Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(24):4726-4735. doi:10.7150/jca.26502 This issue Cite
Review
Spontaneous Cancers, But Not Many Induced Ones in Animals, Resemble Semi-New Organisms that Possess a Unique Programmed Cell Death Mode Different from Apoptosis, Senescent Death, Necrosis and Stress-Induced Cell Death
Mingjun Shi1 ![]() , Haiyan Zhou2, Mingjuan Lei3, Lichan Chen3, Lucas Zellmer4, Yan He5, Wenxiu Yang6, Ningzhi Xu7
, Haiyan Zhou2, Mingjuan Lei3, Lichan Chen3, Lucas Zellmer4, Yan He5, Wenxiu Yang6, Ningzhi Xu7 ![]() , Dezhong Joshua Liao5,6
, Dezhong Joshua Liao5,6 ![]()
1. Department of Pathophysiology, Guizhou Medical University, Guiyang 550025, Guizhou Province, China.
2. Clinical Research Center, Guizhou Medical University Hospital, Guiyang 550004, Guizhou Province, China
3. Hormel Institute, University of Minnesota, Austin, MN 55912, USA
4. Masonic Cancer Center, University of Minnesota, 435 E. River Road, Minneapolis, MN 55455, USA
5. Key Lab of Endemic and Ethnic Diseases of the Ministry of Education of China at Guizhou Medical University, Guiyang 550004, Guizhou Province, China
6. Department of Pathology, Guizhou Medical University Hospital, Guiyang 550004, Guizhou province, China
7. Laboratory of Cell and Molecular Biology & State Key Laboratory of Molecular Oncology, National Cancer Center/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100021, China
Received 2018-4-5; Accepted 2018-9-11; Published 2018-11-25
Abstract
There are four basic cell death modes in animals, i.e. physiological senescent death (SD) and apoptosis as well as pathological necrosis and stress-induced cell death (SICD). There have been numerous publications describing “apoptosis” in cancer, mostly focused on killing cancer cells using radio- or chemo-therapy, with few on exploring how cancer cells die naturally without such treatments. Spontaneous benign or malignant neoplasms are immortal and autonomous, but they still retain some allegiance to their parental tissue or organ and thus are still somewhat controlled by the patient's body. Because of these properties of immortality, semi-autonomy, and semi-allegiance to the patient's body, spontaneous tumors have no redundant cells and resemble “semi-new organisms” parasitizing the patients, becoming a unique tissue type possessing a hitherto unannotated cell death mode besides SD, apoptosis, necrosis and SICD. Particularly, apoptosis aims to expunge redundant cells, whereas this new mode does not. In contrast to spontaneous tumors, many histologically malignant tumors induced in experimental animals, before they reach an advanced stage, regress after withdrawal of the inducer. This mortal and non-autonomous nature disqualifies these animal lesions as authentic neoplasms and as semi-new organisms but makes them a good tissue type for apoptosis studies. Ruminating over cell death in spontaneous cancers and many inauthentic tumors induced in animals from these new slants makes us realize that “whether cancer cells undergo apoptosis” is not an easy question with a simple answer. Our answer is that cancer cells have an uncharacterized programmed cell death mode, which is not apoptosis.
Keywords: apoptosis, programmed cell death, necrosis, cancer, tumor, in vivo, senescence
Introduction
There have been dozens of different modes of cell death described in the literature, as has been adumbrated by the Nomenclature Committee on Cell Death (NCCD) [1,2] and by us as well [3]. Of these various terms, “apoptosis” and “necrosis” are the best known and the ones most widely used in the literature, although, by reading its 2015 incarnation of cell death nomenclature [2], we construe that the NCCD has, since a few years ago, had less interest in using the word “apoptosis”. “Apoptosis” is generally accepted as a procedure of programmed cell death, with the word “programmed” to indicate its nature of active or suicidal death via a pre-determined procedure. In stark contrast, “necrosis”, which has appeared in pathology textbooks with a much longer history, is a passive decease, i.e. a cell death caused by various killing factors. However, the reality is that many quite different types of cell death have all been described as “apoptosis” in the biomedical literature [3], and even necrosis has been described to appear with some features of programming, somewhat like apoptosis [1,2]. One extreme is that the involution type of cell death occurring physiologically in animals is described, such as by us [3-6], as apoptosis, with the massive cell death occurring during the post-lactating (post-weaning) involution of the mammary glands as an example. Another extreme, which is the opposite to the involution type of cell death in physiological situations, is the decimation of cancer cells caused by radiotherapy or chemotherapy both in patients and in cell culture systems. This fact, i.e. that many quite different modes of cell death occurring in physiological or pathological situations are all put under the umbrella of “apoptosis”, clearly bespeaks that either apoptosis has not yet been well defined, or it has been well-defined as we all assume but many peers do not bother to follow the definition and use the word “apoptosis” at their convenience to describe whatever modes of cell demise they have observed. In other words, either apoptosis has no widely accepted criteria or many students do not follow the criteria. Once a death does not really show certain presumable iconic features of “apoptosis”, such as lack of caspase activation [7], “atypical apoptosis” or similar idioms are used to describe it. This current situation in the literature has already created infinite confusion, and probably is the reason why some seasoned students formed the NCCD. One of the areas suffering from this confusion is cancer research, especially in the aspect of cancer therapy that aims to kill cancer cells in patients. Mulling over more deeply programmed cell death in untreated cancers in humans and in many outgrowing lesions in experimental animals from some new angles that have rarely been thought from previously, we realize that “whether cancer cells undergo apoptosis in vivo” is actually not an easy question with a simple answer, as expounded in this essay.
Senescent death and apoptosis are the two physiological types of programmed cell death
In any normal multicellular animal, all cells have a lifespan and will eventually die of aging [8-10], although the lifespans of different cell types vary greatly. “Senescence” is often described as a main mechanism of aging [11-19], although senescence itself is usually defined as permanent growth arrest that does not necessarily lead to the death of the cell [13,17-20]. Therefore, we define “cells die from aging” as “senescent death” (SD) [3], which to us is a physiological event, to differentiate it from senescence itself.
In those animals that are evolutionarily-high on the “life-tree”, there are some cells that are no longer useful after a certain embryonic stage or at certain physiological situations. These archaic cells need to be eliminated, but their elimination should not mar their surrounding normal tissue and certainly not the whole animal. This evolutionarily developed mechanism for purging obsolete cells from a tissue or organ is apoptosis by our definition [3-6], as we construe that this is the cell death mode described by Kerr et al in their trailblazing study in which the word “apoptosis” was created [21]. Kerr et al used this word, created by a professor of ancient Greek, to liken the cell death to “the dropping off as of leaves from a tree”, as recapitulated by Savill [22], which emphasizes that the death is physiological and occurs to individual cells like leaves via an endogenous program. In humans, typical examples of so-defined apoptosis include the cellular deaths occurring during digit individualization in embryo [13,23], post-pubertal involution of the thymus [24-28], postpartum involution of the uterus [29-32], post-lactation (post-weaning) involution of the mammary glands [33-36], atrophy of germline cells in the testes or ovaries in aged men or women [37-42], etc. Therefore, it is those normal but no-longer useful cells that die, which is an icon of apoptosis distinguishable from other cell decease modes, such as SD in which it mainly is those useful cells that die. Unfortunately, this iconic feature of apoptosis that clearly points out “who dies” has not been emphasized in the recent literature, although, ever since 20 years ago, Savill has kept pointing out that it is those “unwanted” cells that die of apoptosis [22,43-46].
All cell types in an animal have a physiological total number. For those cell types that retain a lifelong replicative ability, if the number is decreased, the body will turn on a mechanism of compensatory proliferation, usually dubbed as regeneration, to restore the physiological cell number and thus the physiological function of the tissue or organ [3]. Apoptosis does not set off regeneration because it aims to eliminate useless, thus redundant, cells. However, the “physiological total cell number” of a cell type is a relative concept and may vary in different situations. For instance, the levels of sex hormones are decreased in elderly people, making the reproductive tissues construe that some of their cells are no longer useful and need to be eliminated via a process coined as “atrophy” [37-42], with apoptosis as its essence. Cell or tissue atrophy, which in our opinion resembles involution, can occur in various situations that are still within the physiological range. For example, fasting from food may lead to atrophic death of some adipocytes and even some muscle cells via autophagy [47,48]. Moreover, the dying or already-dead cells will be swiftly removed via engulfment by so-called scavenger cells, which encompass macrophages and other cell types that have phagocytic ability [3-6]. This means that apoptosis involves at least two players, i.e. the suicidal cell and the scavenger, and, in turn, involves complicated communications between the two. Unfortunately, this requirement of “two players” as an indispensable criterion to define apoptosis has rarely been emphasized in the recent literature. Because the dying or dead cells are swiftly disposed of by scavengers before they decompose to immunogenic debris to trigger immune reactions, apoptosis is not associated with inflammation that may gut the host tissue or organ.
Stress-induced cell death is pathological, like necrosis, but is often misconstrued as apoptosis
Unlike in apoptosis, in necrosis it is usually those useful but damaged or ill cells that decease, such as those white blood cells that are infected and decimated by bacteria, and therefore necrosis is often followed by regeneration of the same cell type to restore the physiological cell number. Actually, in many situations of chronic infection, death of useful cells is a constant event and the capacity of regeneration is insufficient to compensate for the cell loss. Cells of connective tissue, mainly fibroblasts, will then step in and proliferate to help heal the wound, which in pathology is referred to as granulation or scar formation, such as liver fibrosis and cirrhosis seen in many patients with chronic hepatitis [49,50]. Moreover, necrosis is a passive death mode in which scavenger cells are not mobilized until the cell corpses have decomposed to immunogenic dregs. In other words, the cells that are about to die do not request scavengers to help in the death procedure, and thus involvement of scavengers is not part of necrosis but, rather, is part of an ensuing inflammation instigated by the already dead cells or their cellular debris (table 1).
Similarities and disparities among four basic cell death modes
| Death mode | Who dies | Prog. | Nature | Situation | Inflam. | R & H | Scar | Scavenger | Cell-cell communication | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| w Scav. | w Sybling | w Stromal | |||||||||
| SD | Normal and useful cell | Yes | Suicide | Physiologic | No | Yes/No | No | Involved | Maybe | Maybe | No |
| Apoptosis | Normal but useless cell | Yes | Suicide | Physiologic | No | No | No | Involved | Required | No | No |
| SICD | Ill but useful cells | Yes | Ind-Sui. | Pathologic | Maybe | Yes | Maybe | Involved | Maybe | Required | Maybe |
| Necrosis | Ill but useful cells | No | Homicide | Pathologic | Maybe | Yes | Maybe | Uninvolved | No | Required | Maybe |
Not: Prog., programmed; Ind-Sui., induced suicide; Inflam., inflammation; R &H, regeneration and wound healing; w Scav., the dying cell communicates with scavenger; w Sybling, the dying cell communicates with normal cells of the same type (sybling cells); w Stromal, the dying cell communicates with connective tissue cells.
Besides necrosis, there is another pathological cell death mode dubbed by us as “stress-induced cell death”, or SICD, which probably is a much larger and more common cell death category than the others [3,6]. SICD mainly affects useful cells and is a programed event; these two features together confer on it traits of both apoptosis and necrosis (table 1) [3,6]. Actually, for this reason, SICD is often misconstrued as apoptosis or necrosis or, sometimes, is called by peers as programmed necrosis [51], aponecrosis [52], necroptosis [53,54] or other parlances to indicate their mixed properties. This misconstruction occurs not only because in SICD it is those useful cells that die, but also because the stress as the prime mover of the cell demise can be an endogenous one, such as an irreparable genetic mutation, which is not as potent as bacterial or viral infection that kills the cells instantly via necrosis. In SICD, the stress, especially the endogenous one, causes the death by eliciting an endogenous demise program as occurs in apoptosis. For instance, when a cell has a genetic mutation, initially the cell stalls its replication for time to repair the mutation. However, when it realizes that the mutation is irreparable, it will turn on a suicidal program to kill itself, in order to avoid passing the mutation to progeny cells to be hereditary [3,5,6]. This death is a typical SICD. Major similarities and disparities among physiological SD and apoptosis as well as pathological necrosis and SICD have been described by us elsewhere in detail [3,6] and are adumbrated in table 1.
Spontaneous tumors resemble semi-new organisms, which complicates the apoptosis issue
Tumorigenesis incepts with immortalization of a normal cell. The immortal cell will then replicate continuously to form a benign or malignant tumor. The immortality does not mean that tumor cells can live forever; it means that tumor cells, including those benign ones, have acquired an ability to replicate endlessly [55]. This in turn has two meanings: 1) By unrelenting replication of its cells, the tumor as a whole resembles an independent, bacterium-like organism that keeps replicating its cells for maintaining itself (Fig 1). 2) The tumor is autonomous, i.e. governing itself and caring only about its own survival, thus, again, resembling an independent organism [6,56-59]. Considering a tumor to be an independent organism is also possible because the tumor can survive forever as cell lines in culture, even after the patient has died, as exemplified by the Hela cell line that was established in 1951 from the cervical cancer of the late patient Henrietta Lacks [60]. Because many benign tumor cells are well differentiated and morphologically resemble their surrounding normal cells, benign tumors are often diagnosed by their altered histology (architecture), and not by their cellular morphology. Therefore, autonomy, just like immortality, is often disassociated with cellular morphology but is probably related to histological changes.
Illustration of the dual properties of a tumor that make it a semi-new and semi-old organism. This duality of tumor cells shown as the overlapping area of the two circles complicates things
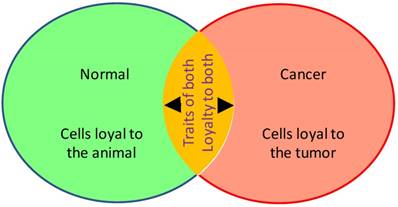
In contrast to tumor cells, all normal cells in a human being are mortal, meaning that they have a death program encoded by some cellular component(s), likely the nuclear and/or mitochondrial genome(s), and will eventually die. Because of this evolutionarily built-in death program, all cells in a human being become allegiant to the human's body, which gives the body the authority to require some cell types with short lifespans to commit suicidal SD, apoptosis or SICD for the interest of other cell types with longer lifespans and in turn for the ultimate interest of the body. For instance, white blood cells are first mobilized by the human's body to fight against infectious bacteria or viruses and die easily during the infection. Autonomy of tumor cells means that they have abandoned their allegiance to the patient's body and become allegiant to the tumor as an organism, and may kill the host organism, i.e. the patient, if such killing is necessary for the tumor's survival.
For twofold reasons a tumor as a “newly developed organism” is evolutionarily-lower on the life-tree, making carcinogenesis an atavism, i.e. a process of reverse evolution: First, cancer cells morphologically resemble embryonic cells because they are morphologically less-differentiated than their adult counterparts. For this reason, pathologists traditionally use a set of embryological phraseologies to describe cancer cells, such as “well differentiated”, “poorly differentiated”, “undifferentiated”, etc. In association with this morphological trait, cancer cells often express high levels of some proteins that normally are only expressed during embryonic stages. For instance, hepatocellular carcinoma cells often express the alpha fetoprotein that is highly expressed by fetal liver, but basically not by the liver in adults [61,62]. Second, as aforementioned, the immortality nature of a tumor requires its cells to replicate relentlessly to maintain the tumor as an “organism”, somewhat similar to the maintenance of a bacterial strain by incessant replication of the bacterial cells. This asexual mechanism for organismal maintenance connotes that a tumor is evolutionarily-lower than the host animal. However, in most cases, even very pernicious tumors retain some differentiated traits and a certain allegiance to the host tissue or organ, and are thus more or less under the control of the patient's body (Fig 1). Benign tumors are well-differentiated and retain more allegiance to the patient's body. Therefore, a tumor as a whole is actually not fully autonomous and is not a completely independent organism.
In their initial study in which the word “apoptosis” was created, Kerr et al. observed phagocytosis of tumor cells by macrophages or neighboring tumor cells [21]. This phenomenon could be described in two antithetical ways: On the one hand, if the tumor is regarded as another organism parasitizing the host (the patient), then the prey and the predator belong to two different organisms. Therefore, engulfment of tumor cells by macrophages is not part of apoptosis because in apoptosis both the prey and the predator belong to the same organism, but rather it resembles engulfment of bacterial cells by macrophages during an infection [3,6]. On the other hand, for two reasons a tumor can still be regarded as a tissue of the patient: First, although malignant cells may bear a lot of mutations, in general the genome of tumor cells is still much the same as that of the patient's cells. Second, most tumor cells retain some differentiation and some physiological function of the parental tissue, and still have some allegiance to the patient's body. Therefore, it is still reasonable to construe that both the macrophages and the tumor cells belong to the same organism. Again, the “semi-new and semi-original organism” nature of tumors, as well as their nature of “semi-autonomy and semi-allegiance to the patient's body” (Fig 1 and table 2), complicate things and put tumors into a unique tissue type that possesses a fifth, hitherto unannotated, mode of cell death besides apoptosis, SD, SICD and necrosis that all occur to the cells of the same single organism.
Summary of key apoptosis properties of spontaneous tumors and many induced tumors
| Lesion origin | Property | How it pertains to apoptosis |
|---|---|---|
| Spontaneous human or animal tumors | Immortality | Immortal tumor cells do not seem to have death program (s) |
| Cell redundancy | Mild nutritional insufficiency may decrease total number of cells | |
| Autonomy | New, bacterium -like organism s. Cells have no lifespan? | |
| Inducer- dependent animal “tumors” | Mortality | Mortal cells have death program (s) and are not neoplastic |
| Cell redundancy | The host organ/tissue has a total number limit | |
| Not a new organism | Part of the host organism , with a lifespan. |
Spontaneous tumors are a unique tissue type unconcerned about cell redundancy
In the absence of treatment, tumor cells inside the patient's body live happily without caring about how their growth produces pain, or even death for the host patient, meaning that the tumor cells live in a separate physiological situation if we just look at the tumor itself and regard it as an independent organism. In this sort of physiological situation, whether a tumor as a sort of independent organism would have excessive cells is a key issue for determining whether it has an apoptotic mechanism, since apoptosis is defined as a mechanism to purge away excessive cells in a physiological manner. The glaring fact that tumor lumps continue enlarging their sizes bespeaks that tumors do not bother to remove excessive cells via apoptosis. In other words, tumors seem to have no reason to retain an apoptotic mechanism during their atavism from their parental normal cells. However, when a tumor has expanded to a certain size, the blood supply may not be sufficient, especially when its angiogenesis lags behind the quick proliferation of its tumor cells. While severe insufficiency of blood nourishment will likely cause necrosis, slight insufficiency may goad tumor cells into atrophy, which somewhat resembles the atrophy of some gonadal cells in elder people and thus may be regarded as apoptosis-like death in the tumor as an organism. Unfortunately, the literature has little discourse on these aspects and on tumor cell death in various physiological situations, such as in the absence of treatment or when blood supply is slightly insufficient, partly because several modes of cell death usually coexist in a tumor tissue and it is hard to distinguish one from the others. For example, tumor cells, especially those heinous ones, bear a lot of irreparable genetic alterations, some of which may function as endogenous stressors to the cells to trigger SICD [3,6].
Contemplating cell death in a tumor tissue from the above-described angles, which has rarely been done so in the literature, provides us new slants and tells us that “whether tumor cells undergo apoptosis in physiological situations” is not as a simple question as many pundits presume and has not been clearly addressed yet. In our opinion, spontaneous tumors in human and animals are unique for its lack of redundant cells, which is another reason to consider them as a unique tissue type that possesses a unique programmed cell death mode different from apoptosis, as apoptosis aims to expunge redundant cells. However, like apoptosis, cell death via this so-far unannotated mechanism is a physiological event to the tumor as an organism.
The tumors' remaining allegiance to the patients may drive apoptosis of normal cells
Studies on determination of the functional similarity between benign tumor cells and their normal counterparts are exiguous. Nevertheless, the aforementioned morphological similarity suggests to us that cells of some benign tumor types, such as uterine leiomyoma, have some functional similarity to their normal counterparts. We surmise that these similarities may render tumor cells loyal to the host tissues or organs. If this conjecture is correct, then when a tumor is expanding, the host tissue or organ should sense that it has more and more redundant cells which need to be eliminated via apoptosis. However, this momentum for apoptosis may be allocated more to the normal cells than to the tumor cells, because normal cells are more loyal to the host. Therefore, we speculate that many normal cells will die of apoptosis when the tumor is expanding. Expansion of cancers may also cause apoptosis of normal surrounding cells via the same mechanism, as most malignant tumors still retain some morphological and functional similarities to their parental tissue types, with the undifferentiated or anaplastic tumors imposing the smallest amount of this momentum for apoptosis. To our knowledge, this possible mechanism for a tumor to cause death of normal cells in the host tissue or organ by creating impetus for apoptosis has so far received little attention and deserves experimental corroboration. Moreover, whether this impetus also causes death of tumor cells and, if yes, whether this type of cell death is apoptosis or SICD, remain as unaddressed questions.
Many “cancers” induced in animals have not yet evolved to neoplasms and thus are good tissues for apoptosis study
There have been a sheer number of animal models of carcinogenesis in which chemicals, hormones, irradiations, genetic manipulations, or a combination of some of these are used as inducers to produce tumors in animals. The tumors so induced manifest malignant morphology and even behaviors, and thus are pathologically diagnosed as cancers. However, once the tumor inducer has been withdrawn, many of the tumors will hastily disappear via regression of the tumor cells; the tumors can be sustained in the absence of the inducer only until a very advanced stage [63,64]. This phenomenon, although rarely mentioned in the literature of recent years, was already documented in the German literature by Fischer in 1906 (B. Fischer, Münch. med. Wochnschr., 1906; 42: 2041), to our knowledge. According to Davis [65,66] as well as Vasiliev and Cheung [67], Fischer showed that subcutaneous injection of Scarlet Red into the ears of rabbits could induce papillomas, but the tumors would regress if administration of Scarlet Red was discontinued. This seminal finding was confirmed by Helmholz in 1907 and by Werner in 1908, according to Davis [65]. During 1914-1924, Yamagiwa et al were able to induce papillomas and some papilocarcinomas in the ears of rabbits with metastasis to lymph nodes by painting the ears with tar, but, again, most tumors, including some malignant ones, would regress if tar-painting was discontinued [68,69]. From 1930s to recent decades, similar inducer-dependency had been reported to tumors induced by different other chemical carcinogens, such as 3-methylcholanthrene [70,71], 3:4-benzyprene [72] and 7,12-dimethylbenz[α]anthracene [73-76]. Chronic treatments of rats or mice with estrogens can induce benign and malignant tumors in the mammary glands and several other organs, but, ever since 1930s, it has been known that the overt tumors, before they reach a very advanced stage, will regress upon withdrawal of the hormone [77-86]. Thyroid neoplasms can be induced in mice by treatment with thiouracil or other goitrogenic compounds that block thyroid hormone secretion and in turn induce secretion of thyroid stimulating hormone from the pituitary. These thyroid tumors often metastasize to the lungs and lymph nodes but still depend on the inducer [87-89]. c-myc, k-ras mutant, or Xmrk transgene can induce malignant tumors in the target organs, but turning the transgene off will lead to regression of the overt tumors [63,90-102]. Conversely, conditional knockout of the tumor suppressor gene p53 can beget tumor formation, but reactivation of the p53 leads to regression of the tumors [103-109]. There are still many animal models of carcinogenesis that have not yet been evaluated for the effect of inducer withdrawal, but we surmise that many of these undetermined models may show similar inducer-dependency until a terminal stage. In our opinion, the reason for the lesions to manifest neoplastic morphology and even behavior in the presence of the inducer is because the inducer coerces the lesions' cells to do so, but not is because genetic alterations responsible for such morphology and behavior have already occurred to transform the cells to authentic neoplasms [63,64].
In the abovementioned animal models, the mechanism for the tumor cell regression upon withdrawal of the inducer has not yet been well explored appertaining to the true nature of the cell death. In our opinion, sustenance of the tumor cells by the inducer is akin to that of germline cells in gonads by sex hormones, whereas withdrawal of the inducer is reminiscent of the hormone depletion by castration. In other words, the tumor cells likely regress via involution or atrophy upon withdrawal of the inducer, and hence is an authentic apoptosis. Since the outgrowths are still mortal and non-autonomous, they cannot be regarded as quasi-new organisms, and thus phagocytosis of the lesion's cells by macrophages, as we observed in the MMTV-c-myc induced mouse mammary tumors [110,111], cab be regarded as part of an apoptotic process. Therefore, besides those aforementioned physiological involution or atrophy of cells or tissues, these animal lesions, because they are not authentic neoplasms, is a good tissue type for studying in vivo mechanisms of apoptosis upon withdrawal of the inducer, making these animal models useful without being misleading. Likely, the existence of these histologically malignant but mortal and non-autonomous cells sends out a signal to the host that the organ or tissue has excessive cells. What remains as enthralling but unaddressed questions is why and how the tissue or organ or even the animal's body decides that it is the cells of the outgrowth, but not their normal counterparts, that make trouble and should be eliminated.
As a caveat that needs to be given, although withdrawal of the inducer can cause complete regression of the overt tumors in many animal models, tumors can swiftly reappear upon reintroduction of the inducer, such as turning on the transgene again or treatment with the chemical or the hormone again [74,84,85,98,112-114]. It is unclear whether, upon the reintroduction of the inducer, it is remnant tumor cells that quickly repopulate or it is other cells that hastily populate, to form the recurrent tumors. If it is the former, it insinuates that withdrawal of the inducer cannot completely extinguish the lesion's cells via apoptosis. This in turn connotes that the physiological cell number of a tissue or organ is only a rough figure, and a small number of excessive cells may be below the detection limit by the tissue or organ and thus will not instigate the tissue or organ to turn on the apoptotic mechanism. Nevertheless, the memory of the inducer by some cells, either remnant ones or others, may be attributed to some yet unidentified genetic mutations, which distinguish these cells from normal cells. Whether the tissue or organ culls these histologically malignant cells for apoptosis based on these mutations remains as an unasked but spellbinding question. Nevertheless, it is obvious to us that the tissue or organ bearing the inauthentic tumor has mechanism(s) to smartly identify the truly redundant and trouble-maker cells for elimination, because it is the lesion's cells that are exterminated upon the inducer withdrawal. Therefore, “no-longer useful”, “obsolete”, “archaic” and “outmoded' are probably words that are too simple to describe those cells that will undergo apoptosis in these inauthentic tumors.
Several relevant questions need to be addressed as well in the future
It is basic knowledge to pathologists that malignant tumors often have a higher cell death rate than their surrounding normal tissue. As said in 1941 by Rous, a Nobel laureate, “that cancer cells are often sick cells and die young is known to every pathologist” [115]. Tumors can still enlarge because there are many more tumor cells that are proliferating [116]. These cellular deaths are traditionally called necrosis in old pathology textbooks as they are assumed to be due to insufficient blood nourishment, although some of the cellular deaths are likely to be SICD. To address whether apoptosis occurs in a tumor tissue, especially a malignant one, as seen in cellular involution and atrophy, several relevant questions also need to be addressed:
- Whether or not bacterial cells undergo aging is still a question in debate [8,117], since bacterial cells keep dividing symmetrically as a means to maintain their strains. Similarly, since immortality of a tumor is maintained by constant symmetrical division of its cells, a series of questions can be raised as to whether tumor cells also have a lifespan and thus a death program that allows them to age and die of SD. These questions can also be asked another way around as to whether tumor cells do not have a lifespan and an SD program, as tumors are immortal.
- Whether only those cells that have a lifespan have a death program and can undergo apoptosis, while those that no longer have a lifespan cannot. This question is raised because many cancer savants study “apoptosis” or “programmed cell death” of cancer cells with an attempt to target it for cancer therapy. Since tumorigenesis incepts with reprogramming the death program of a normal cell to make it immortal, tumor cells should not have a lifespan and thus should not have a death program, according to our understanding of the definition of “immortality”. However, one may argue that the reprogramming confers a new death program onto the immortal cell, allowing it to die of SD or apoptosis. If immortal cancer cells still have a death program, although it is no longer the same as the one in the normal cells, it is meaningful to study it, since learning about it may help us in killing cancer cells. Therefore, the key issues here are what “immortality” means and whether it conflicts with “death program”, since one may argue that “immortality” is referred to the tumor as an organism but not to individual tumor cells which may still have their “death programs”. All these schools of thoughts need to be presented for debate.
- Whether or not tumors as a quasi-new organism parasitizing the patients have obsolete cells. This is raised because by our definition it is obsolete cells that die in authentic apoptosis whereas all tumors, if untreated, keep enlarging.
- Whether the remaining-allegiance of tumor cells to the host patient would spur death of some tumor cells as inferred in an above section, and, if yes, whether this type of death can be considered as apoptosis.
Conclusions
In our opinion, there are four basic cell death modes, i.e. SD, apoptosis, necrosis and SCID [3]. There have been numerous published studies describing “apoptosis” of cancer cells both in cell cultures and in patients or animals, with cancer therapy overarching these studies, but in our opinion therapies as potent stressors decimate cancer cells only via necrosis and SICD. Few studies have been centralized on the mechanisms for cell death inside spontaneous cancers without a treatment. Spontaneous tumors, benign or malignant, are immortal and autonomous, which makes them quasi-new organisms independent of the host tissue or organ. In the meantime, spontaneous tumors still retain, more or less, allegiance to the patient's body. The immortality, the “semi-new and semi-original organism” nature, and the nature of “semi-autonomy and semi-loyalty to the patient's body” equip spontaneous tumors with a fifth, hitherto unrealized and unannotated, cell death mode besides SD, apoptosis, necrosis and SICD. Unlike apoptosis, this so far uncharacterized mode of cell death does not aim to expunge excessive cells from tumor tissues. However, many histologically malignant tumors induced in animals by chemicals, hormones or genetic manipulations are not authentic neoplasms and not semi-new organisms, because they swiftly regress upon withdrawal of the inducer and thus are still mortal and non-autonomous. These features make these animal lesions a good tissue type for study of apoptosis. Meditating on programmed cell death in authentic and inauthentic cancers in various physiological situations, such as in the absence of therapy, from the abovementioned angles provides us with new views and makes us realize that “whether cancer cells undergo apoptosis in vivo” is not an easy question with a simple answer. Our oversimplified answer for it is that untreated cancer cells still retain a mechanism of programmed cell death, but it is not the apoptotic mechanism described by many peers, mainly because cancers are immortal and unconcerned about cell redundancy, unlike normal tissues or organs. Those apoptotic mechanisms described in the mainstream literature are not for authentic apoptosis, but are for SICD. Actually, much insufficient attention has been put, and thus little has been known, on authentic apoptosis in vivo. Delving into the answers for the questions or issues raised this essay will be bewitching tasks with great guerdons.
Abbreviations
MMTV: mouse mammary tumor virus long terminal repeats; NCCD: Nomenclature Committee on Cell Death; SD: senescent death; SICD: stress-induced cell death.
Acknowledgements
We would like to thank Fred Bogott, M.D., Ph.D, former chair of Emergency Department of Austin Medical Center, Mayo Clinic at Austin of Minnesota, USA, for his excellent English editing of this manuscript.
Funding
This work was supported by a grant from Chinese Natural Science Foundation to D. Joshua Liao (grant No. 81660501).
Authors' contributions
MS and HZ drafted the manuscript. LC prepared figures and participated in discussion. ML and YH participated in discussion. NX, WY and DJL formulated the concepts. LZ performed English editing and participated in discussion. DJL finalized the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV. et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107-120
2. Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D. et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 2015;22:58-73
3. Liu X, Yang W, Guan Z, Yu W, Fan B, Xu N. et al. There are only four basic modes of cell death, although there are many ad-hoc variants adapted to different situations. Cell Biosci. 2018;8:6-doi 10.1186/s13578-018-0206-6
4. Liao DJ. The scavenger cell hypothesis of apoptosis: apoptosis redefined as a process by which a cell in living tissue is destroyed by phagocytosis. Med Hypotheses. 2005;65:23-28
5. Liu B, Xu N, Man Y, Shen H, Avital I, Stojadinovic A. et al. Apoptosis in Living Animals Is Assisted by Scavenger Cells and Thus May Not Mainly Go through the Cytochrome C-Caspase Pathway. J Cancer. 2013;4:716-723
6. Zhang J, Lou XM, Jin LY, Zhou RJ, Liu SQ, Xu NZ. et al. Necrosis, and then stress induced necrosis-like cell death, but not apoptosis, should be the preferred cell death mode for chemotherapy: clearance of a few misconceptions. Oncoscience. 2014;1:407-422
7. Zellmer L, Han YP, Chen LC, Xu NZ, Liao DJ. Does the cytochrome c-caspase pathway of cell death occur physiologically in animals? Journal Tumor Med Prev. 2017;1:JTMP.MS.ID.555557.pdf
8. Jia QW, Chen XH, Jia YP, Dou XX, Ezeogu L, Xu NZ. et al.: is type 2 diabetes one of such aging phenomena that lack an irreversible structural change? J Diabetes Metab 2105, 6: 543-doi: 10.4172/2155-6156.1000543.
9. Maklakov AA, Rowe L, Friberg U. Why organisms age: Evolution of senescence under positive pleiotropy? Bioessays. 2015;37:802-807
10. Rubin H. Cell aging in vivo and in vitro. Mech Ageing Dev. 1997;98:1-35
11. Passaro F, Testa G. Implications of Cellular Aging in Cardiac Reprogramming. Front Cardiovasc Med. 2018;5:43-doi 10.3389/fcvm.2018.00043
12. Shakeri H, Lemmens K, Gevaert AB, De Meyer GRY, Segers V. Cellular senescence links aging and diabetes in cardiovascular disease. Am J Physiol Heart Circ Physiol. 2018 -doi: 10.1152/ajpheart.00287.2018
13. Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep. 2014;15:1139-1153
14. Falandry C, Bonnefoy M, Freyer G, Gilson E. Biology of cancer and aging: a complex association with cellular senescence. J Clin Oncol. 2014;32:2604-2610
15. Sikora E, Bielak-Zmijewska A, Mosieniak G. Cellular senescence in ageing, age-related disease and longevity. Curr Vasc Pharmacol. 2014;12:698-706
16. Tan FC, Hutchison ER, Eitan E, Mattson MP. Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology. 2014;15:643-660
17. van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439-446
18. Vicencio JM, Galluzzi L, Tajeddine N, Ortiz C, Criollo A, Tasdemir E. et al. Senescence, apoptosis or autophagy? When a damaged cell must decide its path-a mini-review. Gerontology. 2008;54:92-99
19. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194-1217
20. Sapieha P, Mallette FA. Cellular Senescence in Postmitotic Cells: Beyond Growth Arrest. Trends Cell Biol. 2018 -pii: S0962-8924(18)30059-X. doi: 10.1016/j.tcb.2018.03.003
21. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239-257
22. Savill J. Apoptosis and the kidney. J Am Soc Nephrol. 1994;5:12-21
23. Hernandez-Martinez R, Covarrubias L. Interdigital cell death function and regulation: new insights on an old programmed cell death model. Dev Growth Differ. 2011;53:245-258
24. Boehm T, Swann JB. Thymus involution and regeneration: two sides of the same coin? Nat Rev Immunol. 2013;13:831-838
25. Gui J, Mustachio LM, Su DM, Craig RW. Thymus Size and Age-related Thymic Involution: Early Programming, Sexual Dimorphism, Progenitors and Stroma. Aging Dis. 2012;3:280-290
26. Aw D, Palmer DB. The origin and implication of thymic involution. Aging Dis. 2011;2:437-443
27. Calder AE, Hince MN, Dudakov JA, Chidgey AP, Boyd RL. Thymic involution: where endocrinology meets immunology. Neuroimmunomodulation. 2011;18:281-289
28. Hince M, Sakkal S, Vlahos K, Dudakov J, Boyd R, Chidgey A. The role of sex steroids and gonadectomy in the control of thymic involution. Cell Immunol. 2008;252:122-138
29. Leslie KE. The events of normal and abnormal postpartum reproductive endocrinology and uterine involution in dairy cows: a review. Can Vet J. 1983;24:67-71
30. Morrow DA, Roberts SJ, McEntee K. A review of postpartum ovarian activity and involution of the uterus and cervix in cattle. Cornell Vet. 1969;59:134-154
31. Gier HT, Marion GB. Uterus of the cow after parturition: involutional changes. Am J Vet Res. 1968;29:83-96
32. Morrow DA, Roberts SJ, McEntee K. Pospartum ovarian activity and involution of the uterus and cervix in dairy cattle. II. Involution of uterus and cervix. Cornell Vet. 1969;59:190-198
33. O'Brien J, Martinson H, Durand-Rougely C, Schedin P. Macrophages are crucial for epithelial cell death and adipocyte repopulation during mammary gland involution. Development. 2012;139:269-275
34. Luke CJ, Silverman GA. Necrotic cell death: harnessing the Dark side of the Force in mammary gland involution. Nat Cell Biol. 2011;13:197-199
35. Watson CJ. Post-lactational mammary gland regression: molecular basis and implications for breast cancer. Expert Rev Mol Med. 2006;8:1-15
36. Strange R, Li F, Saurer S, Burkhardt A, Friis RR. Apoptotic cell death and tissue remodelling during mouse mammary gland involution. Development. 1992;115:49-58
37. de la Grandmaison GL, Marchaut J, Watier L, Mediouni Z, Charlier P. Frequency and nature of testicular and paratesticular lesions in forensic autopsies. Med Sci Law. 2013;53:208-212
38. Pop OT, Cotoi CG, Plesea IE, Enache SD, Popescu FC, Enache MA. et al. Correlations between intralobular interstitial morphological changes and epithelial changes in ageing testis. Rom J Morphol Embryol. 2011;52:339-347
39. Harbitz TB. Testis weight and the histology of the prostate in elderly men. An analysis in an autopsy series. Acta Pathol Microbiol Scand A. 1973;81:148-158
40. Ishii T, Sternby NH. Pathology of centenarians. II. Urogenital and digestive systems. J Am Geriatr Soc. 1978;26:391-396
41. Baker HW, Hudson B. Changes in the pituitary-testicular axis with age. Monogr Endocrinol. 1983;25:71-83
42. Motta PM, Heyn R, Makabe S. Three-dimensional microanatomical dynamics of the ovary in postreproductive aged women. Fertil Steril. 2002;78:360-370
43. Savill J, Gregory C, Haslett C. Cell biology. Eat me or die. Science. 2003;302:1516-1517
44. Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784-788
45. Ren Y, Savill J. Apoptosis: the importance of being eaten. Cell Death Differ. 1998;5:563-568
46. Savill J. Apoptosis. Phagocytic docking without shocking. Nature. 1998;392:442-443
47. Anding AL, Baehrecke EH. Autophagy in Cell Life and Cell Death. Curr Top Dev Biol. 2015;114:67-91
48. Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ. 2015;22:367-376
49. Weiskirchen R, Tacke F. Liver Fibrosis: From Pathogenesis to Novel Therapies. Dig Dis. 2016;34:410-422
50. Friedman SL. Hepatic fibrosis - overview. Toxicology. 2008;254:120-129
51. Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 2016;352:aaf2154 doi: 10.1126/science.aaf2154
52. Formigli L, Papucci L, Tani A, Schiavone N, Tempestini A, Orlandini GE. et al. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J Cell Physiol. 2000;182:41-49
53. Desai J, Mulay SR, Nakazawa D, Anders HJ. Matters of life and death. How neutrophils die or survive along NET release and is "NETosis" = necroptosis? Cell Mol Life Sci. 2016;73:2211-2219
54. Hanson B. Necroptosis: A New Way of Dying? Cancer Biol Ther. 2016;17:899-910
55. Zhang J, Lou X, Zellmer L, Liu S, Xu N, Liao DJ. Just like the rest of evolution in Mother Nature, the evolution of cancers may be driven by natural selection, and not by haphazard mutations. Oncoscience. 2014;1:580-590
56. Duesberg P, Mandrioli D, McCormack A, Nicholson JM. Is carcinogenesis a form of speciation? Cell Cycle. 2011;10:2100-2114
57. Huuhtanen RL, Blomqvist CP, Bohling TO, Wiklund TA, Tukiainen EJ, Virolainen M. et al. Expression of cyclin A in soft tissue sarcomas correlates with tumor aggressiveness. Cancer Res. 1999;59:2885-2890
58. Knauss S, Klein A. From aneuploidy to cancer: the evolution of a new species? J Biosci. 2012;37:211-220
59. Vincent MD. Cancer: beyond speciation. Adv Cancer Res. 2011;112:283-350
60. SCHERER WF, SYVERTON JT, GEY GO. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J Exp Med. 1953;97:695-710
61. Sauzay C, Petit A, Bourgeois AM, Barbare JC, Chauffert B, Galmiche A. et al. Alpha-foetoprotein (AFP): A multi-purpose marker in hepatocellular carcinoma. Clin Chim Acta. 2016;463:39-44
62. Terentiev AA, Moldogazieva NT. Alpha-fetoprotein: a renaissance. Tumour Biol. 2013;34:2075-2091
63. Ma Y, Jia Y, Chen L, Ezeogu L, Yu B, Xu N. et al. Weaknesses and Pitfalls of Using Mice and Rats in Cancer Chemoprevention Studies. J Cancer. 2015;6:1058-1065
64. He Y, Yuan C, Chen L, Liu Y, Zhou H, Xu N. et al. While it is not deliberate, much of today's biomedical research contains logical and technical flaws, showing a need for corrective action. Int J Med Sci. 2018;15:309-322
65. Davis JS. III. The Effect of Scarlet Red, in Various Combinations, upon the Epitheliation of Granulating Surfaces. Ann Surg. 1910;51:40-51
66. Davis JS. X. A Further Note on the Clinical Use of Scarlet Red and its Component, Amidoazotoluol, in Stimulating the Epitheliation of Granulating Surfaces. Ann Surg. 1911;53:702-719
67. VASILIEV JM, CHEUNG AB. Evolution of epithelial proliferation induced by scarlet red in the skin of normal and carcinogen-treated rabbits. Br J Cancer. 1962;16:238-245
68. Fujiki H. Gist of Dr. Katsusaburo Yamagiwa's papers entitled "Experimental study on the pathogenesis of epithelial tumors" (I to VI reports). Cancer Sci. 2014;105:143-149
69. Yamagiwa K, Ichikawa K. Experimental study of the pathogenesis of carcinoma. CA Cancer J Clin. 1977;27:174-181
70. Huggins C, BRIZIARELLI G, SUTTON H Jr. Rapid induction of mammary carcinoma in the rat and the influence of hormones on the tumors. J Exp Med. 1959;109:25-42
71. Mider GB, Morton JJ. Skin tumors following a single application of methylcholanthrene in C57 brown mice. Am J Pathol. 1939;15:299-302
72. Cabot S, Shear N, Shear MJ, Perrault A. Studies in carcinogenesis: XI. Development of skin tumors in mice painted with 3:4-benzpyrene and creosote oil fractions. Am J Pathol. 1940;16:301-312
73. Haslam SZ, Bern HA. Histopathogenesis of 7,12-diemthylbenz(a)anthracene-induced rat mammary tumors. Proc Natl Acad Sci U S A. 1977;74:4020-4024
74. McGuire WL, Chamness GC, Costlow ME, Shepherd RE. Hormone dependence in breast cancer. Metabolism. 1974;23:75-100
75. Mobbs BG. Uptake of (3H)oestradiol by dimethylbenzanthracene-induced rat mammary tumours regressing spontaneously or after ovariectomy. J Endocrinol. 1969;44:463-464
76. YOUNG S, COWAN DM. Spontaneous regression of induced mammary tumours in rats. Br J Cancer. 1963;17:85-89
77. Burrows H. Carcinoma mammae occurring in a male mouse under continued treatment with oestrin. Am J Cancer. 1935;24:613-616
78. Geschickter CF, Lewis D, Hartman CG. Tumors of the breast related to the oestrin hormone. Am J Cancer. 1934;21:828-859
79. Mceuen CS. Occurrence of cancer in rats treated with oestrone. Am J Cancer. 1938;34:184-195
80. Cutts JH, Froude GC. Regression of estrone-induced mammary tumors in the rat. Cancer Res. 1968;28:2413-2418
81. Cutts JH. Enzyme activities in regressing estrone-induced mammary tumors of the rat. Cancer Res. 1973;33:1235-1237
82. Huggins C. Endocrine-induced regression of cancers. Cancer Res. 1967;27:1925-1930
83. Huggins C. Endocrine-induced regression of cancers. Am J Surg. 1978;136:233-238
84. Noble RL, Cutts JH. Mammary tumors of the rat: a review. Cancer Res. 1959;19:1125-1139
85. Wang C, Lisanti MP, Liao DJ. Reviewing once more the c-myc and Ras collaboration: converging at the cyclin D1-CDK4 complex and challenging basic concepts of cancer biology. Cell Cycle. 2011;10:57-67
86. Liao DJ, Dickson RB. Roles of androgens in the development, growth, and carcinogenesis of the mammary gland. J Steroid Biochem Mol Biol. 2002;80:175-189
87. BIELSCHOWSKY F. Chronic iodine deficiency as cause of neoplasia in thyroid and pituitary of aged rats. Br J Cancer. 1953;7:203-213
88. FURTH J. Conditioned and autonomous neoplasms: a review. Cancer Res. 1953;13:477-492
89. MORRIS HP, DALTON AJ, GREEN CD. Malignant thyroid tumors occurring in the mouse after prolonged hormonal imbalance during the ingestion of thiouracil. J Clin Endocrinol Metab. 1951;11:1281-1295
90. Sun L, Nguyen AT, Spitsbergen JM, Gong Z. Myc-induced liver tumors in transgenic zebrafish can regress in tp53 null mutation. PLoS One. 2015;10:e0117249
91. Li Z, Huang X, Zhan H, Zeng Z, Li C, Spitsbergen JM. et al. Inducible and repressable oncogene-addicted hepatocellular carcinoma in Tet-on xmrk transgenic zebrafish. J Hepatol. 2012;56:419-425
92. Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell. 1999;4:199-207
93. Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S. et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112-1117
94. D'Cruz CM, Gunther EJ, Boxer RB, Hartman JL, Sintasath L, Moody SE. et al. c-MYC induces mammary tumorigenesis by means of a preferred pathway involving spontaneous Kras2 mutations. Nat Med. 2001;7:235-239
95. Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ. et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249-3262
96. Arvanitis C, Felsher DW. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol. 2006;16:313-317
97. Li Y, Li H, Spitsbergen JM, Gong Z. Males develop faster and more severe hepatocellular carcinoma than females in krasV12 transgenic zebrafish. Sci Rep. 2017;7:41280
98. Tilli MT, Furth PA. Conditional mouse models demonstrate oncogene-dependent differences in tumor maintenance and recurrence. Breast Cancer Res. 2003;5:202-205
99. Tran PT, Fan AC, Bendapudi PK, Koh S, Komatsubara K, Chen J. et al. Combined Inactivation of MYC and K-Ras oncogenes reverses tumorigenesis in lung adenocarcinomas and lymphomas. PLoS One. 2008;3:e2125-doi 10.1371/journal.pone.0002125
100. Nguyen AT, Emelyanov A, Koh CH, Spitsbergen JM, Parinov S, Gong Z. An inducible kras(V12) transgenic zebrafish model for liver tumorigenesis and chemical drug screening. Dis Model Mech. 2012;5:63-72
101. Uchiyama K, Watanabe D, Hayasaka M, Hanaoka K. A novel imprinted transgene located near a repetitive element that exhibits allelic imbalance in DNA methylation during early development. Dev Growth Differ. 2014;56:653-668
102. Zheng W, Li Z, Nguyen AT, Li C, Emelyanov A, Gong Z. Xmrk, kras and myc transgenic zebrafish liver cancer models share molecular signatures with subsets of human hepatocellular carcinoma. PLoS One. 2014;9:e91179-doi 10.1371/journal.pone.0091179
103. Tonelli C, Morelli MJ, Sabo A, Verrecchia A, Rotta L, Capra T. et al. Genome-wide analysis of p53-regulated transcription in Myc-driven lymphomas. Oncogene. 2017;36:2921-2929
104. Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323-1334
105. Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L. et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661-665
106. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V. et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656-660
107. Anders K, Kershaw O, Larue L, Gruber AD, Blankenstein T. The immune system prevents recurrence of transplanted but not autochthonous antigenic tumors after oncogene inactivation therapy. Int J Cancer. 2017;141:2551-2561
108. Dolezal JM, Wang H, Kulkarni S, Jackson L, Lu J, Ranganathan S. et al. Sequential adaptive changes in a c-Myc-driven model of hepatocellular carcinoma. J Biol Chem. 2017;292:10068-10086
109. Wang Y, Suh YA, Fuller MY, Jackson JG, Xiong S, Terzian T. et al. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest. 2011;121:893-904
110. Liao DJ, Dickson RB. Cell death in MMTV-c-myc transgenic mouse mammary tumors may not be typical apoptosis. Lab Invest. 2003;83:1437-1449
111. Liao DJ, Dickson RB. c-Myc in breast cancer. Endocr Relat Cancer. 2000;7:143-164
112. Briand P. Hormone-dependent mammary tumors in mice and rats as a model for human breast cancer (review). Anticancer Res. 1983;3:273-281
113. Noble RL. The development of prostatic adenocarcinoma in Nb rats following prolonged sex hormone administration. Cancer Res. 1977;37:1929-1933
114. Noble RL. Prostate carcinoma of the Nb rat in relation to hormones. Int Rev Exp Pathol. 1982;23:113-159
115. Rous P, Kidd JG. CONDITIONAL NEOPLASMS AND SUBTHRESHOLD NEOPLASTIC STATES: A STUDY OF THE TAR TUMORS OF RABBITS. J Exp Med. 1941;73:365-390
116. Lou X, Zhang J, Liu S, Xu N, Liao DJ. The other side of the coin: The tumor-suppressive aspect of oncogenes and the oncogenic aspect of tumor-suppressive genes, such as those along the CCND-CDK4/6-RB axis. Cell Cycle. 2014;13:1677-1693
117. Baig UI, Bhadbhade BJ, Watve MG. Evolution of aging and death: what insights bacteria can provide. Q Rev Biol. 2014;89:209-223
Author contact
![]() Corresponding authors: Mingjun Shi, Department of Pathophysiology, Guizhou Medical University, Guiyang, Guizhou Province 550025, China. Email: smjtyfedu.cn OR Dr. Ningzhi Xu, Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, China. Email: xuningzhiac.cn OR Dr. D. Joshua Liao, Department of Pathology, Guizhou Medical University Hospital, Guiyang, Guizhou 550004, China. Email: djliaoedu.cn
Corresponding authors: Mingjun Shi, Department of Pathophysiology, Guizhou Medical University, Guiyang, Guizhou Province 550025, China. Email: smjtyfedu.cn OR Dr. Ningzhi Xu, Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, China. Email: xuningzhiac.cn OR Dr. D. Joshua Liao, Department of Pathology, Guizhou Medical University Hospital, Guiyang, Guizhou 550004, China. Email: djliaoedu.cn