Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2018; 9(24):4774-4782. doi:10.7150/jca.25811 This issue Cite
Research Paper
Glucocorticoid-Inducible Kinase 2 Promotes Bladder Cancer Cell Proliferation, Migration and Invasion by Enhancing β-catenin/c-Myc Signaling Pathway
Jin-Bo Chen1*, Meng Zhang2*, Xiao-Long Zhang3, Yu Cui1, Pei-Hua Liu1, Jiao Hu1, Hui-Huang Li1, Hang Jin1, Long-Fei Liu1, Min-Feng Chen1, He-Qun Chen1, Chao-Zhao Liang2 ![]() , Xiong-Bing Zu1
, Xiong-Bing Zu1 ![]()
1. Department of Urology, Xiangya Hospital, Central South University, NO. 87 Xiangya Road, Changsha, Hunan 410008, P.R. China
2. Department of Urology, the First Affiliated Hospital of Anhui Medical University, NO. 218 Jixi Road, Hefei, Anhui 230022, P.R. China
3. Department of Experimental Research, Sun Yat-Sen University Cancer Center, NO.651 East Dongfeng Road, Guangzhou, Guangdong 510060, P.R. China
*These authors contributed equally to this work.
Received 2018-2-28; Accepted 2018-10-21; Published 2018-12-10
Abstract
Background: Bladder cancer is one of the most common malignancies in urologic system. The glucocorticoid-inducible kinase 2 (SGK2) expression and function were largely unknown in cancers. Current study was aimed to investigate the role of SGK2 in bladder cancer and its potential mechanisms.
Methods: SGK2 expression was quantified by western blot (WB) in multiple bladder cancer cell lines (T24, 5637, J82 and UMUC3) compared with normal urothelial cell line (SVHUC). SGK2 knocking down and overexpression model were established by lentivirus transfection. MTT, colony formation, wound healing and transwell assay were used to assess the tumor cell proliferation, migration and invasion abilities, respectively. In addition, molecular function analysis was performed using FunRich software V3. Immunoprecipitation (IP) assay was applied to investigate the interaction between SGK2 and β-catenin at protein level. TCGA database was retrieved to verify the association between these genes and clinical tumor stage as well as prognosis among bladder cancer patients.
Results: SGK2 expression was significantly upregulated in multiple bladder cancer cell lines compared with SVHUC at protein level. Cell proliferation, migration and invasion abilities were significantly decreased after knocking down SGK2 in J82 and UMUC3 cell lines. Inversely, cell aggressive phenotypes were significantly increased after overexpressing SGK2 in T24 cell line. Furthermore, functional analyses of SGK2 based on TCGA database showed that SGK2 related genes were involved in receptor activity, ATP binding, DNA repair protein, trans-membrane receptor activity and lipid binding. In addition, protein interaction analysis identified c-Myc was significantly enriched in SGK2 positively associated genes. The prediction was validated by WB and IP assay that SGK2 could directly bind with β-catenin at protein level to regulate their downstream gene c-Myc expression in bladder cancer to influence tumor progression. And clinical data generated from TCGA database also identified these downstream genes were significantly associated with tumor stage and survival status of bladder cancer patients.
Conclusion: Taken together, our findings suggest SGK2 promotes bladder cancer progression via mediating β-catenin/c-Myc signaling pathway, which may serve as a potential therapeutic target for bladder cancer patients.
Keywords: glucocorticoid-inducible kinase 2, bladder cancer, c-Myc, β-catenin
Introduction
Bladder cancer (BCa) is one of the most common malignant genitourinary tumors, with high morbidity and mortality rates [1]. At initial diagnosis, approximately 75% of patients have non-muscle invasive bladder cancer (NMIBC) associated with a favorable prognosis. However, about 25% patients present with muscle-invasive or metastatic disease associated with a relatively poor prognosis [2]. Early diagnosis with individualized treatment and follow-up is critical to a successful clinical outcome [3]. Despite great efforts in diagnosis and treatment methods, patients with bladder cancer suffer from high recurrence rates and poor clinical outcomes for advanced disease [4]. Therefore, it is of great significance to identify specific molecular biomarkers for early diagnosis and clinical therapeutics in bladder cancer patients.
The glucocorticoid-inducible kinase (SGK) family has three isoforms: SGK1, SGK2 and SGK3. SGK2 and SGK3 share 80% homology with SGK1 in their kinase domains, which can be activated by 3'-phosphoinositide-dependent kinase-1 (PDK1), placing them under the regulation of the phosphoinositide 3'-kinase (PI3K) signaling pathway [5]. Previous studies reported that SGK1 and SGK3 played vital roles during the progression of various cancers [6, 7]. However, very few studies investigated the function of SGK2 in cancer and its role in bladder carcinoma remains unclear. Similar to SGK1 and SGK3, SGK2 also can activate certain sodium and potassium channels, indicating it may be involved in the physiological and pathological processes regulation [8]. Thus, in the present study, we investigated the role of SGK2 in bladder cancer and its potential mechanisms by in vitro study and TCGA clinical analysis.
Materials and Methods
Cell culture and reagents
Human bladder cancer cell lines T24, 5637, J82 and UMUC3 as well as normal epithelial cell line SVHUC were purchased from the Shanghai Institute of Cell Biology, Chinese Academy of Sciences. All cells were cultured with Dulbecco's Modified Eagle's Medium (DMEM; Gibco Company, Grand Island, NY, USA) with 10% fetal bovine serum (FBS; Gibco Company) and 1 % penicillin/streptomycin (Invitrogen) and at 37°C with 5% CO2. MG132 and CHX (cycloheximide) were obtained from Sigma-Aldrich (MO, USA).
Lentivirus packaging and plasmid transfection
The target plasmids include as follows: sh-vector, shSGK2, oe-vector, oeSGK2. The above plasmids with the packaging plasmid psAX2 and envelope plasmid pMD2G, were transfected into 293T cells by calcium chloride transfection method, respectively. The shSGK2 sequences were as follows: Sense: GCACCTGAAGTGCTTCGGAAA, anti-sense: TTTCCGAAGCACTTCAGGTGCT. After 48h later, the virus supernatant was directly added to cells in 6-well plate (or immediately frozen in -80 ℃ freezer for future use) maintaining for 24h, and after 48h culture, we collected the cell protein to test infection efficiency.
Western blot analysis
Proteins were isolated from these transfected cells. Concentration was quantified by Bradford assay. Using SDS-PAGE protein electrophoresis, the PVDF membranes were blocked with 5% skim milk and incubated overnight at 4°C with primary antibodies GAPDH (1:1000, sc-47724, Santa Cruz Biotechnology, CA, USA), SGK2 (1:500, sc-100355, Santa Cruz Biotechnology, CA, USA), c-Myc (1:500, sc-40, Santa Cruz Biotechnology, CA, USA), β-catenin (1:500, sc-7963, Santa Cruz Biotechnology, CA, USA) and washed 3 times with TBST. Then, membranes were incubated with goat anti- mouse IgG secondary antibodies (1:5000; Santa Cruz Biotechnology, CA, USA) and re-washed TBST 3 times. The ECL system (Thermo Fisher Scientific) was applied for western blot band detection.
MTT assay
Cells at 60%-80% confluence were plated at a concentration of 5 × 103 cells/well in 24-well plates and incubated for 24 h before treatment. At day 0, 2, 3, 4, MTT (0.5mg/ml) was added to each well. After 2 h further incubation, removed the medium and added DMSO 500ul into each well. Gently shaking in room temperature for 5 minutes and transferred 100ul/each into 96-well plate. Then, we measured the 570 nm optical density (OD570) for each well (ELX-800, Bio-Tek).
Colony formation assay
The cell suspension with 500 cells/group were placed into a 6cm plate. Cells were cultured with 3 ml media at 37°C incubator. After 2 weeks of cultivation cells were gently washed with PBS, fixed with formalin and stained with 0.1% crystal violet, the cloning efficiency was determined.
Wound healing assay
Cells from each group were seeded in 6-well plates at around 90% confluence. Subsequently, we utilized a 200ul pipette tip to create symmetrical wounds. After washing with PBS twice, cells were incubated with non-serum DMEM medium for 24 h. Migration pictures were taken at 0h and 24 h after drawing the wound. The wound distance of 6 random fields per well at 40× magnification was measured by Image J software. Each experiment was performed in triplicate.
Transwell assay
Migration and invasion assays were conducted using 8-μm transwell chambers (Corning Company, NY, USA). For invasion assay, we firstly added 100 ul Matrigel mixed with non-serum medium to the upper chambers (Matrigel, 1:20; BD Biosciences), and incubated in the 37°C for 4 h. Then, 5 × 104 cells suspension in 150ul non-serum medium were seeded into the upper chambers. 800 ul of 10% FBS medium was added into the lower chambers. For migration assay, 2 × 104 cells were seeded into the upper chambers. At 24 h later, transwells were gently washed with PBS, fixed with formalin, removed upper chamber cells with swabs and stained with 0.1% crystal violet. Invaded cells were counted in 3 random fields per well under a 100x microscope. Each experiment was performed in triplicate.
Immunoprecipitation (IP) assay
IP was performed according to the protocol. Briefly, after cold PBS washing, the UMUC3 cells (3*15 cm plate dishes) were scraped into lysis buffer containing protease inhibitors for 30 min on ice. After ultrasound, sample lysis was centrifuged at 14,000×g for 20 min at 4 °C. The supernatants were incubated with normal IgG or anti-SGK2 antibody and protein A/G-beads. The precipitated complexes were washed with lysis buffer and boiled for 5 min in SDS sample buffer. β-catenin antibody were used for western blot analysis.
Molecular function of SGK2 in bladder cancer based on TCGA database
The expression profiles of bladder cancer were generated from The Cancer Genome Atlas (TCGA) database [https://xenabrowser.net/datapages/?cohort=TCGA%20Bladder%20Cancer%20(PRAD)], containing 426 tumor samples and 19 normal controls. Using R package version 2.10.0 software, we extracted 1000 genes that positive or negative related to SGK2 at gene expression level, respectively. In addition, gene enrichment and protein interaction tests were conducted based on FunRich V3 software.
Statistical analysis
The SPSS 19.0 (Chicago, IL, USA) and GraphPad 6.0 software were used to data analysis. Results were expressed as the means ± standard deviation (SD). The t-test and one-way analysis of variance (ANOVA) were performed. P < 0.05 was considered to have statistical significance.
Results
Gene expression and cell line selection
To investigate the role of SGK2 in bladder cancer, we retrieved TCGA database to verify its mRNA expression differences between tumor and para-tumor tissues, and identified that SGK2 expression in tumor group was not significantly increased compared with para-tumor group (P > 0.05) (Figure 1A). In addition, we also checked the Oncomine database, to further verify the association between SGK2 copy number variations (CNVs) and tumor status, and identified that CNVs was positively related to high pathological and clinical stages, as well as the recurrence status (Figure S1).
SGK2 expression in bladder cancer patients and cell lines. (A). The SGK2 mRNA expression between bladder tumor vs. para-tumor tissues. Patients' data was retrieved from TCGA database. (B). Western blot (WB) showed SGK2 protein expression in several bladder cancer cell lines (T24, 5637, J82, UMUC3) compared with normal epithelial cell line SVHUC. (C). WB showed the SGK2 knocking down efficacy in UMUC3 and J82 cell lines. (D). WB showed the SGK2 overexpression efficacy in T24 cell line.
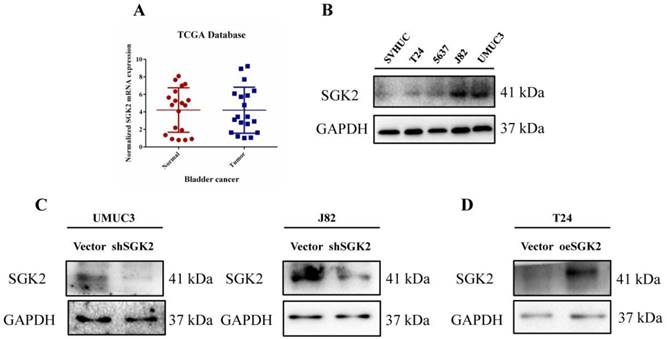
As SGK2 belongs to protein kinases family, we predicted it may play an important role during bladder cancer progression mainly at protein level. Therefore, we tested the expression levels of SGK2 in several commonly used bladder cancer cell lines (T24, 5637, J82, UMUC3) compared with normal urothelial cell line SVHUC. We identified that SGK2 was significantly increased in UMUC3 and J82 cell lines compared with SVHUC (Figure 1B). Therefore, we adopted these two cell lines for following knocking down SGK2 function assays (Figure 1C). And gain-of-function cell models were established in T24 cell line (Figure 1D).
SGK2 promotes bladder cancer cell growth and colony formation (CF) abilities in vitro
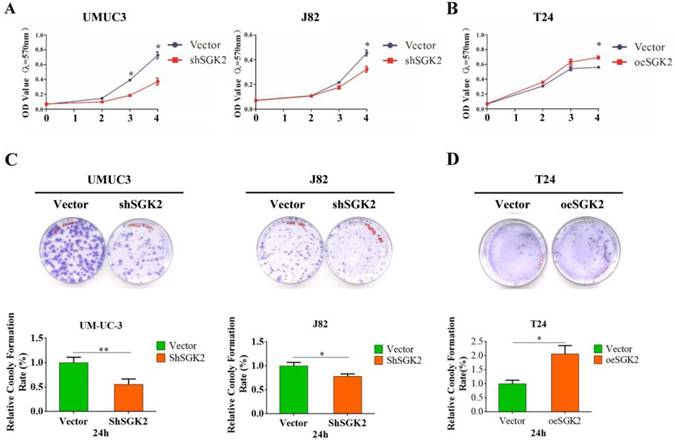
To gain insight into the function of SGK2 in bladder cancer progression, we applied MTT assays to test cell viability after silencing SGK2 expression in UMUC3 and J82 cell lines, and results revealed a significantly decreased cell growth (Figure 2A). Inversely, cell growth was significantly increased after overexpressing SGK2 in T24 cell line compared with vector control group (Figure 2B). Further, CF assay was utilized to determine the SGK2's function for CF abilities. Results showed shSGK2 in UMUC3 and J82 cells significantly reduced CF ability (Figure 2C) and oeSGK2 in T24 increased CF ability (Figure 2D).
SGK2 promotes bladder cancer cell growth and colony formation (CF) abilities in vitro. (A). MTT assays indicated cell growth was inhibited after knocking down SGK2 in UMUC3 and J82 cell lines (P < 0.05). (B). Cell growth was significantly increased after overexpressing SGK2 in T24 cell line using MTT assay (P < 0.05). (C). The colony formation (CF) assay indicated knocking down SGK2 significantly decreased CF ability in UMUC3 (P < 0.01) and J82 cells (P < 0.05). (D). CF ability was significantly increased after overexpressing SGK2 in T24 cell line (P < 0.05). Data are representative of three independent experiments. * indicated P < 0.05. ** indicated P < 0.01.
SGK2 promotes bladder cancer cells migration and invasion abilities in vitro
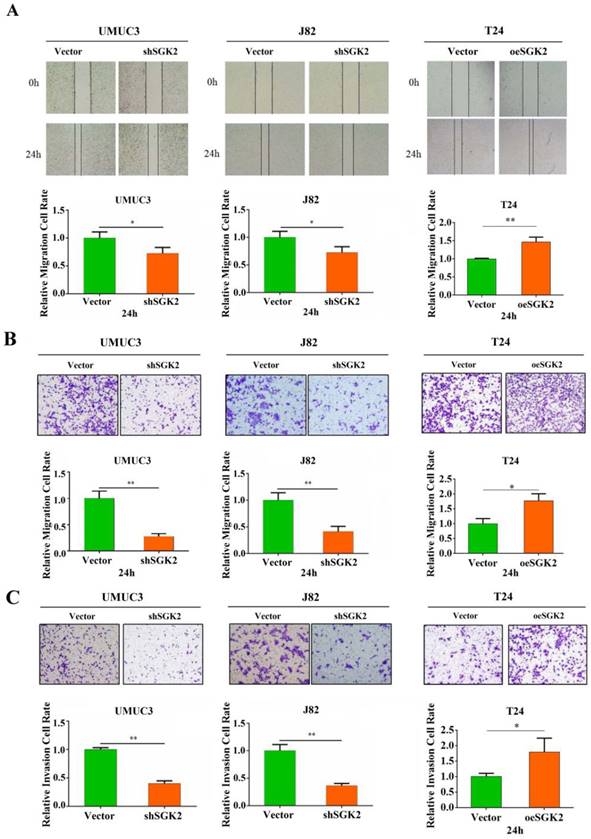
To investigate the effects of SGK2 expression on migration and invasion of bladder cancer cells, wound healing and transwell assay were applied. The results showed that knocking down SGK2 expression in J82 and UMUC3 cell lines significantly decreased cell migration and invasion abilities. Inversely, these abilities were significantly increased after SGK2 overexpression in T24 cell line (Figure 3A-3C).
SGK2 promotes bladder cancer cells migration and invasion abilities in vitro. (A). Wound healing assays indicated cell migration was inhibited after knocking down SGK2 in UMUC3 and J82 cell lines (P < 0.05). Inversely, cell migration was significantly increased after overexpressing SGK2 in T24 cell line (P < 0.01). (B). Transwell assays indicated cell migration was inhibited after knocking down SGK2 in UMUC3 and J82 cell lines (P < 0.01). Inversely, cell migration was significantly increased after overexpressing SGK2 in T24 cell line (P < 0.05). (C). Transwell assays indicated cell invasion was inhibited after knocking down SGK2 in UMUC3 and J82 cell lines (P < 0.01). Inversely, cell invasion was significantly increased after overexpressing SGK2 in T24 cell line (P < 0.05). Data are representative of three independent experiments. * indicated P < 0.05. ** indicated P < 0.01.
Molecular interaction and functional analysis of SGK2
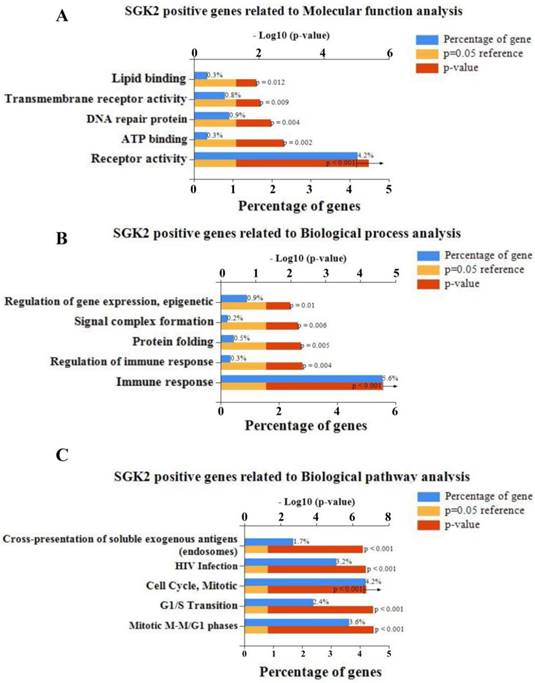
To predict the molecular interaction and function of SGK2 in bladder cancer, a gene co-expression network was constructed according to Pearson-correlation coefficients based on TCGA database. Next, the top 1000 SGK2-positive/negative related genes were classified according to GO term using FunRich software V3. Molecular function analysis indicated positive related genes of SGK2 were mainly enriched in receptor activity, ATP binding, DNA repair protein, trans-membrane receptor activity and lipid binding (Figure 4A), and negative genes were mainly enriched in ATPase activity, metallopeptidase activity, galactosyltransferase activity, GTPase activator activity and deaminase activity (Figure S2A). In addition, for biological process analysis, we found that SGK2-positive related genes were enriched in immune response, regulation of immune response, protein folding, signal complex formation and regulation of gene expression epigenetic (Figure 4B), and SGK2-negative related genes were mainly involved in energy, metabolism, ion transport, amino acid and derivative metabolism and fatty acid metabolism (Figure S2B). Moreover, biological pathway analysis was also conducted, and details were presented in Figure 4C and Figure S2C, for both positive and negative SGK2 related genes, respectively.
Molecular function analysis of SGK2. (A). Molecular function analysis suggested that positive related genes of SGK2 were mainly enriched in receptor activity, ATP binding, DNA repair protein, transmembrane receptor activity and lipid binding. (B). Biological process analysis indicated SGK2 positive related genes were enriched in immune response, regulation of immune response, protein folding, signal complex formation and regulation of gene expression epigenetic. (C). Biological pathway analysis indicated SGK2 positive related genes were enriched in Mitotic M-M/G1 phases, G1/S Transition, Cell cycle mitotic, HIV infection, Cross-presentation of soluble exogenous antigens.
To further evaluate the interactive relationships among genes that are positive or negative related to SGK2, we mapped these genes to protein network through FunRich software and identified that c-Myc was the key enriched interactive protein in SGK2-positive gene network (Figure S3), while GRB2 was in SGK2-negative gene network (Figure S2D). When retrieved references [9-10], we identified that c-Myc belonged to β-catenin/c-Myc pathway, and plays a more critical role during tumorigenesis rather than GRB2.
SGK2 regulates the protein expression of β-catenin/c-Myc pathway
Based on molecular function results, we further using WB assay to test whether β-catenin/c-Myc pathway involved in bladder cancer progression by acting as the downstream targets of SGK2. The result showed the protein levels of β-catenin and c-Myc were significantly decreased after down-regulating SGK2 expression in UMUC3 and J82 cells (Figure 5A). On the contrary, the protein levels of these genes were significantly increased after overexpressing SGK2 in T24 cell line (Figure 5B).
SGK2 regulates c-Myc/β-catenin gene expression. (A). WB results showed the protein levels of β-catenin and c-Myc were significantly decreased after down-regulating SGK2 expression in UMUC3 and J82 cells. (B). WB results showed the protein levels of β-catenin and c-Myc were significantly increased after overexpressing SGK2 in T24 cell. (C). Immunoprecipitation (IP) assay indicated SGK2 could directly interact with β-catenin protein in T24 cell. (D). MG132 (10 µM) abrogated shSGK2 induced inhibition of β-catenin protein expression in UMUC3 cell. (E). A time-dependent decrease in β-catenin protein expression in T24-vector and T24-oeSGK2 cell lines exposed to CHX (10 µg/mL). However, the decrease speed was marked slower in T24-oeSGK2 than T24-vector group. (F). The expression of c-Myc and CTNNB1 gene (encoding β-catenin protein) were significantly associated with tumor stage of bladder cancer (P < 0.05). (G). c-Myc expression was significantly associated with overall survival (OS), recurrence-free survival (RFS) in bladder cancer patients (P < 0.05). Upper and middle indicated protein expression analysis and lower indicated gene expression analysis.
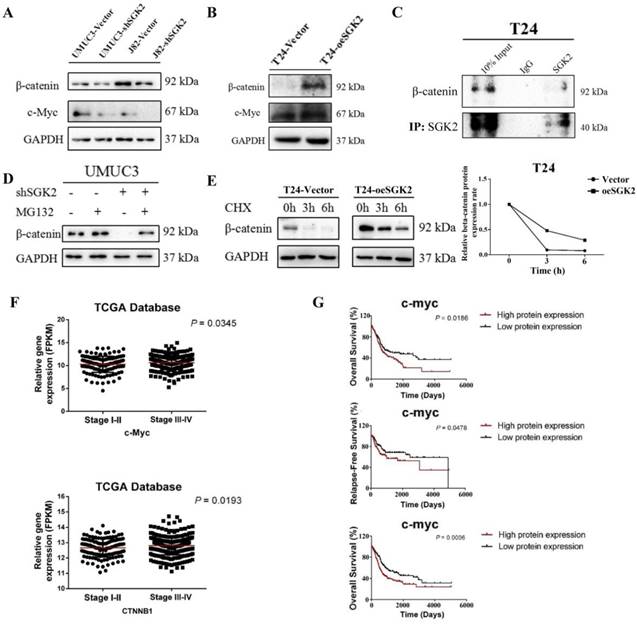
To further dissect the mechanism that how SGK2 affect c-Myc and β-catenin, we first searched some publications which showed β-catenin (encoded by CTNNB1 gene) was an upstream of c-Myc [9]. Thus, we mainly focused on the regulation between SGK2 and β-catenin. As previously studies found, the kinase could regulate the expression of downstream genes by directly phosphorylation or bind with them, influencing their protein stability [10]. Here, we applied the immunoprecipitation (IP) assay and our result indicated SGK2 could directly interact with β-catenin at protein level (Figure 5C). To further investigate whether SGK2 could affect the β-catenin protein stability, 10 µM MG132 (a proteasomal inhibitor) assay was applied, and he result revealed that MG132 abrogated shSGK2 induced inhibition of β-catenin protein expression in UMUC3 cell (Figure 5D). To further confirm that SGK2 affect β-catenin protein synthesis, T24-vector and T24-oeSGK2 cells were pretreated with CHX (10 µg/mL) for 0h, 3h, 6h. The result showed a time-dependent decrease in β-catenin protein expression in these two cell lines exposed to CHX. However, the decrease speed was marked slower in T24-oeSGK2 than T24-vector group (Figure 5E). These results support the hypothesis that the SGK2 could directly bind with β-catenin to regulate its expression through stabilizing its protein, and thus regulate the downstream gene c-Myc to influence bladder cancer.
In addition, we also retrieve from TCGA database to verify the relationship between expression of these genes and overall survival (OS), recurrence-free survival (RFS), tumor stage of bladder cancer patients. And we identified that c-Myc expression was significantly associated with OS, RFS and tumor stage of bladder cancer (Figure 5F & 5G). Also, the CTNNB1 gene, which encoded β-catenin protein, was significantly associated with tumor stage of bladder cancer (Figure 5F). Taken together, results suggested that SGK2 could modulate β-catenin/ c-Myc pathway to influence bladder cancer aggressive phenotypes, and these downstream genes could be served as therapeutic targets or prognostic markers for bladder cancer patients.
Discussion
Bladder cancer is one of the most common malignancy among urinary system tumors, with an estimated 429,800 new incidence and 165,100 mortality occurred in 2012 worldwide [11]. It is easy to recurrence and progression, which needs lifetime monitoring after diagnosis, bringing great burden to the society [12]. However, there is no specific and efficient targeted therapy till now. Therefore, finding potential biomarkers and identifying its underlying mechanisms may aid us to discover novel therapeutics to better fight against bladder cancer.
Previous studies indicated SGK family was highly homologous with the AKT kinase family, which shared similar upstream activators and downstream targets [13]. Hoang B et al found the SGK kinase activity in multiple myeloma cells could protect against endoplasmic reticulum (ER) stress apoptosis via SEK/JNK signaling [14]. Results from Shanmugam I et al suggested SGK1 is an androgen-regulated gene which played a vital role in AR-dependent survival and gene expression [15]. Yao Y et al proved lnc-SGK1 promoted Th2 and Th17 differentiation in human gastric cancer by SGK1/Jun B signaling [16]. Towhid ST et al discovered the small molecule EMD638683, which was a SGK1 inhibitor, promoted radiation-induced suicidal death of colon tumor cells [17]. Gasser JA et al uncovered SGK3 was amplified in breast cancer and activated PIK3CA downstream in a dependent manner of the phosphoinositide phosphatase INPP4B [18]. Liu H et al verified miR-212-3p could directly target SGK3 and inhibit glioblastoma cell proliferation [19]. However, very few studies investigated the role and potential mechanisms of SGK2 in cancer.
Here, our in vitro study demonstrated that silencing SGK2 in bladder cancer significantly decreased cell proliferation, migration and invasion. Inversely, when overexpressing SGK2, cell proliferation, migration and invasion were significantly increased. In addition, we analyzed the TCGA database to verify these genes which are positive or negative related to SGK2. FunRich software was also used to predict the interaction within these positive and negative genes, and identified that c-Myc was enriched in positive genes connecting with others. Since there was a publication suggested that SGK2 can interact with β-catenin pathway to promote hepatocellular carcinoma progression [20]. Therefore, we detected c-Myc and β-catenin expression, and identified these proteins are significantly increased or decreased after overexpressing or knocking down SGK2, respectively. In addition, results from IP assay as well as protein stability assays using MG132 and CHX indicated SGK2 could interact with and stabilize β-catenin protein. And according to references that c-Myc could be regulated by β-catenin, which was the key factor of β-catenin pathway. Thus, we predicted that SGK2 could regulate bladder cancer progression via β-catenin/c-Myc pathway.
Many studies reported c-Myc and β-catenin were oncogenes for tumor initiation and progression [21-25]. In our current study, we also checked the relationship between these genes and tumor stage as well as prognosis of bladder cancer based on the TCGA database. We identified that c-Myc expression was significantly associated with OS, RFS and tumor stage of bladder cancer. Also, the CTNNB1 gene, which encode β-catenin protein, was significantly associated with tumor stage of bladder cancer. These results suggested their potential roles as prognostic markers for bladder cancer patients.
Conclusion
To sum up, we identify SGK2 protein expression is elevated in bladder cancer cells lines. And SGK2 promotes bladder cancer cell proliferation, migration, invasion as well as colony formation abilities presumably via modulating β-catenin/c-Myc expression. Thus, SGK2 serves as an oncogene in the progression of bladder cancer and may be a potential therapeutic target for the treatment of bladder cancer patients.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81572523, 81802827), the Hunan Province Funds for Distinguished Young Scientists of China (2016JJ1026), the Fundamental Research Funds for the Central Universities of Central South University (2016zzts121) and the Natural Science Foundation of Guangdong Province, China (2017A030313800).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kamat AM, Hahn NM, Efstathiou JA, Lerner SP, Malmstrom PU, Choi W. et al. Bladder cancer. Lancet. 2016;388:2796-810
2. Youssef RF, Lotan Y. Predictors of outcome of non-muscle-invasive and muscle-invasive bladder cancer. ScientificWorldJournal. 2011;11:369-81
3. Tan YG, Eu E, Lau Kam On W, Huang HH. Pretreatment neutrophil-to-lymphocyte ratio predicts worse survival outcomes and advanced tumor staging in patients undergoing radical cystectomy for bladder cancer. Asian journal of urology. 2017;4:239-46
4. Rayn KN, Hale GR, Grave GP, Agarwal PK. New therapies in nonmuscle invasive bladder cancer treatment. Indian journal of urology. 2018;34:11-9
5. Bhalla V, Soundararajan R, Pao AC, Li H, Pearce D. Disinhibitory pathways for control of sodium transport: regulation of ENaC by SGK1 and GILZ. American journal of physiology-renal physiology. 2006;291:F714-21
6. Talarico C, Dattilo V, D'Antona L, Menniti M, Bianco C, Ortuso F. et al. SGK1, the New Player in the Game of Resistance: Chemo-Radio Molecular Target and Strategy for Inhibition. Cellular physiology and biochemistry. 2016;39:1863-76
7. Bruhn MA, Pearson RB, Hannan RD, Sheppard KE. AKT-independent PI3-K signaling in cancer - emerging role for SGK3. Cancer management and research. 2013;5:281-92
8. Lang F, Cohen P. Regulation and physiological roles of serum- and glucocorticoid-induced protein kinase isoforms. Science's STKE. 2001;2001:re17
9. Higgs MR, Lerat H, Pawlotsky JM. Hepatitis C virus-induced activation of beta-catenin promotes c-Myc expression and a cascade of pro-carcinogenetic events. Oncogene. 2013;32:4683-93
10. Zhang D, Fei F, Li S, Zhao Y, Yang Z, Qu J. et al. The role of beta-catenin in the initiation and metastasis of TA2 mice spontaneous breast cancer. Journal of cancer. 2017;8:2114-23
11. Antoni S, Ferlay J, Soerjomataram I, Znaor A, Jemal A, Bray F. Bladder Cancer Incidence and Mortality: A Global Overview and Recent Trends. European urology. 2017;71:96-108
12. Dy GW, Gore JL, Forouzanfar MH, Naghavi M, Fitzmaurice C. Global Burden of Urologic Cancers, 1990-2013. European urology. 2017;71:437-46
13. Bruhn MA, Pearson RB, Hannan RD, Sheppard KE. Second AKT: the rise of SGK in cancer signalling. Growth factors. 2010;28:394-408
14. Hoang B, Shi Y, Frost PJ, Mysore V, Bardeleben C, Lichtenstein A. SGK Kinase Activity in Multiple Myeloma Cells Protects against ER Stress Apoptosis via a SEK-Dependent Mechanism. Molecular cancer research. 2016;14:397-407
15. Shanmugam I, Cheng G, Terranova PF, Thrasher JB, Thomas CP, Li B. Serum/glucocorticoid-induced protein kinase-1 facilitates androgen receptor-dependent cell survival. Cell death & differentiation. 2007;14:2085-94
16. Yao Y, Jiang Q, Jiang L, Wu J, Zhang Q, Wang J. et al. Lnc-SGK1 induced by Helicobacter pylori infection and highsalt diet promote Th2 and Th17 differentiation in human gastric cancer by SGK1/Jun B signaling. Oncotarget. 2016;7:20549-60
17. Towhid ST, Liu GL, Ackermann TF, Beier N, Scholz W, Fuchss T. et al. Inhibition of colonic tumor growth by the selective SGK inhibitor EMD638683. Cellular physiology and biochemistry. 2013;32:838-48
18. Gasser JA, Inuzuka H, Lau AW, Wei W, Beroukhim R, Toker A. SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Molecular cell. 2014;56:595-607
19. Liu H, Li C, Shen C, Yin F, Wang K, Liu Y. et al. MiR-212-3p inhibits glioblastoma cell proliferation by targeting SGK3. Journal of neuro-oncology. 2015;122:431-9
20. Liu J, Zhang G, Lv Y, Zhang X, Ying C, Yang S. et al. SGK2 promotes hepatocellular carcinoma progression and mediates GSK-3beta/beta-catenin signaling in HCC cells. Tumour biology. 2017;39:1010428317700408
21. Wen H, Ma H, Li P, Zheng J, Yu Y, Lv G. Expression of far upstream element-binding protein 1 correlates with c-Myc expression in sacral chordomas and is associated with tumor progression and poor prognosis. Biochemical and Biophysical Research Communications. 2017;491:1047-54
22. Jiang Y, Mao C, Yang R, Yan B, Shi Y, Liu X. et al. EGLN1/c-Myc Induced Lymphoid-Specific Helicase Inhibits Ferroptosis through Lipid Metabolic Gene Expression Changes. Theranostics. 2017;7:3293-305
23. Huang J, He Y, McLeod HL, Xie Y, Xiao D, Hu H. et al. miR-302b inhibits tumorigenesis by targeting EphA2 via Wnt/ beta-catenin/EMT signaling cascade in gastric cancer. BMC cancer. 2017;17:886
24. Wang W, Wen Q, Luo J, Chu S, Chen L, Xu L. et al. Suppression Of beta-catenin Nuclear Translocation By CGP57380 Decelerates Poor Progression And Potentiates Radiation-Induced Apoptosis in Nasopharyngeal Carcinoma. Theranostics. 2017;7:2134-49
25. Chen J, Zhao J, Chen X, Ding C, Lee K, Jia Z. et al. Hyper activation of beta-catenin signalling induced by IKKε inhibition thwarts colorectal cancer cell proliferation. Cell Proliferation. 2017:50 doi: 10.1111/cpr.12350
Author contact
![]() Corresponding authors: Xiong-Bing Zu, M.D., Ph.D., Department of Urology, Xiangya Hospital, Central South University, NO.87 Xiangya Road, Changsha, Hunan, P.R. China, E-mail: whzuxbcom or Chao-Zhao Liang, M.D., Ph.D., Department of Urology, the First Affiliated Hospital of Anhui Medical University, NO.218 Jixi Road, Hefei, Anhui 230022, P.R. China. E-mail: liang_chaozhaocom. Tel: +86-731-8975-3011 Fax: +86-731-8975-3280.
Corresponding authors: Xiong-Bing Zu, M.D., Ph.D., Department of Urology, Xiangya Hospital, Central South University, NO.87 Xiangya Road, Changsha, Hunan, P.R. China, E-mail: whzuxbcom or Chao-Zhao Liang, M.D., Ph.D., Department of Urology, the First Affiliated Hospital of Anhui Medical University, NO.218 Jixi Road, Hefei, Anhui 230022, P.R. China. E-mail: liang_chaozhaocom. Tel: +86-731-8975-3011 Fax: +86-731-8975-3280.