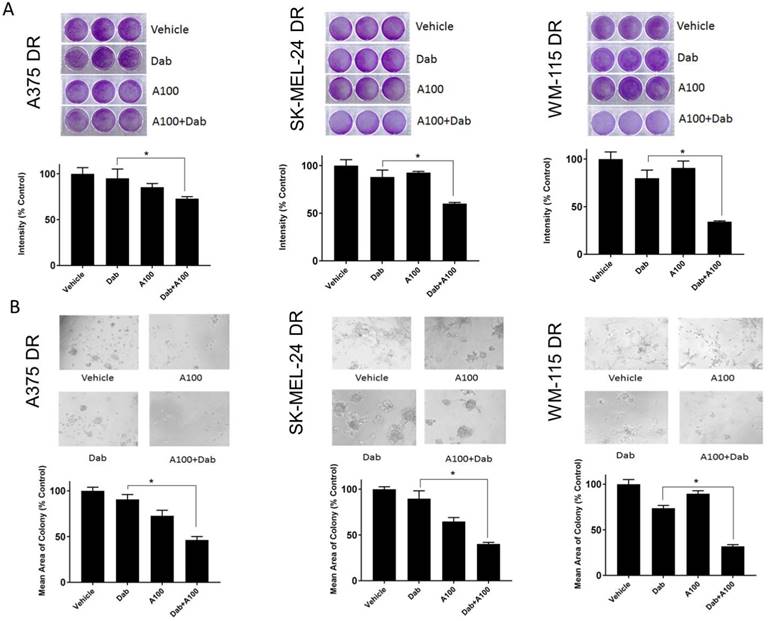
A100 sensitizes dabrafenib resistant melanoma cells to a BRAF inhibitor
The effect of A100 was assessed on the growth of dabrafenib resistant cell lines in the presence or absence of dabrafenib. The crystal violet data indicated that A100 in combination with dabrafenib significantly suppressed cell proliferation of dabrafenib resistant cell lines (A375DR, SK-MEL-24DR and WM-115DR) compared to individual agents (Figure 7A). A100 when treated in combination with dabrafenib suppressed colony formation of dabrafenib resistant cell lines grown in a basement membrane of matrigel (Figure 7B) illustrating that A100 restores sensitivity to BRAF inhibition in cells rendered BRAF inhibitor resistant.
A100 sensitizes dabrafenib resistant melanoma cells to BRAF inhibition. (A) A375DR, SK-MEL-24DR and WM115DR (50,000 cells/well) were plated in triplicate and allowed to grow in the absence or presence of 0.8 µM dabrafenib (Dab) and/or 3.2 µM A100 as indicated. Medium and inhibitors were replenished every 2 days. After growth of 4 to 6 days when vehicle treated cells were greater than 80% confluent, cells were stained with crystal violet. Intensities analyzed by odyssey infrared imaging system and represented using bar graph. Statistical analysis by one-way ANOVA (*p < 0.05, n=3 independent experiments performed in triplicate). (B) A375DR, SK-MEL-24DR and WM115DR cells were seeded on matrigel and allowed to grow in the absence or presence of 0.8 µM dabrafenib (dab) and/or 3.2 µM A100 as indicated. Medium and inhibitors were replenished every 3 days. Images were captured after 9 days. Acini size was quantified using ImageJ software. Each bar graph represents the mean acini size + SEM of 5 random fields (*p < 0.05). Data are representative of three independent experiments.
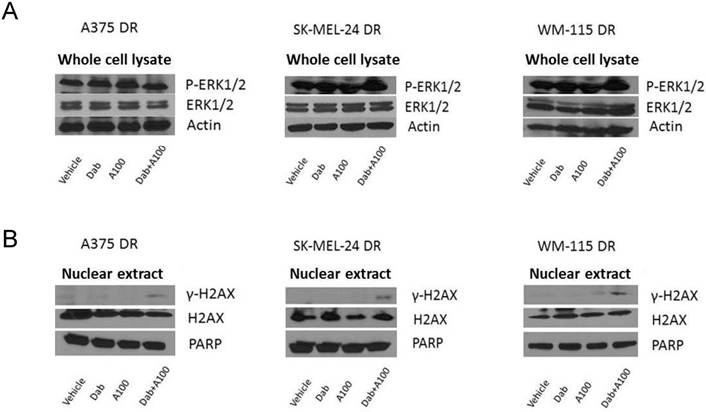
The combination of a ROS activated pro-drug, A100, and BRAF inhibitor induce DNA damage in dabrafenib resistant cell lines
Dabrafenib-resistant cells showed persistent activation of ERK even in the presence of dabrafenib (Figure 8A). DNA damage in response to A100 (3.2 µM) and/or dabrafenib (0.8 µM) was analyzed by measuring nuclear γ-H2AX levels by immunoblotting. A100 in combination with dabrafenib induced γ-H2AX levels whereas single agent A100 or dabrafenib failed to induce γ-H2AX (Figure 8B). These data indicate that even with persistent MAPK signaling, the combination of a BRAF inhibitor and a ROS activated pro-drug are sufficient to induce a sensor of DNA double strand breaks.
A100 in combination with dabrafenib induces DNA damage in dabrafenib-resistant cell lines. (A) A375DR, SK-MEL-24DR and WM115DR cells were treated with 0.8 µM dabrafenib (dab) and/or 3.2 µM A100 as indicated. Whole cell lysates were prepared post 4 hours treatment and separated in a 7.5% SDS gel followed by immunoblot analysis with indicated antibodies. Actin served as loading control. (B) Nuclear extracts were prepared after 24 hour treatment and separated in a 7.5% SDS gel followed by immunoblot analysis with indicated antibodies. PARP was used as nuclear loading control.
Discussion
Treatment strategies for melanoma patients with BRAF mutations have been well investigated. Approximately 15% of patients with BRAF mutant metastatic melanoma exhibit primary resistance to BRAF and MEK inhibitors as these patients do not achieve tumor regression. Patients that do have a reduction in tumor growth invariably recur within 6 months to one year of treatment initiation [24, 25]. Therefore, current research is focused on pursuing methods of overcoming resistance to BRAF and MEK inhibition. Dabrafenib, a drug approved by the FDA in 2013, has been used as a single agent treatment for patients with BRAFV600E mutation advanced melanoma [26]. Later in 2014, a combination of MEK and BRAF inhibitor trametinib and dabrafenib therapy became FDA approved [27]. The combination has improved survival in metastatic melanoma for approximately one year; unfortunately, resistance continues to be problematic [28].
Our study is focused on A100 that can be used as a ROS-activated prodrug which can cause DNA damage in melanoma cells. There are several pieces of evidence suggesting that the increased ROS in cancer cells can induce agents which could act as prodrugs for site specific activation in the tumor microenvironment because of the presence of ROS. Such an approach can make a cytotoxic agent behave as a targeted chemotherapeutic drug [29]. However, there are very few reports of ROS-activated prodrug in melanoma. The study by Cohen et al. demonstrated that H2O2-activated MMP inhibitor to act as a prodrug by protecting the hydroxyl group of the zinc binding group with a boronic ester [30]. The same group also designed and synthesized a new type of nitrogen mustard prodrug which is activated in the presence of high ROS present in cancer cells triggered by H2O2 to release the active chemotherapy agent. Another study shows that amionoferrocene-based prodrug reacts with H2O2 to induce quinone methides and iron ions which catalyze the generation of hydroxyl radical [31]. The prodrug is toxic toward human promyelocytic leukemia and human glioblastoma-astrocytoma but not towards non-malignant fibroblast.
Based on publications demonstrating that BRAF inhibitors increase ROS [10, 32], we postulated that the addition of a ROS activated pro-drug to a BRAF inhibitor would result in greater inhibition of growth in BRAF mutant melanomas. A100 is a newly identified ROS activated amine-containing compound used previously for targeting myeloma [14, 15]. Vadukoot et al. designed and synthesized new hydrogen peroxide-activated agents that can be activated in some high ROS- accumulated myeloid leukemia cell lines [15]. The specific molecular target(s) of A100 is unknown and requires further exploration. In our work examining A100 in BRAF mutant melanoma, we observed differences in sensitivity of cells to dabrafenib or A100 when cells were grown in two-dimensional culture (Figure 2A) versus a reconstituted basement membrane (Figure 2B) which is likely due to the inherent differences in growth conditions present in a basement membrane versus growth on plastic. We found that treatment with the combination of A100 and dabrafenib resulted in statistically significant reduction in colony size compared to dabrafenib treatment in parental and BRAF inhibitor resistant cells grown on a basement membrane of matrigel (Figure 2B and Figure 7B).
We observed increased ROS levels in BRAF inhibitor resistant cells (Figure 5). The higher ROS levels in dabrafenib resistant melanoma cells suggest that chronic treatment with the BRAF inhibitor dabrafenib causes an increase in ROS levels. The increased levels of ROS could lead to further activation of A100. However, BRAF inhibitor resistant cells were not markedly more sensitive to the ROS activated pro-drug A100 compared to BRAF inhibitor sensitive cells. The dabrafenib resistant cells additionally demonstrated increased levels of the antioxidant enzymes SOD2 and PRDX1. SOD2 is a mitochondrial superoxide scavenger and H2O2 regulator that can be repressed or activated in cancers subject to context-dependent stimuli [33]. PRDX1 has peroxidase activity and can eliminate superoxide, scavenging oxygen free radicals. PRDX1 has been implicated in tumor cell proliferation and metastasis [34, 35]. The elevated levels of these antioxidant enzymes in BRAF inhibitor resistant cells underscore the complexity of differences between BRAF inhibitor sensitive and resistant cells.
We observed that, as expected, dabrafenib inhibits MAPK signaling in BRAF inhibitor sensitive but not resistant cells (Figure 3A, 8A). Strikingly, the combination of BRAF inhibitor and A100 induce DNA damage and repair system as assessed by ɣ-H2AX levels (Figure 3B, 8B). ɣ-H2AX is a marker of activated DNA damage and is overexpressed in many malignancies and their precursor lesions [16]. Interestingly, A100 as a single agent was not sufficient to induce ɣ-H2AX in BRAF inhibitor resistant cells (Figure 7B) but the combination of BRAF inhibitor with A100 did induce ɣ-H2AX. How dabrafenib in combination with A100 results in increased levels of a sensor of DNA damage still needs to be mechanistically explored.
Overall, this study suggests that the combination of a ROS activated pro-drug (A100) with BRAF inhibitor (dabrafenib) could be a potential strategy to treat BRAF-mutant melanoma patients. This is based on the ability of A100 to restore sensitivity to BRAF inhibition in cells rendered resistant to BRAF inhibition and the induction of DNA damage response with co-treatment of ROS activated agent and BRAF inhibitor.
Acknowledgements
Research was funded by University of Cincinnati Pilot Translational Research and Innovative Core Grant Program. The comparative proteomics data were collected on a nanoLC-MSMS system purchased in part by a National Institutes of Health sponsored Shared Instrumentation grant (S10 RR027015) to KDG. We thank Dr. Zalfa Abdel-Malek for beneficial and thoughtful discussions and providing donor melanocytes.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Holderfield M, Deuker MM, McCormick F, McMahon M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14:455-67
2. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J. et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-16
3. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M. et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-65
4. Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D. et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30-9
5. Ascierto PA, McArthur GA, Dreno B, Atkinson V, Liszkay G, Di Giacomo AM. et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol. 2016;17:1248-60
6. Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453-62
7. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931-47
8. AbdulSalam SF, Thowfeik FS, Merino EJ. Excessive Reactive Oxygen Species and Exotic DNA Lesions as an Exploitable Liability. Biochemistry. 2016;55:5341-52
9. Azouzi N, Cailloux J, Cazarin JM, Knauf JA, Cracchiolo J, Al Ghuzlan A. et al. NADPH Oxidase NOX4 Is a Critical Mediator of BRAF(V600E)-Induced Downregulation of the Sodium/Iodide Symporter in Papillary Thyroid Carcinomas. Antioxid Redox Signal. 2017;26:864-77
10. Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC. et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell. 2013;23:302-15
11. Cesi G, Walbrecq G, Zimmer A, Kreis S, Haan C. ROS production induced by BRAF inhibitor treatment rewires metabolic processes affecting cell growth of melanoma cells. Mol Cancer. 2017;16:102
12. Wang H, Jiang D, Liu J, Ye S, Xiao S, Wang W. et al. Compound K induces apoptosis of bladder cancer T24 cells via reactive oxygen species-mediated p38 MAPK pathway. Cancer Biother Radiopharm. 2013;28:607-14
13. Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23:2838-49
14. Thowfeik FS, AbdulSalam SF, Wunderlich M, Wyder M, Greis KD, Kadekaro AL. et al. A ROS-Activatable Agent Elicits Homologous Recombination DNA Repair and Synergizes with Pathway Compounds. Chembiochem. 2015;16:2513-21
15. Vadukoot AK, AbdulSalam SF, Wunderlich M, Pullen ED, Landero-Figueroa J, Mulloy JC. et al. Design of a hydrogen peroxide-activatable agent that specifically targets cancer cells. Bioorg Med Chem. 2014;22:6885-92
16. Wasco MJ, Pu RT, Yu L, Su L, Ma L. Expression of gamma-H2AX in melanocytic lesions. Hum Pathol. 2008;39:1614-20
17. Bennett MR, Pleasant L, Haffner C, Ma Q, Haffey WD, Ying J. et al. A Novel Biomarker Panel to Identify Steroid Resistance in Childhood Idiopathic Nephrotic Syndrome. Biomark Insights. 2017;12:1177271917695832
18. Wang Y, Yang F, Gritsenko MA, Wang Y, Clauss T, Liu T. et al. Reversed-phase chromatography with multiple fraction concatenation strategy for proteome profiling of human MCF10A cells. Proteomics. 2011;11:2019-26
19. Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47:497-510
20. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S. et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603-7
21. Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM. et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855-67
22. Zou X, Ratti BA, O'Brien JG, Lautenschlager SO, Gius DR, Bonini MG. et al. Manganese superoxide dismutase (SOD2): is there a center in the universe of mitochondrial redox signaling? J Bioenerg Biomembr. 2017;49:325-33
23. Ding C, Fan X, Wu G. Peroxiredoxin 1 - an antioxidant enzyme in cancer. J Cell Mol Med. 2017;21:193-202
24. Manzano JL, Layos L, Buges C, de Los Llanos Gil M, Vila L, Martinez-Balibrea E. et al. Resistant mechanisms to BRAF inhibitors in melanoma. Ann Transl Med. 2016;4:237
25. Winder M, Viros A. Mechanisms of Drug Resistance in Melanoma. Handb Exp Pharmacol. 2017
26. Gibney GT, Zager JS. Clinical development of dabrafenib in BRAF mutant melanoma and other malignancies. Expert Opin Drug Metab Toxicol. 2013;9:893-9
27. Maverakis E, Cornelius LA, Bowen GM, Phan T, Patel FB, Fitzmaurice S. et al. Metastatic melanoma - a review of current and future treatment options. Acta Derm Venereol. 2015;95:516-24
28. Welsh SJ, Rizos H, Scolyer RA, Long GV. Resistance to combination BRAF and MEK inhibition in metastatic melanoma: Where to next? Eur J Cancer. 2016;62:76-85
29. Peng X, Gandhi V. ROS-activated anticancer prodrugs: a new strategy for tumor-specific damage. Ther Deliv. 2012;3:823-33
30. Major Jourden JL, Cohen SM. Hydrogen peroxide activated matrix metalloproteinase inhibitors: a prodrug approach. Angew Chem Int Ed Engl. 2010;49:6795-7
31. Hagen H, Marzenell P, Jentzsch E, Wenz F, Veldwijk MR, Mokhir A. Aminoferrocene-based prodrugs activated by reactive oxygen species. J Med Chem. 2012;55:924-34
32. Corazao-Rozas P, Guerreschi P, Jendoubi M, Andre F, Jonneaux A, Scalbert C. et al. Mitochondrial oxidative stress is the Achille's heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget. 2013;4:1986-98
33. Kim YS, Gupta Vallur P, Phaeton R, Mythreye K, Hempel N. Insights into the Dichotomous Regulation of SOD2 in Cancer. Antioxidants (Basel). 2017:6
34. Cong N, Huang W, Yuan JP, Li GZ, Zhai GS, Li BS. Peroxiredoxin1 promotes cell proliferation, migration and invasion of colorectal cancer via p38MAPK signaling. Eur Rev Med Pharmacol Sci. 2018;22:1922-8
35. Li HX, Sun XY, Yang SM, Wang Q, Wang ZY. Peroxiredoxin 1 promoted tumor metastasis and angiogenesis in colorectal cancer. Pathol Res Pract. 2018
Author contact
![]() Corresponding author: Joan T. Garrett, James L. Winkle College of Pharmacy, University of Cincinnati, 231 Albert Sabin Way, Cincinnati, Ohio 45267-0514. Phone: 513-558-0741; Fax: 513-558-4372; Email: joan.garrettedu
Corresponding author: Joan T. Garrett, James L. Winkle College of Pharmacy, University of Cincinnati, 231 Albert Sabin Way, Cincinnati, Ohio 45267-0514. Phone: 513-558-0741; Fax: 513-558-4372; Email: joan.garrettedu