Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(1):1-10. doi:10.7150/jca.27472 This issue Cite
Research Paper
The Efficiency of Verteporfin as a Therapeutic Option in Pre-Clinical Models of Melanoma
Jason W. Lui1,2,3, Sixia Xiao2, Kelsey Ogomori2, Jon E. Hammarstedt4, Elizabeth C. Little2, Deborah Lang3 ![]()
1. Committee on Development, Regeneration, and Stem Cell Biology, University of Chicago, Chicago, Illinois, U.S.A.
2. Section of Dermatology, University of Chicago, Chicago, Illinois, U.S.A.
3. Department of Dermatology, Boston University, Boston, Massachusetts, U.S.A.
4. Department of Orthopaedic Surgery, Allegheny General Hospital, Pittsburgh, Pennsylvania, U.S.A.
Received 2018-5-25; Accepted 2018-10-23; Published 2019-1-1
Abstract
Yes Associated Protein 1 (YAP) and Transcriptional coactivator with PDZ-Binding Motif (TAZ) have gained notoriety for their ability to drive tumor initiation and progression in a wide variety of cancers, including melanoma. YAP and TAZ act as drivers of melanoma through its interaction with the TEAD family of transcription factors. Verteporfin is a benzoporphyrin derivative that is used clinically for photodynamic treatment of macular degeneration. Recently it has emerged as a potential inhibitor of YAP/TAZ-TEAD interaction independent of light activation. In this study we determine if verteporfin has clinical potential by testing this compound on human melanoma cell cultures and in a clinically significant mouse model, BrafCA; Tyr-CreERT2; Ptenf/f, which parallels human melanoma in terms of disease progression, genetics, and histopathology. In culture, Verteporfin treatment induces a rapid drop in YAP and TAZ protein levels and cell numbers. In the transgenic model, utilizing drug levels that correspond to previously determined safe doses in human patients and with a dosing regimen calculated in this study, Verteporfin did not inhibit melanoma initiation or progression in comparison to mock treated controls. Taken together, our study suggests that although Verteporfin induces YAP/TAZ degradation in melanoma cell lines, Verteporfin was not effective as a YAP/TAZ-TEAD specific inhibitor of melanoma in our studies that aimed to mimic conditions found in clinic in terms of treatment regimen and disease model.
Keywords: melanoma, YAP, TAZ, Verteporfin, hippo pathway, mouse models
Introduction
Melanoma is a tumor with an aggressive nature, high degree of metastasis and a rising incidence rate [1]. Although significant discoveries have expanded therapeutic options in recent years, there are still many clinical challenges toward treating this disease. Yes Associated Protein 1 (YAP) and Transcriptional coactivator with PDZ-Binding Motif (TAZ) are two transcriptional coactivators that have been implicated to drive many different cellular processes that favor cellular proliferation, drug resistance, and metastasis in a wide variety of cancers, including melanoma [2, 3]. YAP and TAZ were originally discovered as crucial members of the organ size controlling Hippo Pathway, where a central core kinase cascade regulates the location (and subsequently the transcriptional activity) of YAP and TAZ. In addition to the Hippo Pathway, recent studies have also shown YAP and TAZ to be regulated by a wide variety of Hippo independent signals [3]. Both YAP and TAZ do not contain a DNA binding domain, so formation of a complex with transcription factors to drive expression of downstream target genes is required. Traditionally their main partners in driving cancer progression and survival are the TEAD family of transcription factors [4]. Thus, it is possible that inhibition of YAP/TAZ- TEAD interaction could prove to be a viable therapeutic strategy against melanoma.
Verteporfin (market name Visudyne) is a benzoporphyrin derivative that has been traditionally used in the clinic for photodynamic treatment of macular degeneration [5]. A recent in vitro screen yielded Verteporfin as a candidate inhibitor of YAP-TEAD interaction independent of light activation [6]. Since then, many studies have shown that Verteporfin inhibits tumor volume, growth, and YAP expression in a wide variety of xenograft models [7]. While xenograft models in melanoma are a valuable tool for studying human melanoma cells and the process of metastasis in an in vivo environment, this mouse model is poorly predictive of clinical efficiency [8]. Transgenic models, with intact microenvironments and immune systems, are a better predictor of translational outcomes for human patients [9, 10]. Prior to these studies, Verteporfin use has not been examined in cutaneous melanoma. Therefore, the ability of Verteporfin to inhibit melanoma growth and survival was tested. The response to Verteporfin by a panel of human melanoma cell lines in culture in terms of YAP and TAZ protein levels, cell growth, migration, and cellular morphology was measured. In addition, Verteporfin was tested as a therapeutic agent for melanoma in a pre-clinical transgenic model, BrafCA; Tyr-CreERT2; Ptenf/f mice, following a determined dosing regimen at clinically relevant drug levels.
Materials and Methods
Cell culture and growth curves
Human melanoma lines A375, LOX IMVI, A375-P, A375-M, mel-537, mel-624, SKMEL5, SKMEL23 and SKMEL28 (ATCC, Manassas, VA and University of Chicago Comprehensive Cancer Center Core Facilities) were cultured in DMEM with 10% FBS (Sigma-Aldrich). A375M and A375P refer to selected cell lines derived from A375 cells that exhibit low and high levels of metastasis for the A375 cell line in vivo [11]. Morphology, melanoma-marker testing, and histological analysis were used to verify melanoma cell identity and lack of mycoplasma contamination. For cellular growth curves, Cells were initially seeded at 10-20% and images in 5 random locations were taken daily. Cell numbers were calculated by averaging the daily cell counts for each of the 5 images per group over the course of 0 to 8 days in various conditions (Verteporfin treatments, DMSO treatments, YAP/TAZ knockdowns and transfection with siScramble controls). All growth curve experiments were performed minimally in triplicate. Normalization of curves was performed by calculating fold change levels over starting cell numbers from day 0.
Western analysis
Cells were lysed in RIPA buffer and 30 μg total protein was separated on 4-15% Bis-Tris gels and subsequently transferred to nitrocellulose membranes. The membranes were then probed overnight with 1:1000 YAP/TAZ antibody (Cell Signaling) and 1:10000 GAPDH (Cell Signaling). Membranes were washed with 1X TBS-T three times for 20 minutes and incubated with 1:4000 goat anti-rabbit IgG-HRP (Santa Cruz) and developed with Clarity Western ECL substrate according to the manufacturer's instructions (Bio-Rad).
Verteporfin timecourse
Cells were treated with Verteporfin (Sigma- Aldrich) at concentrations of 1, 2, 5 μM, or DMSO carrier alone. Cell lysates were collected at various time points (30 minutes, 2 hours, 3 hours, 24 hours) and analyzed for YAP and TAZ levels via Western blotting. Verteporfin treated samples were compared to DMSO vehicle control treated cells.
SiRNA treatment
Cells were seeded at 50-70% confluency in 6-well plates and subsequently transfected with 5μl of a 20μM siRNA stock solution against YAP1, WWRT1 (TAZ), and/or siScramble (Thermofisher ID Number S20366-YAP1, S24787 - WWTR1, 4390844 - siScramble) using Lipofectamine 3000 (Thermo Fisher) according to the manufacturer's instructions. Cell lysates were collected 2 days post-siRNA transfection.
Scratch assays
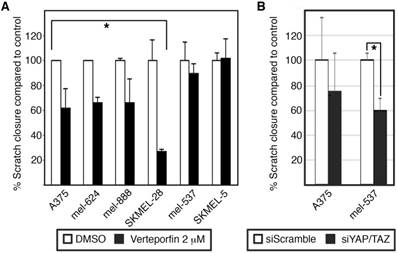
Scratch assays were performed and analyzed following prior methods [12, 13]. Cells were seeded at full confluency in 6 well plates. After siRNA transfection a wound was created using a 10 μL sterile tip. The media was then replaced with DMEM (10% FBS) with or without 2 μM Verteporfin. Photographs were taken immediately after wound creation and 24 hours post scratch. The open area was measured in arbitrary units using the ruler tool in Adobe Photoshop CS6. Percent closure for each picture pair (0 hr and 24 hr) was calculated as (gap at 24 hr/gap at 0 hr X 100). For each group (experimental and control) and cell line, the experiments were performed minimally in triplicate. For graphs shown in Figure 3, the level of closure for each control cell line at 24 hr is set to 100% to normalize the data across cell lines.
Both Verteporfin treatment and siRNA inhibition of YAP and TAZ inhibit melanoma cell migration. (A) Wound healing assay in Verteporfin (2μM) treated melanoma cells. The percent of wound closure was measured by dividing the width of the initial wound with the width of the wound 24 hours post scratch for the various groups comparing Verteporfin treated cells to control in 6 different melanoma lines. DMSO was used as a vehicle control. (B) Wound healing assay in YAP/TAZ siRNA inhibited melanoma cells. The percent of wound closure 24 hours post wound was measured comparing YAP/TAZ knockdown cells to siScrambled control in A375 and mel-537 cells. For both panels, an asterisk (*) indicates p<0.05, or p<0.005 for SKMEL-28 cells.
Cell length and morphology
Cells were treated with Verteporfin, siYAP/TAZ, or DMSO alone (2 μg/ml). Cell length was defined as the length from the tip of the longest dendrite to the cell body to the tip of the second longest dendrite. Length was then measured in arbitrary units for 50 cells in each group using the ruler tool in Photoshop CS6.
Mouse Verteporfin treatments
BrafCA; Tyr-CreERT2; Ptenf/f mice were previously described [14]. For localized melanoma induction, topical administration of 1-2 μl of 1.9 mg/ml (5mM) 4-hydroxytamoxifen (4HT) was applied on three consecutive days to 12 week old BrafCA; Tyr-CreERT2; Ptenf/f mice. The mice were subjected to 4 and 6 mg/kg Verteporfin intraperitoneal injections every other day for the course of the study starting the first day of melanoma induction. An equivalent amount of DMSO was used as a vehicle control. Tumors were collected 42 days post induction.
Mouse Verteporfin kinetics
The following protocol was modified from previous methods [15]. Wildtype mice (6 mice/group) were subjected to 0, 2, 4, 6 mg/kg Verteporfin intraperitoneal injections. Mouse tissue samples were collected 6, 24, 48, and 72 hours post IP-injections. The samples were then homogenized in 2% SDS, diluted tenfold with a chloroform-methanol binary mixture (1:2 v/v), and centrifuged for 10 min at 3000 RPM. The resulting supernatant was then measured using fluorescent spectroscopy with excitation settings at 400 nm and emission at 550-750 nm.
Immunohistochemistry
Skin tumor samples were harvested from mice 42 days post induction. Tumor samples were fixed with formalin and subsequently paraffin embedded. Tissue was cut into 5μm slices and rehydrated through an ethanol to water wash series. Antigen retrieval was performed by boiling the sections in Tris-EDTA buffer for 30 minutes and subsequently placed in cold ddH2O for 10 minutes. Sections were blocked using 5% normal horse serum in 1X TBS and probed for YAP/TAZ (Cell Signaling, 1:200) at 4 degrees Celsius overnight. Sections were washed with 1X TBS-T and incubated with DyLight 594 Anti-Rabbit IgG (diluted in blocking buffer 1:200, Vector Labs) at room temperature for an hour. Three additional washes with washing buffer for 5 minutes were performed prior to mounting with Vectashield Anti-fade Mounting Medium with DAPI (Vectashield).
Densitometric analyses of YAP/TAZ expression
To quantify levels of expression of YAP and TAZ in experimental and control group tumor specimens, 10 images were taken from 3 independent slides where YAP and TAZ expression were detected by immunofluorescence analysis. For each image, densitometry of the resultant fluorescence was performed with ImageJ software (ImageJ version 1.47 public domain software; National Institutes of Health, Bethesda, MD, USA). For normalization of the values, each densitometric reading for YAP/TAZ (red channel) was divided by the value for the nuclear DAPI staining from the blue channel. The data presented are densitometric readings from the averages of three independent slides per group.
Statistical analyses
Significance of the differences between the control and experimental groups was determined with Student's t-test and Chi-square analysis with a confidence interval of 95%. All values stated as significant have p values of less than or equal to 0.05 unless indicated. All experiments were performed minimally in triplicate.
Results
YAP and TAZ are expressed in melanoma cells, and this expression is reduced by Verteporfin
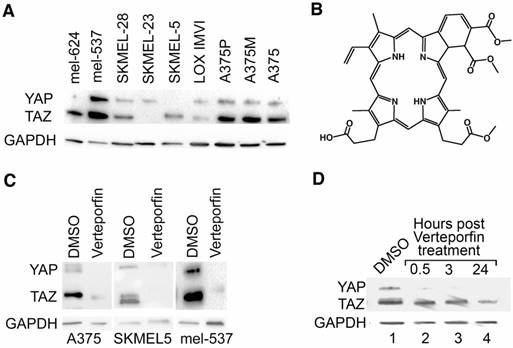
To initially determine the effect of Verteporfin on YAP and TAZ, 9 human melanoma cell lines were analyzed for the presence of YAP and TAZ proteins. 7 lines expressed varying degrees of both YAP and TAZ protein, while 2 lines expressed either YAP or TAZ (Figure 1A). Three lines (A375, SKMEL5, mel-537) were chosen for further analysis based on their varying degrees of YAP and TAZ expression. The benzoporphyrine derivative, Verteporfin (Figure 1B), was identified as an inhibitor of YAP and TAZ function [6]. Verteporfin treatment (2 μM) lead to a decrease in both YAP and TAZ protein levels that was detectable as early as 30 minutes and up to 24 hours (Figures 1C,1D).
Verteporfin treatment decreases YAP and TAZ protein levels in melanoma cells. (A) YAP and TAZ are expressed in melanoma cells. Western blot analysis probing for YAP and TAZ, with GAPDH as a loading control, was performed in a panel of cell lines (Lanes 1-9). (B) Chemical structure diagram of Verteporfin, as modified from information from manufacturer (Sigma-Alrich). (C) YAP and TAZ protein levels drop upon Verteporfin treatment. A375, SKMEL5, and mel-537 cells were treated with Verteporfin at 2 μM. DMSO was used as a vehicle control. 2 hours after treatment, cell lysates were collected for Western blot analysis. (D) A375 cells were subjected to Verteporfin treatment (2 μM) and cell lysates were collected 30 minutes, 3 hours, and 24 hours post treatment for Western analysis testing for YAP and TAZ expression, with GAPDH as a loading control.
Both Verteporfin treatment and direct RNA-interference of YAP and TAZ inhibits cell population expansion in melanoma cells
YAP and TAZ, as downstream effectors of the HIPPO signaling pathway, are implicated in controling cellular proliferation and organ size in humans and flies [16, 17]. To determine if Verteporfin affects melanoma cell growth, cell numbers were recorded over a time course with exposure to the drug or with carrier alone (DMSO). Verteporfin treatment at 2 different concentrations (2 and 5 μM) led to a reduced rate of melanoma cell proliferation over a time-course of several days as compared to vehicle control (Figure 2A). While each cell line exhibited multiple population doublings in control groups, the rate of cell proliferation in Verteporfin-treated groups did not significantly rise above starting cell numbers in the three lines tested. Here, we find that Verteporfin significantly inhibits cell growth in A375, mel-624, and mel-537 melanoma cells at concentrations of 2 and 5 μM.
Verteporfin treatment and direct RNA-interference of YAP and TAZ inhibits cell population expansion. (A) Verteporfin treatment reduces the rate of melanoma cell proliferation. A375, mel-624, and mel-537 cells were subjected to Verteporfin treatment (2 and 5 μM) and cell counts were taken over the course of 5 days. There are significant differences between control and experimental groups at 5 days of treatment (p<0.05). Cells from at least five independent fields (100X magnification, with approximately 10-500 cells/field depending on experimental group and conditions) for each experiment were counted, and experiments were performed in triplicate. Graphs are plotted as fold change in overall cell numbers (y axis) over time (x axis). (B) Gene-specific targeting of YAP, TAZ, or both in melanoma cells. Western analysis probing for YAP and TAZ, with GAPDH as a loading control, was performed in A375 (top panel) and mel-537 (lower panel) cell lines. Expression of both YAP and TAZ is inhibited by >90% of control levels as measured by densitometry for both cell lines (A375 and mel-537). (C) Gene-specific siRNA against YAP and TAZ lead to reduced melanoma cell numbers when compared to cells transfected with siScramble negative control siRNAs. There is a significant difference in cell numbers at day 3 (A375) or Day 6 (mel-537)(p<0.05). Cell counts were performed following methods described in (A).
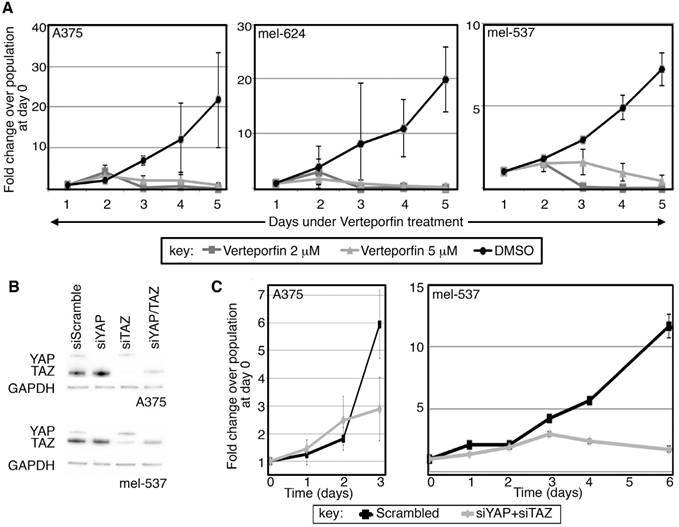
Since Verteporfin treatment led to a reduction of cellular growth and YAP/TAZ protein levels, and that Verteporfin has been previously described to inhibit YAP and TAZ function [6], inhibition of YAP and TAZ through siRNA targeting was performed to determine if the resulting cellular phenotype would replicate that of Verteporfin treatment. Multiple YAP and TAZ specific siRNAs were tested for specificity and efficiency in inhibition (Figure S1), and siTAZ2 and siYAP4 were utilized for all following experiments. These siRNAs inhibit >90% of each protein, as well as both factors when combined (Figure 2B). In parallel to the findings for Verteporfin, inhibition of both YAP and TAZ lead to a significant reduction in the rate of melanoma cell proliferation as compared to the siScramble control group (Figure 2C). Block of both YAP and TAZ expression resulted in cell numbers of <50% and <10% in A375 and mel-537 cells, respectively, in comparison to siScramble control groups, as determined by quantification of cell numbers in at least 5 cell fields/group (100X magnification). Both Verteporfin treatment and inhibition of YAP and TAZ by siRNA resulted in a significant attenuation in cell population expansion in A375 and mel-537 cells.
Cellular response between Verteporfin treatment and direct RNA-interference of YAP and TAZ is divergent in terms of cellular morphology
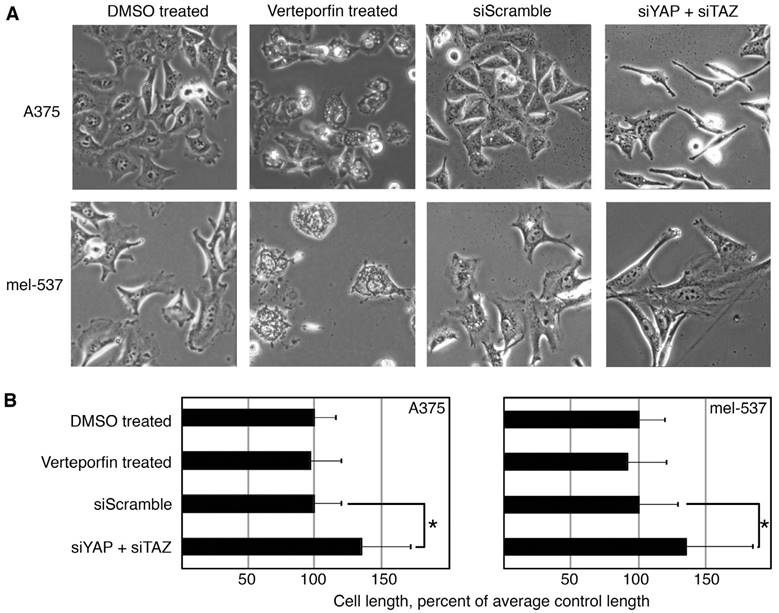
Other functional tests, including migration assays and cell length quantification, were performed on cells treated with Verteporfin and YAP/TAZ siRNA treated cells. To test migration, wound healing assays were performed, and the scratch area was measured right after wound creation (time 0). At set times after the procedure (depending on cell line) the distance of cells migrating into the area was measured. Two sets of experiments were run, one with Verteporfin and DMSO as a control (Figure 3A), and siYAP/TAZ and siScramble as a control (Figure 3B). To normalize findings between cell lines, gap closure of control cells at 24 hours post scratch formation is set at 100% control closure levels. Both experiments showed similar but not identical trends. Verteporfin inhibited migration in 4/6 cell lines. One of the lines with a significant attenuation in migration by Verteporfin, A375, had a similar trend with siYAP/ TAZ, albeit not to significant levels. In addition, mel-537 cells did not demonstrate any significant migratory change with Verteporfin treatment, but migration of this cell line was significantly inhibited with siRNA interference of YAP and TAZ.
Cell morphology and length was also analyzed between Verteporfin treated cells and cells with YAP and TAZ inhibition with siRNA. Overall, the cells had morphological differences between all groups. In comparison to controls, Verteporfin treated cells were rounded and appeared to have vesicles, while the siYAP/TAZ treated cells were longer and linear/bipolar rather than epithelioid/polygonal (Figure 4A). The siYAP/TAZ treated cells were significantly longer than control cells, while Verteporfin treated cells were not. The overall cell length was measured as the distance from the distal points from the longest dendritic-like process to the cell body and then again to the next longest process (Figure 4B). After siRNA induced YAP/TAZ knockdowns, both A375 and mel-537 cells increased processes to 134.2% ± 36.8% and 135.1 ± 48.8% respectively as compared to all other groups (DMSO, Verteporfin, siScramble, n = 200 cells/group, p<0.05). While there were similar trends between Verteporfin treated cells and cells with YAP/TAZ inhibition due to siRNA interference, there were also some differences in the cellular response including migration, morphology, and cell length.
Verteporfin treatment does not phenocopy dual YAP/TAZ knockdown. (A, B) YAP and TAZ loss through siRNA inhibition, but not Verteporfin treatment, induced elongated dendritic extensions. Dendritic extension length was measured in Verteporfin treated (2μM) or YAP/TAZ siRNA inhibited in A375 and mel-537 cells 2 days post treatment. DMSO vehicle and siScramble treatments were used as controls. Examples of overall cellular morphology (A) and quantification of dendritic extensions (B) where average control cell length is set at 100% cell length. For each group, at least 200 cells/group was measured. Only the siYAP + siTAZ cell group demonstrated a significant length difference when compared to its matched control group siScramble (both cell lines p<0.05 as indicated by an asterisk (*)).
Verteporfin treatment of a mouse model of melanoma does not result in a significant change in tumor initiation, progression, or overall tumor size
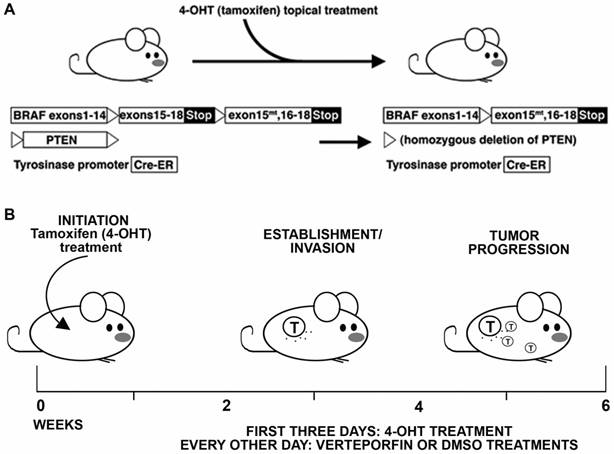
Verteporfin treatment was tested in an in situ transgenic model of melanoma. For this pre-clinical study, parameters were set utilizing a model that has similar tumor progression and genetics to the human disease, and with drug dosing that follows levels outlined for prior human clinical trials and usage in patients. For the mouse model, the BrafCA; Tyr-Cre ERT2; Ptenf/f transgenic mice, where topically administered tamoxifen induces expression of a mutant Braf allele and deletes Pten expression [14], was selected to induce in situ melanoma formation (Figure 5). This model mimics human disease genetically, histologically, and in the kinetics of disease progression. After three treatments of tamoxifen topically, the model typically develops pigmented lesions in approximately two weeks that quickly progresses to large nodular tumors within the following 2-4 weeks [14, 18]. In terms of dose, amounts were derived from prior studies for human use, and equivalent levels were given to the mice that were determined to be safe in humans in the clinic, following guidelines outlined by the Treatment of Age-related Macular Degeneration with Photodynamic Therapy (TAP) study group [19] and in clinical trials for eye disease and cancer [20, 21]. In these studies, it was determined that optimal dose was 6 mg per square meter of body surface area (but up to 12 mg/m2 was tolerated). Following prior methods for conversion between mg/m2 to mg per kg of body weight [22, 23], this converts to 0.2-0.4 mg/kg for humans, or 2-4 mg/kg for mice.
Summary schematic of BrafCA; Tyr-CreERT2; Ptenf/f murine melanoma model experiments. (A) Treatment and transgenes in the BrafCA; Tyr-CreERT2; Ptenf/f model. 4-hydroxytamoxifen (4HT) is applied topically, which deletes PTEN while simultaneously inducing expression of a constitutively active BRAF, inducing melanoma in the mouse. (B) Schematic of timeline for experimental procedure and kinetics of tumor initiation and progression.
To properly test Verteporfin as a therapeutic in vivo, we tested the kinetics of Verteporfin in the C57B6 mouse strain. Following previously published methods [15] we first identified optimum absorbance for detecting Verteporfin in tissues (680 nm) as well a corresponding spectral peak for background and low Verteporfin absorbance (540 nm). Readings were normalized by dividing Verteporfin readings at 680 nm by background levels at 540 nm (Figure S2A). To test penetrance and durability of Verteporfin in mouse tissues, mice were treated with three doses of drug (2, 4, and 6 mg/Kg) and liver and skin tissues were collected over a time-course of 6, 24, 48 and 72 hours. Verteporfin levels in both the skin and liver dropped quickly in the first 24 hours but were still present for up to 3 days post injection (Figure S2B). Based on these control experiments, a drug treatment regimen of intraperitoneal injections every other day at 2 different Verteporfin concentrations was performed.
Under the conditions tested, Verteporfin treatment did not inhibit tumor initiation as compared to the DMSO control (Figure 6A). Verteporfin and DMSO treated control mice were observed daily for the presence of pigmented lesions at the location of tamoxifen treatment on shaved back skin. Both experimental and control groups developed nevoid-like growths between 18 to 21 days post tamoxifen induction, with insignificant differences in nevi appearance between the 4 and 6 mg/Kg groups and the control mice using either the Mantel-Cox log-rank test (P = 0.3462) or the Gehan-Breslow-Wilcoxon test (P = 0.5597).
Verteporfin treatment does not inhibit tumor initiation or YAP/TAZ protein levels in murine melanoma model. Topical administration of 1-2 μl of 1.9 mg/ml (5mM) 4-hydroxytamoxifen (4HT) was applied on three consecutive days to 12 week old BrafCA; Tyr-CreERT2; Ptenf/f mice. The mice were subjected to 4 and 6 mg/kg Verteporfin intraperitoneal injections every other day for the course of the study starting the first day of melanoma induction. (A) Verteporfin treatment does not significantly alter initiation of melanocytic tumors from DMSO control treated mice. Mice were observed daily for the presence of pigmented lesions at the location of tamoxifen treatment on shaved backskin. Percent of mice lesion free are graphed by percentage (y axis) over a time course (x axis). (B,C) Verteporfin treatment does not inhibit tumor progression of melanomas in the BrafCA; Tyr-CreERT2; Ptenf/f mice. Examples of gross specimens post dissection are shown (B). Mouse tumors are measured with a caliper externally and volumes were calculated using the formula V= a X b2/2, where a is the largest diameter and b in the smallest [24]. Tumor sizes are normalized to percent of the average size for control (DMSO treated) tumors and graphed by group (C). There was not a significant difference in tumor size between treatment and control groups at 45 days post tumor induction (p=0.262). (D,E) Immunohistochemical stain for YAP/TAZ shows no reduction of YAP/TAZ levels in Verteporfin samples as compared to DMSO control. Tumor samples were fixed with formalin and subsequently paraffin embedded. 5 μM sections were cut and probed for YAP/TAZ. For the graph in (E), fluorescent staining for YAP/TAZ (red channel) was quantified by densitometry measurements using image analysis (ImageJ) from 3 samples and 10 separate images per sample, and the values were normalized to DAPI nuclear stain fluorescence (blue channel). There was not a significant difference in the fluorescent staining between Verteporfin treated and control DMSO treated groups (p=0.326).
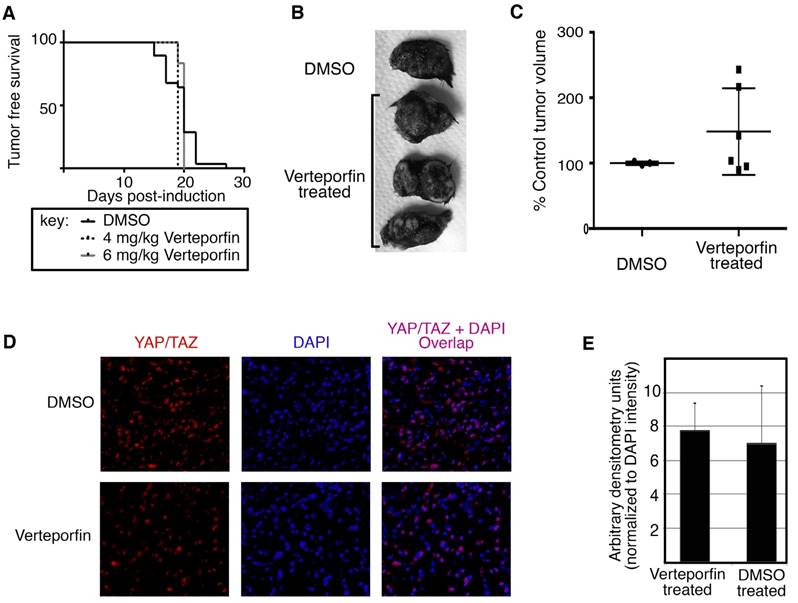
In our system, there was no significant difference in the progression of tumors in terms of overall tumor size. After tumor initiation, the pigmented lesions develop into nodular melanoma in this mouse model in approximately 4 weeks after tamoxifen induction (Figure 5). At 45 days, the tumors grew to nodules of approximately 1cm3 and mice were sacrificed and tissue was harvested. The gross appearance of tumors was similar between groups (example shown in Figure 6B). Mouse tumors were measured with a caliper externally and volumes were calculated using the formula V= a X b2/2, where a is the largest diameter and b in the smallest [24]. Tumor sizes are normalized to percent of the average size for control (DMSO treated) tumors. There was not a significant difference in tumor size between treatment and control groups (p=0.262, Figure 6C).
Levels of YAP/TAZ were detected in the collected tumors. Immunohistochemical staining for YAP/TAZ shows no reduction of YAP/TAZ levels in Verteporfin samples as compared to DMSO control (Figure 6D). To quantify the immunofluorescent staining, YAP/TAZ (red channel) was measured by densitometry using image analysis (ImageJ) from 3 samples and 10 separate images per sample, and the values were normalized to DAPI nuclear stain fluorescence (blue channel). The difference in the fluorescent staining between experimental and control groups was not significant (p=0.326, Figure 6E).
In summary, we provide evidence that although Verteporfin induces both a rapid drop in YAP/TAZ protein levels and a reduction in melanoma cell numbers in culture, it does not inhibit melanoma tumor initiation and progression in vivo in BrafCA, Tyr-CreERT2, Ptenf/f mice. These data suggest that a role for Verteporfin as a candidate for melanoma therapeutics is limited.
Discussion
YAP and TAZ make attractive targets for melanoma therapy due to their roles in melanoma progression. Recent reports implicating Verteporfin as a molecule that targets and disrupts the function of these proteins make this small molecule inhibitor an attractive melanoma therapeutic candidate. While our data and others support an inhibitory role of Verteporfin on YAP/TAZ proteins in culture and in orthotopic models, this drug was not effective in the BrafCA; Tyr-CreERT2; Ptenf/f transgenic model of melanoma. The work presented here demonstrates some in culture effects of Verteporfin on tumor cells that mirror YAP/TAZ inhibition by siRNA (Figure 2). However, there are also some notable differences. Most notably, there was a significant divergence in outcomes on cellular phenotype and cell length. While siRNA targeting YAP and TAZ induce an elongated bipolar cell morphology, this was not seen with Verteporfin treatment over a wide span of drug concentrations (0.1 ng to 10 μM) (Figure 4 and data not shown). An even more significant difference between our studies and prior published works is the reporting of significant in vivo anti-tumor properties of Verteporfin while our findings do not. While the findings in these other papers record significant findings, we believe that the lack of response Verteporfin treatment in this report (Figure 6) is due to differences in our experimental approach. The rationale for this conclusion is 1) the mouse models used in the studies, 2) the methods used to calculate proper dosage of drug, and 3) drug delivery route. Our studies, as designed, should more faithfully predict how Verteporfin would function in clinic as a drug treatment of melanoma.
To conduct the experiments presented here, a transgenic model of melanoma was employed since the in situ initiation, tumor progression, disease kinetics, and histopathology more faithfully mimic human melanoma [14, 25] than the immunodeficient transplant models used for other published studies [26-31]. This difference between models may reveal some insight into the mechanism of action of Verteporfin. While we saw no effect against melanoma tumors in our model (Figure 6) there is no doubt that there is a measureable and significant consequence of Verteporfin treatment in the transplant models. It may be that some (or all) of the responsive cells are not the tumor cells at all but the supportive vasculature. Verteporfin is demonstrated to inhibit angiogenesis and lymphangiogenesis in both eye diseases and cancer models [32-34]. The disparity in findings may be due to different vascular requirements of tumors formed in situ in a native environment versus a large mass of transplanted tumor cells. Another clue is the impressive findings of Verteporfin use in uveal melanoma models [26, 31]. One of these studies, unlike the other published works that rely on xenograft immunodeficient mouse models, uses a clinically relevant orthotopic transplant to the vascular rich environment of the eye [31]. It is not clear if the results are the consequence of YAP/TAZ inhibition of the transplant cells or from Verteporfin influencing the surrounding vascular cells, and if YAP/TAZ is or is not the direct target within these vascular cells. Indeed, there are reports that find the action of Verteporfin to be YAP/TAZ independent and our studies support these findings [35, 36].
Another major difference in the experiments presented here and other studies are the levels of Verteporfin used to treat the mice, as well as the delivery of the drug. As we focused on designing a murine treatment plan that would faithfully predict how Verteporfin could function clinically as a melanoma therapeutic, we decided to utilize the mouse equivalent dosing of Verteporfin that is used in clinic. This is in stark contrast to previously published works, which use 200-2000X the levels of drug over that what is tested as safe in humans. In support of this, many of the published studies report a decrease of weight of the experimental mice in comparison to controls. While this may be due, as proposed, to reduced tumor burden, it may also be a sign of cachexia. The dosing in the studies presented here follow levels deemed safe for humans clinically [19] and calculated to the equivalent dosing in mice. In addition, while other studies inject Verteporfin or related compounds directly into or proximal to the tumor site, the studies presented in this report relied on systemic treatment. The rationale for this approach is that for clinical efficiency, it is necessary for compounds to travel through the body in a biologically active form and reach tumors. Melanoma may present with a primary cutaneous site, but often there are multiple metastases and these secondary tumors are linked to the morbidity and mortality of this cancer [37]. We find that the drug reaches distal sites (Figure S2), but may be an inactive metabolite, since YAP/TAZ levels are unaffected in the tumors (Figure 6). While our studies do not support Verteporfin as an option for melanoma therapy, our studies and others do support that Verteporfin has potential beyond its current use as a photodynamic therapeutic.
Abbreviations
BRAF: Serine/Threonine-Protein Kinase B-Raf; Cre-ER: Cre recombinase/estrogen receptor chimeric protein; DMSO: Dimethyl sulfoxide; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; PTEN: Phosphatase and tensin homolog protein; TAZ: Transcriptional coactivator with PDZ-Binding Motif; Tyr: Tyrosinase; YAP: Yes Associated Protein 1.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by grants from the National Institutes of Health, grant numbers NIH R01CA130202, R01AR062547 and R01CA184001 (to D.L.), the American Cancer Society, grant number RSG-CSM-121505 (to D.L.), the Wendy Will Case Foundation, and the Friends of Dermatology-Chicago, and the University of Chicago Molecular and Cellular Biology training grant T32GM007183 (to J.W.L.), the American Skin Association Daneen and Charles Stiefel Investigative Scientist Award, the Falanga Scholar endowment (Boston University), and the Department of Dermatology at Boston University.
Ethical approval
All animal studies presented strictly follow procedures as outlined in the Interdisciplinary Principles and Guidelines for the Use of Animals in Research, Testing, and Education by the New York Academy of Sciences, and are in accordance with the University of Chicago Institutional Animal Care and Use Committee (IACUC).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Reed KB, Brewer JD, Lohse CM, Bringe KE, Pruitt CN, Gibson LE. Increasing incidence of melanoma among young adults: an epidemiological study in Olmsted County, Minnesota. Mayo Clinic proceedings. 2012;87:328-34
2. Nallet-Staub F, Marsaud V, Li L, Gilbert C, Dodier S, Bataille V. et al. Pro-invasive activity of the Hippo pathway effectors YAP and TAZ in cutaneous melanoma. J Invest Dermatol. 2014;134:123-32
3. Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the Roots of Cancer. Cancer Cell. 2016;29:783-803
4. Zhao B, Ye X, Yu J, Li L, Li W, Li S. et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962-71
5. Kim JE, Finlay GJ, Baguley BC. The role of the hippo pathway in melanocytes and melanoma. Frontiers in oncology. 2013;3:123
6. Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA. et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26:1300-5
7. Gibault F, Corvaisier M, Bailly F, Huet G, Melnyk P, Cotelle P. Non-Photoinduced Biological Properties of Verteporfin. Current medicinal chemistry. 2016;23:1171-84
8. Merlino G, Flaherty K, Acquavella N, Day CP, Aplin A, Holmen S. et al. Meeting report: The future of preclinical mouse models in melanoma treatment is now. Pigment Cell Melanoma Res. 2013;26:E8-E14
9. Day CP, Merlino G, Van Dyke T. Preclinical mouse cancer models: a maze of opportunities and challenges. Cell. 2015;163:39-53
10. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D. et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457-61
11. Kozlowski JM, Hart IR, Fidler IJ, Hanna N. A human melanoma line heterogeneous with respect to metastatic capacity in athymic nude mice. J Natl Cancer Inst. 1984;72:913-7
12. Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329-33
13. Lee H, Kim JS, Kim E. Fucoidan from seaweed Fucus vesiculosus inhibits migration and invasion of human lung cancer cell via PI3K-Akt-mTOR pathways. PLoS One. 2012;7:e50624
14. Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr. et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544-52
15. Busetti A, Soncin M, Jori G, Rodgers MA. High efficiency of benzoporphyrin derivative in the photodynamic therapy of pigmented malignant melanoma. Br J Cancer. 1999;79:821-4
16. Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA. et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120-33
17. Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421-34
18. Hooijkaas AI, Gadiot J, van der Valk M, Mooi WJ, Blank CU. Targeting BRAFV600E in an inducible murine model of melanoma. Am J Pathol. 2012;181:785-94
19. TAP Study Group. Photodynamic therapy of subfoveal choroidal neovascularization in age-related macular degeneration with verteporfin: one-year results of 2 randomized clinical trials-TAP report. Treatment of age-related macular degeneration with photodynamic therapy (TAP) Study Group. Arch Ophthalmol. 1999;117:1329-45
20. Bressler NM, Bressler SB. Photodynamic therapy with verteporfin (Visudyne): impact on ophthalmology and visual sciences. Invest Ophthalmol Vis Sci. 2000;41:624-8
21. Huggett MT, Jermyn M, Gillams A, Illing R, Mosse S, Novelli M. et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. Br J Cancer. 2014;110:1698-704
22. Freireich EJ, Gehan EA, Rall DP, Schmidt LH, Skipper HE. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey, and man. Cancer Chemother Rep. 1966;50:219-44
23. Schein PS, Davis RD, Carter S, Newman J, Schein DR, Rall DP. The evaluation of anticancer drugs in dogs and monkeys for the prediction of qualitative toxicities in man. Clin Pharmacol Ther. 1970;11:3-40
24. Carlsson G, Gullberg B, Hafstrom L. Estimation of liver tumor volume using different formulas - an experimental study in rats. J Cancer Res Clin Oncol. 1983;105:20-3
25. Becher OJ, Holland EC. Genetically engineered models have advantages over xenografts for preclinical studies. Cancer Res. 2006;66:3355-8 discussion 8-9
26. Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M. et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell. 2014;25:831-45
27. Jiang N, Hjorth-Jensen K, Hekmat O, Iglesias-Gato D, Kruse T, Wang C. et al. In vivo quantitative phosphoproteomic profiling identifies novel regulators of castration-resistant prostate cancer growth. Oncogene. 2015;34:2764-76
28. Nguyen LT, Tretiakova MS, Silvis MR, Lucas J, Klezovitch O, Coleman I. et al. ERG Activates the YAP1 Transcriptional Program and Induces the Development of Age-Related Prostate Tumors. Cancer Cell. 2015;27:797-808
29. Slemmons KK, Crose LE, Rudzinski E, Bentley RC, Linardic CM. Role of the YAP Oncoprotein in Priming Ras-Driven Rhabdomyosarcoma. PLoS ONE. 2015;10:e0140781
30. Song S, Ajani JA, Honjo S, Maru DM, Chen Q, Scott AW. et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014;74:4170-82
31. Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z. et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell. 2014;25:822-30
32. Hou Y, Le VNH, Clahsen T, Schneider AC, Bock F, Cursiefen C. Photodynamic Therapy Leads to Time-Dependent Regression of Pathologic Corneal (Lymph) Angiogenesis and Promotes High-Risk Corneal Allograft Survival. Invest Ophthalmol Vis Sci. 2017;58:5862-9
33. Tatar O, Shinoda K, Adam A, Eckert T, Eckardt C, Lucke K. et al. Effect of verteporfin photodynamic therapy on endostatin and angiogenesis in human choroidal neovascular membranes. Br J Ophthalmol. 2007;91:166-73
34. Wei H, Wang F, Wang Y, Li T, Xiu P, Zhong J. et al. Verteporfin suppresses cell survival, angiogenesis and vasculogenic mimicry of pancreatic ductal adenocarcinoma via disrupting the YAP-TEAD complex. Cancer Sci. 2017;108:478-87
35. Dasari VR, Mazack V, Feng W, Nash J, Carey DJ, Gogoi R. Verteporfin exhibits YAP-independent anti-proliferative and cytotoxic effects in endometrial cancer cells. Oncotarget. 2017;8:28628-40
36. Zhang H, Ramakrishnan SK, Triner D, Centofanti B, Maitra D, Gyorffy B. et al. Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci Signal. 2015;8:ra98
37. Zbytek B, Carlson JA, Granese J, Ross J, Mihm MC Jr, Slominski A. Current concepts of metastasis in melanoma. Expert Rev Dermatol. 2008;3:569-85
Author contact
![]() Corresponding author: Deborah Lang, PhD, Boston University, Department of Dermatology, 609 Albany Street, room J205, Boston, Massachusetts, U.S.A. 02118 Telephone: 617-638-5522; Fax: 617-638-5515; e-mail: deblangedu
Corresponding author: Deborah Lang, PhD, Boston University, Department of Dermatology, 609 Albany Street, room J205, Boston, Massachusetts, U.S.A. 02118 Telephone: 617-638-5522; Fax: 617-638-5515; e-mail: deblangedu