Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(1):223-237. doi:10.7150/jca.26978 This issue Cite
Review
An In-Depth Look at Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT): Clinical Implications from Recent Molecular Findings
Bingjian Lu1,2 ![]() , Haiyan Shi1
, Haiyan Shi1
1. Department of Surgical Pathology, Women's Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang Province, PR China.
2. Center for Uterine Cancer Diagnosis & Therapy Research of Zhejiang Province, Women's Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang Province, PR China.
Received 2018-5-1; Accepted 2018-10-21; Published 2019-1-1
Abstract
Small cell carcinoma of the ovary, hypercalcemic type (SCCOHT) is a highly aggressive cancer in young women. The histogenesis remains unclear although a potential origin of germ cells has been suggested recently. The high throughput next generation sequencing techniques have facilitated the identification of inactivating SMARCA4 mutations as the driver of SCCOHT. These findings may greatly impact on the prevention, diagnosis, molecular classification and treatment of SCCOHTs. The SMARCA4 mutations, typically associated with dual loss of BRG1 and BRM expression, are highly sensitive and specific for the diagnosis of SCCOHT. Germline mutations of SMARCA4 support familial SCCOHT with a critical requirement of genetic counseling and possible prophylactic surgery for carriers. SCCOHT, malignant atypical teratoid/rhabdoid tumors, thoracic sarcomas and some undifferentiated carcinomas harbor rhabdoid morphology and mutations in the SMARC genes, generating an emerging molecular classification of SMARC-mutated tumors. A multi-modality treatment approach consisting of surgery and high dose multi-agent chemotherapy in atypical teratoid/rhabdoid tumors may have potential benefits for SCCOHT patients. Preliminary studies have implicated that the inhibitors targeting EZH2 and the receptor tyrosine kinase, and anti-PD-L1 immunotherapy might be potentially effective for SCCOHT patients. These recent advances on molecular genetics, diagnosis and treatment of SCCOHT address the necessity of multiple institutional collaboration work among oncologist, pathologist, genomic scientist, geneticist, molecular biologist, and pharmacologist.
Keywords: small cell carcinoma, ovary, hypercalcemia, SMARCA4, mutation, diagnosis
Background
Small cell carcinoma of the ovary is a rare, lethal cancer, containing two different subtypes: the pulmonary type (small cell carcinoma of the ovary, pulmonary type (SCCOPT), and hypercalcemic type (small cell carcinoma of the ovary, hypercalcemic type, SCCOHT) [1]. SCCOPT resembles small cell carcinoma of the lung owing to its neuroendocrine differentiation. Less than 20 SCCOPT cases have been reported to date [2, 3]. The age of the patients ranges from 28 to 85 years. SCCOHT differs from SCCOPT both clinically and histopathologically [1]. SCCOHT is the most common undifferentiated ovarian malignant tumor in women under 40 years. It frequently has a variable component of large cells showing “rhabdoid” features, but lacks neuroendocrine features.
In SCCOHT, many aspects including its molecular pathogenesis and histogenesis, remain understudied. Recent advances in next generation sequencing, such as whole exome sequencing and whole genome sequencing, provide powerful tools to identify novel significant genomic alterations in ovarian cancers [4, 5]. Several studies have demonstrated that inactivating mutations of SMARCA4, a gene involving the SWItch/Sucrose Non-Fermentable (SWI/SNF) chromatin remodeling complex, are the likely driver in the majority of SCCOHT cases [6-8]. This finding has greatly changed our recognition on the pathology and clinical practice of this rare disease. Moreover, the expanding data from multiple institutes worldwide have thrown the light on the potential improvement of prognosis with aggressive multi- modality treatment [9-12]. In this review, we will discuss recent advances on SCCOHT, particularly focusing on the molecular genesis and their clinical implications.
An overview of SCCHOT
Dr Robert Scully initially depicted the terrains of SCCOHT nearly 3 decades ago [13]. Approximately 400 cases have been reported in the English literatures to date. Most patients presented with abdominal discomfort and/or distention. Some asymptomatic patients were found by close clinical examination due to their familial history. A small number of patients had symptoms of hypercalcemia, which might result in severe pancreatitis, altered mental status and even unnecessary neck exploration [14-16]. The largest series showed that most tumors were unilateral (148/150, 99%) with a higher proportion in the right ovary (341/457, 66%) [16]. Familial cases were often bilateral [17, 18]. The tumors usually have a large size (average 15.3 cm, 6-26 cm in greatest dimension). Only one case was suggested to be of primary peritoneal origin since the tumors were present on the pelvic/abdominal peritoneum and the normal-sized ovaries were free of tumors grossly [19]. Another pericolonic case might be secondary to a previous ovarian mucinous borderline tumor, intestinal type [20].
The rare undifferentiated cancer has two distinct clinical features, which are helpful clues for the clinical discrimination from common ovarian carcinomas. First, SCCOHT patients are young. In the large cohort of Young et al. [16], the patients ranged from 9 to 43 (average 23.9) years of age. About 92% patients were diagnosed between 10 and 40 years. It should be very cautious to diagnose SCCOHT in women aged over 60 years although only one 71-year patient was reported to date. That case might be a misdiagnosis most likely since the modern diagnostic markers (SMARCA4 immunostaining or mutation analysis) were not available at that time. Second, preoperative calcium levels are elevated in about 62% patients. Thus, the term “hypercalcemic type” has been applied to distinguish it from ovarian small cell carcinoma of the pulmonary type (SCCOPT), a high grade neuroendocrinal carcinoma [2, 3]. Hypercalcemia has been presumed to be associated with tumoral production of parathyroid hormone related peptides, or rarely parathyroid hormone [16, 21, 22]. However, the immunoexpression level of parathyroid hormone related peptides or parathyroid hormone in SCOCHT tissues does not parallel with the serum calcium prior to surgery [23]. The possible underlying explanation is that parathyroid hormone related peptides or parathyroid hormone produced by the tumor cells cannot be totally released from the cytoplasm. Hypercalcemia can return to a normal level after the tumors are totally removed. Nevertheless, the value of serum calcium is unclear in the assessment of cancer relapse or therapeutic response in SCCOHT. Serum CA125 was slightly elevated in 15 SSCOHT patients with available data [9]. A recent study from the National Cancer Data Base (NCDB) between 2004 and 2014 showed a high frequency of elevated CA125 (271/337, 83.9%) [24]. CA125 can potentially be applied as a clinical indicator for SCCOHT although we feel like that the serum level in SCCOHT does not elevate as obviously as that in ovarian epithelial carcinomas.
Clinical behavior and treatment of SCCOHT
SCCOHT is a highly aggressive tumor. Extraovarian spread is seen in approximately half of the cases. Lymph node metastasis is present in 19/34 (55.8%) cases. Tumor stage remains the mainstay in the assessment of prognosis [9-11, 16]. Young et al. [16] reported that one third patients (14/42) at FIGO (International Federation of Gynecology and Obstetrics) stage Ia survived free of disease 1-13 years after surgery whereas almost all patients at an advanced stage died of disease. A study from a single institution showed that 35 of 47 (74.5%) SCCOHT patients recurred (median: 6.5 months, range 2.3-125.4 months), 30 (63.8%) died of disease (median: 14.9 months, 95% CI 11.8-23.5 months), and 6 (12.8%) were alive with disease [9]. The mean overall survival time was 35.3 months in patients at stage I, and only 3.3 months at stage IV. A combined investigation on 293 SCCOHT patients [10] showed that patients at 40 years or older had a worse outcome than younger patients, but there was no significant survival difference between patients with and without germline SMARCA4 mutations. The 5-year overall survival rate in the NCDB cohort was 24.1% for patients with cancer- directed surgery, and only 18% and 12.3% in patients with stage III and IV respectively [24]. Intriguingly, Ghazi A, et al. [25] reported a SCCOHT patient that developed after two months of ovarian stimulation for in vitro fertilization and died one month after her initial symptoms. This unique case suggests the possibility of ovarian stimulation- associated cancer risk and mortal prognosis, which merits further multi-centric clinical investigation.
The rarity of SCCOHT limits the implementation of randomized clinical trial by now. Therefore, there is no consensus of the standard treatment on this lethal cancer [9-11]. The patients can be treated with surgery including unilateral salpingo-oophorectomy with or without hysterectomy, or total abdominal hysterictomy with bilateral salpingo-oophorectomy. Some patients may undergo omentectomy, debulking of extra-ovarian tumors, lymph node dissection, and peritoneal biopsies. Bilateral oophorectomy has been addressed in patients with germline SMARCA4 mutations to date [26]. Fertility-conserving surgery has not been recommended by most surgeons because of its aggressive behavior [27]. However, some gynecologists advocated that the poor survival rates of SCCOHT may justify a less aggressive fertility sparing approach without compromising the outcome [28]. A patient with advanced stage of SCCOHT was alive for 60 months without evidence of disease after the treatment of a fertility-sparing approach and the following induction chemotherapy, interval debulking surgery, and local radiotherapy [28]. The standard chemotherapy for ovarian carcinoma is applied in most SCCOHT patients, but no form is of proven benefit [16]. However, a Germany study reported that 4 of 7 patients with the treatment of conventional chemotherapy achieved a complete response for 7 to 73 months [12]. Two studies in vitro indicated that SCCOHT cell lines were resistant to platinum chemotherapeutic drugs [29, 30] whereas microtubule- stabilizing compounds, particularly epothilone B/ Ca2+, demonstrated an strong anti-proliferation effect in vitro and in xenografts in vivo [30]. Epothilone B/Ca2+ might be incorporated into a multi-modality approach to improve the treatment effect of this rare, lethal cancer although clinical data have been unavailable by now.
Recent data have indicated that a multi-modality treatment approach consisting of surgery, high dose multi-agent chemotherapy with possible stem cell transplantation and radiotherapy is an attractive treatment option for the SCCOHT patients [9-11, 24, 31, 32]. Qin Q, et al. [32] reported that a young girl with stage IIIc SCCOHT had an 8-year disease free survival after a combined treatment of cytoreductive surgery, high-dose consolidative chemotherapy, autologous hematopoietic stem cell transplantation and pelvic radiotherapy. One patient with relapse achieved her second complete response and had survived for 64 months after treatment of high-dose chemotherapy [12]. Witkowski L, et al [10] found that the 5-year survival after surgery was 71% in FIGO stages II-IV patients with high dose chemotherapy-stem cell rescue, and only 25% with conventional chemotherapy alone. However, they failed to demonstrate a survival benefit for patients with adjuvant radiotherapy. A retrospective study on 47 SCCOHT patients also suggested that multi-agent chemotherapy and radiotherapy were associated with a good prognosis [9]. A GCIG (the Gynecological Cancer Intergroup) study demonstrated that the five of six SCCOHT patients at stage I with adjuvant radiotherapy, and only one of four without adjuvant radiotherapy, were alive and disease-free [11]. In the seven patients at stage III or unknown stage, all but one died of cancer despite the salvage treatment with chemotherapy and radiotherapy. In the recent NCDB (national cancer database) cohort [24], the survival in patients with cancer directed surgery, high dose chemotherapy and hematologic transplant procedure was superior to patients with surgery and standard chemotherapy. Moreover, the combination of surgery, chemotherapy and radiotherapy achieved a better survival than patients with surgery and chemotherapy alone.
Histopathology of SCCOHT
The tumors are generally large and unilateral. They have a fleshy white, pale to grey appearance [Figure 1A]. They are predominantly solid. Large areas of hemorrhage, necrosis and cystic degeneration are not uncommon. The most significant microscopic change is a diffuse growth pattern consisting of closely packed small cells and focal areas of follicle-like spaces containing eosinophilic and rarely basophilic fluids [Figure 1B]. The neoplastic cells are predominately small, uniform, and hyperchromatic. The nuclei contain coarsely clumped chromatins and single small nucleoli. The mitotic figures are brisk. The large cells are seen in at least half of all SCCOHTs if the tumors are extensively sampled. They can be focal, predominant or exclusive (so called “large cell variant of SCCOHT”) [16, 33]. The large cells show “rhabdoid” features with eccentric nuclei, prominent nucleoli and glassy eosinophilic cytoplasm [Figure 1C]. Approximate1y 12% cases have small foci of mucinous glands or cysts lined by mature epithelium, signet-ring cells or atypical mucinous cells.
Histopahtology of SCCOHT. The large tumor has a fleshy white to grey appearance with extensive hemorrhage, necrosis and cystic degeneration (A). Microscopically, the tumor has a diffuse growth pattern consisting of closely packed small cells and focal areas of follicle-like spaces containing eosinophilic fluids [Figure 1B]. The large cells characteristically show “rhabdoid” features with eccentric nuclei, prominent nucleoli and glassy eosinophilic cytoplasm [Figure 1C]. The cancer cells are negative for BRG1 immunostaining while the lymphocytes have a retained BRG1 staining [Figure 1D].
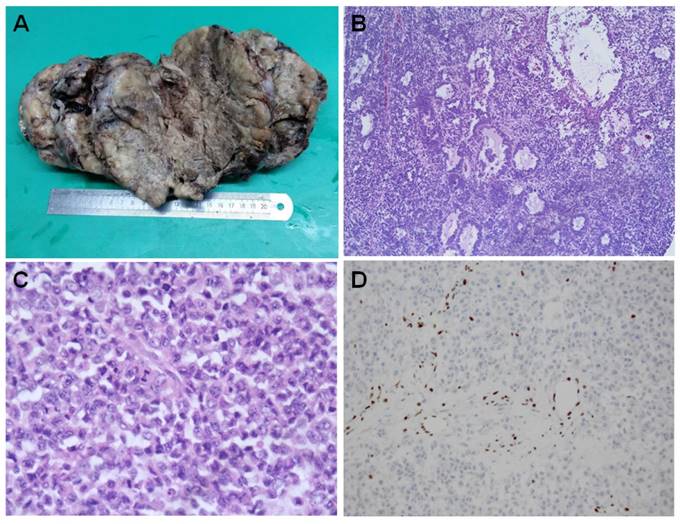
The tumor cells showed diffuse nuclear staining with antibodies against the N-terminal of WT1, and focal with pan-cytokeratin (AE1/AE3), epithelial membrane antigen, CD10 and calretinin, etc [34]. Some tumors can express parathyroid hormone related peptide, or rarely parathyroid hormone. CK5/6, chromogranin, CD99, desmin, alpha inhibin. Thyroid transcription factor 1 (TTF1) is generally negative. The immunostaining profile has limited diagnostic value and cannot classify SCCOHT into any specific types of epithelium. Until recently, loss of SMARC4 protein (also called BRG1) expression has been recognized as a diagnostic hallmark for this rare tumor [Figure 1D] [35-38].
SCCHOT should be differentiated from a wide range of primary or metastatic ovarian tumors including adult and juvenile granulosa cell tumor, small cell neuroendocrine carcinoma (metastatic or pulmonary type), and germ cell tumors, etc. It remains a challenging task to differentiate SCCOHT from juvenile granulosa cell tumors even by expert gynecological pathologists because of their overlapping morphological and immunohistochemical features, particularly when juvenile granulosa cell tumors have hypercalcemia [39-41]. The predominance of monotonous, small, hyperchromatic cells with brisk mitotic activity is the pinpoint directing to the diagnosis of SSCOHT. We feel like that the presence of “rhabdoid” cells also provides another helpful clue in favor of SCCOHT.
Histogenesis of SCCOHT
SCCOHT is included in the catalogue of miscellaneous ovarian tumors in the recent World Health Organization (WHO) Classification of Tumors of the Female Reproductive Organs (2014) [42] since its histogenesis and cell lineage are unclarified by now. Recent studies have indicated that SCCOHT is characterized by germline or somatic mutations in SMARCA4 and other subunits in the SWI/SNF chromatin remodeling complex [6-8]. These mutations are found in a variety of cancers including epithelial carcinoma and mesenchymal neoplasms [43-45]. These mutations can not provide useful information on their histologic origin, either. Variable speculations on the histogenesis have been proposed, but none is fully accepted.
Initially, SCCOHT was regarded as an epithelial malignancy (so the name “carcinoma” was given) since the tumor cells were positive for epithelial markers, such as pan-cytokeratin and epithelial membrane antigen, and harbored abundant dilated rough endoplasmic reticulum and numerous ribosomes under electron microscopy [16, 46 47]. In fact, the diffuse expression of epithelial markers is very uncommon in SCCOHT. Histochemical, immunohistochemical and ultrastructural examinations provide no reliable features of a specific cell lineage in the ovarian epithelial carcinomas. Moreover, no other convincing evidence has been added to support the epithelial origin later on. Flow cytometry on paraffin-embedded tissues indicates that diploidy is typically present in SCCOHT [16, 48, 49] whereas high grade carcinomas are commonly aneuploidy and polyploidy [50-52]. Unlike high grade serous carcinoma [52, 53], a recent genomic study has demonstrated that, SCCOHT harbors a simple genome including few somatic significant mutations and chromosomal alterations [29]. Collectively, great doubts have been cast on the epithelial histogenesis of SCCOHT although the term “carcinoma” is conventionally applied to date.
The young age distribution, uniform small cells and follicle formation raise the possibility of a sex cord derivation. However, transitions to recognizable forms of sex cord tumors, such as Call-Exner bodies or Sertoli tubules, have never been observed in SCCOHT. The relatively uniform cell population without thecal cells or other gonadal stroma is very unlikely in sex cord tumors. The reliable expression of classical sex cord markers, such as alpha inhibin and steroidogenic factor-1, has not been found in SCCOHT yet. Mutations in FOXL2 and Dicers are frequently present in adult granulosa cell tumors and Sertoli-Ledig cell tumors, respectively [54-56], but they have not been identified in SCCOHT by recent high throughput next-generation sequencing studies [6-8]. The sex cord derivation of SCCOHT seems very unlikely. SCCOHT lacks diffuse expression of the classical neuroendocrinal markers, such as chromogranin A, and dense-core granules under electron microscope, therefore, there is no evidence for its neuroendocrinal differentiation.
In 1987, Ulbright et al. [57] proposed the potential germ cell origin of SCCOHT by providing some pieces of evidence. In addition to young age distribution, they found some common microscopic, ultrastructural and immunohistochemical features between SCCOHT and yolk sac tumor. These features included eosinophilic, hyaline globules and extra- cellular basement membrane-like substance under microscopy and electron microscopy, and cytoplasmic staining of laminin and alpha-1-antitrypsin by immunohistochemistry. They also commented on a histologically mimicking testicular tumor with mixed components of seminoma and teratoma. Moreover, the patient had an aggressive clinical course similar to SCCOHT. It was well documented that small foci of mucinous epithelium, ranging from benign, borderline to malignancy, can be found in approximate1y 12% SCCOHT cases [16]. A 33-year-old woman had a morphologically and immunohistochemically compatible SCCOHT in her peri-colonic region 5 years after removal of the ovarian borderline mucinous tumor of the intestinal type [20]. A subset of primary ovarian mucinous neoplasm is believed to be associated with teratoma [58]. As a collar, the coexistence of mucinous epithelium and SCCOHT might be regarded as a clue to support the germ cell origin of SCCOHT. Kupryjanczyk et al. [59] reported two SCCOHT cases with small foci of immature teratoma and one also with small foci of yolk sac tumor after extensive sampling (80 and 120 sections in each). It can be inferred from this report that extensive sampling may increase the chance of identifying teratoma or other germ cell tumor components in SCCOHT. Unfortunately, such laborious work is very unlikely in pathological practice even in large medical centers. Recently, McCluggage et al. [60] also reported two SCCOHT cases, one with dermoid cyst and another with immature teratoma and small foci of yolk sac tumor. Theoretically, all kinds of cancers throughout the body can occur in teratoma, such as primitive neuroectodermal tumor, glioblastoma multiform and malignant melanoma, etc [61]. It is unequivocally acceptable to speculate that SCCOHT may arise from teratoma. Presumably, the overgrowth of SCCOHT may predominate over other components in the teratoma resulting in small foci of teratoma, mucinous epithelium or other transformed malignant germ cell tumors including yolk sac tumor [60]. In addition, recent advances in molecular genetics have indicated that SCCOHT can be regarded as an ovarian malignant teratoid/rhabdoid tumor (MRT) [62]; thereof, SCCOHT is most likely an embryonic tumor originating from neural elements within an immature teratoma or other germ cell tumors [60]. Familial SCCOHT has been recognized for two decades [63, 64]. Theoretically, SCCOHT might occur in patients with familial teratoma if teratoma was the histological origin of SCCOHT. A woman with SCCOHT had a prior history of cystectomy for dermoid cyst in the same ovary. The patient's sister had been treated for an immature ovarian teratoma. This unique case provides a tentative link between SCCOHT and teratoma in the familial setting [65].
Molecular genetics of SCCOHT
Familial SCCOHT characterized by an autosomal dominant transmission implicates the possibility of the underlying mutations in a single gene or a family of genes [63, 64]. Mutations in hereditary diseases might be potential causative molecular events in sporadic cases. Great efforts have been taken on the foundation of comprehensive molecular characterization in SCCOHT. Accumulation of p53 proteins was occasionally reported in SCCOHT [34, 66], but TP53 mutations, the hallmark of high grade serous carcinoma, were extremely rare in SCCOHT. To date, only two patients harbored TP53 mutations: one was a woman with Li-Fraumeni syndrome (germline TP53 mutation) [67]; another was a recurrent SCCOHT case showing TP53 (I254N) and POLE (D287A) mutations [68]. The additional mutations in the recurrent SCCOHT might reflect the molecular evolution in the process of tumor progression. BRCA1- and BRCA2-gemline mutation carriers have significant risk of developing ovarian carcinomas, predominantly including high grade serous carcinoma and rarely other histotypes, such as none-small neuroendocrine carcinoma [69, 70]. However, so far, no SCCOHT case has been found to be associated with BRCA1- or BRCA2-germline mutations. It becomes clear that SCCOHT differs from ovarian carcinoma by displaying a low mutational burden and little chromosomal abnormalities [29, 71]. No activated mutations in classical oncogenes including KRAS or BRAF have been identified in SCCOHT yet. Despite of a low mutation load cancer, SCCOHT harbored an immunogenic microenvironment resembling the landscape of the tumors with immune checkpoint blockade, characterized by a high Programmed Death Ligand-1 (PD-L1) expression (8/11), strongly associated with T-cell infiltration, and increased expression genes related to Th1 and cytotoxic cell function [72]. The inconsistencies between mutation burden and immunogenicity underscored the requirement for additional biomarkers for tumor recognition in SCCOHT.
A pilot study investigated gene expression and mutations of SMARCA4 and INI-1 in two SCCOHT cases since the authors noted the histological resemblance between SCCOHT and atypical teratoid/ rhabdoid tumor (AT/RT) of the central nervous system which harbored an alternative loss of INI-1 or SMARCA4 [73]. The authors found that both tumors had retained INI-1, but lost SMARCA4 expression by immunohistochemistry. Sanger sequencing on DNA from formalin-fixed paraffin-embedded tissues identified that both tumors harbored SMARCA4 mutations (c.2184_2206del and nonsense c.3277C>T in one tumor, and nonsense c.3760G>T in another). This pilot observation has two important implications: the emerging pathogenic role of SMARCA4 mutations in SCCOHT, and the necessity of multiple-institutional collaborations among clinicians, pathologists and molecular biologists to further confirm the pathogenic mutations. However, from this study, it remained to be clarified whether SMARCA4 mutation was the driver of SCCOHT in the absence of familial studies, large case series, comprehensive sequence and functional analysis.
The high throughput, cost-effective next generation sequencing technologies contain a variety of platforms such as whole-exome sequencing and whole genome sequencing [5]. They have greatly contributed to the identification of driver mutations in rare heritable cancers, such as SCCOHT. Multi- institutional collaborations to recruit large cohorts are the key to get the strong power to discriminate driver mutations from the passenger mutations or nonpathogenic normal variations in the background. Three far-reaching SCCOHT collaborations, which applied various next generation sequencing technologies with a large cohort, have further validated that SCCOHT is a monogenic disease caused by SMARCA4 mutations [6-8]. Recently, Lang JD and Hendricks WPD [74] have underpinned that the approaches and methods for the identification of SMACA4 mutations as drivers in SCCOHT “point to the broader relevance of this paradigm for future research in rare cancers”.
Witkowski, L et al. [7] initially performed whole-exome sequencing of tumor and normal DNA from 6 affected individuals of three independent families. They found that SMARCA4 was the only gene that had deleterious germline mutations in the affected women. Moreover, the identical SMARCA4 mutations were found in the germline of the mothers and their daughters in these families, and a second somatic hit, either in loss of heterozygosity or mutations in the second allele, occurred in each tumor. They next used whole-exome sequencing (n=11) and Sanger sequencing (n=15) of additional formalin-fixed paraffin-embedded tumors (3 familial cases, 23 individual cases), confirming that at least one germline or somatic deleterious SMARCA4 mutation was present in 24 of 26 SCCOHT tumors. Moreover, SMARCA4 protein (i.e. BRG1) expression was lost in 38 of 40 tumors. Ramos P, et al. [6] applied whole-genome sequencing, whole-exome sequencing and Sanger sequencing of tumors and germline samples from 12 SCCOHT patients (9 tumors with 4 matched germline samples, 3 additional germline samples) and one cell line. SMARCA4 was the only recurrently mutated gene, bearing inactivating mutations in 6/9 tumors (2 with biallelic inactivation) and the BIN-67 cells, and truncating mutations in 2/7 germline samples. They also found that SMARCA4 protein was absent in 82% (14/17) of SCCOHT tumors and only in 0.4% (2/485) of other primary ovarian tumors. Thereafter, Ramos P, et al. [26] reported the presence of SMARCA4 mutations in all but one of additional 12 tumors and a second cell line (SCCOHT-1). Levine DA and colleagues [8] carried out a custom hybrid capture-based sequencing of matched tumor/germline samples from 12 SCCOHT tumors. The targeted panel, which was developed by Memorial Sloan Kettering Cancer Center, contained 279 cancer genes. This study also confirmed that biallelic SMARCA4 mutations were present in all 12 SCCOHT tumors and loss of protein expression in 7 of 9 cases. In vitro experiments showed that deprived SMARCA4 expression promoted cell growth and re-expression resulted in dose-dependent cell growth inhibition, further supporting that SMARCA4 might function as a tumor suppressor gene in this rare cancer.
After these breakthrough works, a growing body of evidence unanimously provides an etiologic link between SMARCA4 mutations and SCCOHT [10, 17, 18, 26, 75, 76]. A total of 118 (96 none-redundant) pathogenic SMARCA4 mutations have been describeed in 94 patients to date [Table 1] [10, 17, 18, 26, 75-79]. These SMARCA4 mutations do not reveal the presence of hotspots yet. Twenty-six patients harbor germline mutations including 10 familial cases from 5 pedigrees. The specific mutations consist of frameshift (43/118, 36.4%), stop/nonsense (38/118. 32.2%)), splice-site (24/118, 20.3%), missense (7/118, 5.9%) and in-frame deletions (6/118, 5.1%). Most tumors (80/84, 95.2%) with these mutations are associated with loss of SMARCA4 protein (BRG1) expression. Four SCCOHTs with retained or equivocal BRG1 expression may suggest the presence of inactivated proteins considering the biological significance of these mutations (in frame deletion of exons 2, missense 1 and splicing 1). Biallelic mutations of SMARCA4 are present in 24 of 94 (25.5%) SCCOHTs. Fourteen tumors contain biallelic somatic mutations while five have one somatic and another germline mutation. Loss of heterozygosity and epigenomic alterations are very likely for inactivation of one allele in cases having SMARCA4 mutation of one allele (70/94, 74.5%).
SMARCA4 mutations in SCCOHTψ
| Cases [Ref] | Mutations, Protein Changes | Mutation Types | Origin | Protein Expression |
|---|---|---|---|---|
| Ramos, et al. [6, 26] | ||||
| DAH23 | c.2438+1_2438+2insTGA | Splicing site | UN | Loss |
| DAH457 | c.3277C>T, p.Arg1093* | Nonsense | UN | NA |
| DG1006 | c.2855delA, p.Glu952fs | Frameshift | Somatic | Loss |
| c.4672_4675delAGCG, p.Ser1559fs | Frameshift | Somatic | ||
| DG1219 | c.3168+1G>A | Splicing site | Somatic | Loss |
| SCCO-001 | c.1595delC, p.Ala532fs | Frameshift | UN | Loss |
| c.482C>T, p.Ala161Val | Missense | UN | ||
| SCCO-002 | c.1237C>T, p.Gln413* | Nonsense | Somatic | Loss |
| c.2783C>T, p.Leu928Pro | Missense | Somatic | ||
| SCCO-004 | c.613delC, p.Val204fs | Frameshift | Somatic | Loss |
| SCCO-005 | c.3896_3907del12,p.Glu1300_Asn1303del | In-frame Deletion | Somatic | NA |
| SCCO-006 | c.2293_2315del23, p.Trp764fs | Frameshift | Somatic | Loss |
| c.2506G>T, p.Gly836* | Nonsense | Somatic | ||
| SCCO-007 | c.1626delC, p.Ile542fs | Frameshift | UN | Loss |
| c.991C>T, p.Gln331* | Nonsense | UN | ||
| SCCO-008 | c.2935C>T, p.Arg979* | Nonsense | Germline | NA |
| SCCO-009 | c.3539delC, p.Tyr1050fs | Frameshift | UN | Loss |
| SCCO-011 | c.3565C>T, p.Arg1189* | Nonsense | UN | Loss |
| SCCO-014 | c.2001delG, p.Glu667fs | Frameshift | UN | NA |
| c.3481delC, p.Leu1161fs | Frameshift | UN | ||
| SCCO-015 | c.3565C>T, p.Arg1189* | Nonsense | UN | NA |
| SCCO-016 | c.3985_3986insC, p.Arg1329fs | Frameshift | UN | Loss |
| SCCO-017 | c.722_735del15, p.Gly241fs*41 | Frameshift | Germline | Loss |
| SCCO-019 | c.2531_2532delTT, p.Phe844fs | Frameshift | UN | Loss |
| NF10 | c.3277C>T, p.Arg1093* | Nonsense | Germline | Loss |
| NF11 | c.1751_1751delA, p.Lys584Argfs*29 | Frameshift | Somatic | Loss |
| NF12 | c.3512_3513delTG, p.Val1171Aspfs*4 | Frameshift | Somatic | Loss |
| c.3488_3525del38, p.Leu1163Gln*fs65 | Frameshift | Somatic | ||
| NF13 | c.3229C>T, p.Arg1077* | Nonsense | Germline | Loss |
| NF14 | c.2973+1 G>A | Splicing site | Somatic | Loss |
| NF15 | c.2838_2838delC, p.Phe947Leufs*3 | Frameshift | Somatic | Loss |
| NF17 | c.2274+2T>G | Splicing site | Somatic | Loss |
| NF18 | c.3216-2A>G | Splicing site | Somatic | Loss |
| c.1189C>T, p.Arg397* | Nonsense | Somatic | ||
| NF21 | c.2859+1G>C | In-frame Deletion | Somatic | Loss |
| c.196C>T, p.Gln66* | Nonsense | Somatic | ||
| Jelinic P. et al. [8] | ||||
| 101 | c.3546+1G>A | Splicing site | Somatic | Loss |
| 102 | C.4170+1G>A | Splicing site | Somatic | Loss |
| 103 | Exon 25-26 Deletion | In-frame deletion | Somatic | Retained |
| 104 | C.1746+1G>A | Splicing site | Somatic | NA |
| c.2932C>T, p.Arg978* | Nonsense | Somatic | ||
| 105 | c.1626_1626delC, p.Ile542Metfs*71 | Frameshift | Somatic | NA |
| c.991C>T, p.Gln331* | Nonsense | Somatic | ||
| 106 | c.1165_1165delC, p.Ser391Profs*20 | Frameshift | Somatic | Loss |
| c.3277C>T, p.Arg1093* | Nonsense | Somatic | ||
| 107 | c.3896_3907del12, p.Glu1300_Asn1303del | In-frame deletion | Somatic | UN |
| 108 | c.2539C>T, p.Gln847* | Nonsense | Somatic | Loss |
| 109 | c.2287_2288insG, p.Leu762fs | Frameshift | Somatic | Loss |
| c.2506G>T, p.Gly836* | Nonsense | Somatic | ||
| 110 | c.3496C>T, p.Gln1166* | Nonsense | Somatic | Loss |
| 111 | c.3013C>T, p.Arg1005* | Nonsense | Germline | Loss |
| 112 | c.2859+1G>A | Splicing site | Somatic | Equivocal |
| 114 | c.2973+1G>A | Splicing site | Somatic | Loss |
| Lavrut PM, et al. [17] | ||||
| c.301_302delAG, p.Gly102Profs*26 | Frameshift | Germline | Loss | |
| Kupryjanczyk, et al. [73] | ||||
| PJK1182 | c.3760G>T, p.Glu1254* | Nonsense | Germline | Loss |
| PJK1233 | c.3277C>T, p.Arg1093* | Nonsense | UN | Loss |
| c.2184_2206del , p.Gln729fs | Frameshift | UN | ||
| Bailey S, et al. [75] | ||||
| c.1757_1757delA, p.Lys586fs*27 | Frameshift | Somatic | Loss | |
| Laé M, et al. [77] | ||||
| c.3229C>T, p.Arg1077* | Nonsense | Germline | Loss | |
| Le Loarer F, et al. [78] | ||||
| SCCOHT1 | c.300_301delAG, p.Gly102Profs*26 | Frameshift | UN | Loss |
| SCCOHT2 | c.4236_4237delGC, p.Arg1413Glnfs*41 | Frameshift | UN | Loss |
| SCCOHT3 | c.3216-1G>T | Splicing site | UN | Loss |
| SCCOHT4 | c.2322_2322delC, p.Asn774Lysfs*57 | Frameshift | UN | Loss |
| SCCOHT5 | c.3241A>G, p.Lys1081Glu | Missense | UN | Loss |
| SCCOHT6 | c.1542C>T, p.Gln515* | Nonsense | UN | Loss |
| SCCOHT7 | c.3229C>T, p.Arg1077* | Nonsense | UN | Loss |
| SCCOHT8 | c.3216-1G>T | Splicing site | UN | Loss |
| SCCOHT9 | c.3229C>T, p.Arg1077* | Nonsense | UN | Loss |
| SCCOHT10 | c.3951-1G>T | Splicing site | UN | Loss |
| Witkowski L, et al. [7, 10, 18, 79] | ||||
| NF1 | c.1224_1226delGCTinsAG, p.Leu409Glyfs*2 | Frameshift | Germline | Loss |
| NF2 | c.1663C>T, p.Gln555* | Nonsense | Somatic | Loss |
| NF3 | c.3496C>T, p.Gln1166* | Nonsense | Somatic | Loss |
| NF4 | c.3638_3638delA, p.Lys1213Argfs*3 | Frameshift | Germline | Loss |
| NF5 | c.3480_3481insG, p.Leu1161Alafs*15 | Frameshift | Germline | Loss |
| NF6 | c.2129_2129delC, p.Lys711Serfs*63 | Frameshift | Somatic | Loss |
| c.1378C>T, p.Gln460* | Nonsense | Somatic | ||
| NF7 | c.2245_2246insA, p.Met749Asnfs*75 | Frameshift | Somatic | Loss |
| UN1 | c.2362C>T, p.Gln788* | Nonsense | UN | Loss |
| c.561C>G, p.Thr187* | Nonsense | UN | ||
| UN2 | c.3676C>T, p.Gln1226* | Nonsense | UN | Loss |
| UN3 | c.2932C>T, p.Arg978* | Nonsense | Germline | Loss |
| UN4 | c.3531delC, p.Trp1178Glyfs*38 | Frameshift | Somatic | Loss |
| c.4687delG, p.Ile1564Serfs*32 | Frameshift | Somatic | ||
| UN5 | c.2275-1G>T, | Splicing site | Somatic | Loss |
| UN6 | c.2838_2838delC, p.Phe947Leufs*3 | Frameshift | UN | Loss |
| UN7 | c.1141C>T, p.Arg381* | Nonsense | Germline | Loss |
| UN8 | c.2190_2191insG, p.Tyr731Valfs*10 | Frameshift | UN | Loss |
| UN9 | c.1420+1G>T | Splicing site | UN | Retained |
| UN10 | c.2049delC, p.Val684Trpfs*90 | Frameshift | UN | Loss |
| UN11 | c.3244delT, p.Phe1082Leufs*24 | Frameshift | UN | Loss |
| UN12 | c.2766G>A, p.Trp922* | Nonsense | UN | Loss |
| UN13 | c.3546+1G>T | Splicing site | UN | Loss |
| UN14 | c.2915T>C, p.Leu972Pro | Missense | UN | |
| c.3168+1G>C | Splicing site | UN | Loss | |
| UN15 | c.1761+2T>A | Splicing site | UN | Loss |
| UN16 | c.233_237delCCATGinsACC, p.Ser78Tyrfs*3 | Frameshift | UN | Loss |
| FA1a | c.1027_1027delG, p.Val343Cysfs*68 | Frameshift | Somatic | Loss |
| c.4170+1G>A | Splicing site | Germline | ||
| FA1b | c.4170+1G>A | Splicing site | Germline | Loss |
| FA2a | c.643C>T, p.Gln215* | Nonsense | Germline | Loss |
| FA2b | c.1687_1700del14, p.Asn563Glyfs*82 | Frameshift | Somatic | Loss |
| c.643C>T, p.Gln215* | Nonsense | Germline | ||
| FA3a | c.2617-3C>G | Splicing site | Germline | Loss |
| FA4a | c.3239G>A, p.Gly1080Asp | Missense | Germline | Retained |
| FA4b | c.1326delC, p.Ser442Argfs*59 | Frameshift | Somatic | Loss |
| c.3239G>A, p.Gly1080Asp | Missense | Germline | Loss | |
| FA7a | c.2859+1G>C | In-frame Deletion | Germline | Loss |
| FA7b | c.2859+1G>C | In-frame Deletion | Germline | Loss |
| FA8a | c.175C>T, p.Gln59* | Nonsense | Germline | Loss |
| FA8b | c.175C>T, p.Gln59* | Nonsense | Germline | Loss |
| c.2375C>T, p.Leu792Pro | Missense | Somatic | ||
| 2013_Mother | c.3533G>A, p.Trp1178* | Nonsense | Germline | Loss |
| c.2438+1G>T | Splicing site | Somatic | Loss | |
| Case24 | c.1236delC, p.Phe412fs | Frameshift | Somatic | Loss |
| c.2970delA, p.Gly990fs | Frameshift | Somatic | ||
| Case25 | c.2123+1G>A | Splicing site | Somatic | Loss |
| Case27 | c.1119-1G>C | Splicing site | Somatic | Loss |
| Moes-Sosnowska, et al. [76] | ||||
| c.2352insG, p.Lys785Glufs*39 | Frameshift | Germline | Loss | |
Abbreviations: NA=not available; UN=unknown.
Rare exceptions with no SMARCA4 mutations and intact BRG1 expression may bear mutations in other subunits in the SWI/SNF chromatin remodeling complex. Ramos P, et al. [6] found one SCCOHT that had no SMARCA4 mutation, but instead exhibited loss of SMARCB1 (INI1). One SCCOHT was found to have an ARID1A (an accessory subunit of the SWI/SNF complex) mutation rather than SMARCA4 mutations [71].
Clinical implications from molecular genetics
Chromatin remodeling is one of the key mechanisms in dynamic regulation of gene expression. Some different proteins/protein complexes, such as the SWI/SNF chromatin remodeling complex, participate in this process. In human, three core subunits (INI1/SMARCB1, BAF155/SMARCC1, and BAF170/SMARCC2) and two helicases/ATPases (BRM/SMARCA2 and BRG1/SMARCA4) are essential in maintaining the chromatin remodeling activity of the SWI/SNF complex [80]. The SWI/SNF complex uses the energy of ATPase hydrolysis to alter interactions between DNA and histone proteins, and the nucleosome positioning and restructuring that makes gene promoter regions more accessible to transcription factors and key cellular proteins [81]. They play key roles in the biological process of DNA transcription, replication, repair and recombination [82].They can interact with a number of tumor suppressors such as p15, p16, p21 and Rb, etc [82, 83]. The SWI/SNF dysfunction may promote the oncogenic activities of EZH2 (enhancer of zeste homolog 2) and the receptor tyrosine kinase (RTK) [84, 85].
Broad transcriptional dysregulation through the SWI/SNF dysfunction has become evident in human cancers. An estimated 20% of human cancers displayed mutations in one or more of the SWI/SNF subunits [84]. These large set of “SWI/SNFomas” are particularly associated with pediatric cancers, such as brain tumors, medulloblastoma, AT/RT, and leukemia [86-88] as well as a small subset of adult cancers including non-small cell lung cancer, gastrointestinal or uterine undifferentiated carcinoma, and Burkitt lymphoma [43-45, 89-91]. Given the substantial portion of “SWI/SNFomas” in human cancers, the identification of SMARCA4 mutations in virtually all SCCOHT tumors may confer a broader translational relevance covering tumor classification, diagnosis, prevention and intervention [Figure 2].
Clinical implications from SMARCA4 mutations in SCCOHT
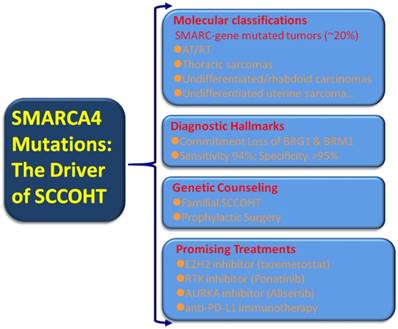
Molecular classification: expanding the rhabdoid tumors/SMARC-gene mutated tumors
SMARCB1 (INI1) represents the most extensively investigated subunit of the SWI/SNF complex [82]. INI1 immunostaining is a highly useful tool in the diagnosis of a number of cancers in surgical pathology. The expanding “SMARCB1 deficient neoplasia”, characterized by inactivation mutations of SMARCB1 and loss of INI1 expression, is a heterogenous group of tumors with substantial variations in histology and clinical behavior [92]. AT/RT, which frequently occurs in the central nervous system, kidney and soft tissues in children, is the prototype of SMARCB1 deficient neoplasia. They are consisted of large to medium-sized anaplastic cells with abundant eosinophilia cytoplasm (so called rhabdoid features). SMARCB1 mutations/loss of INI1 expression is the hallmark of AT/RT [93, 94], however, SMARCA4 inactivation/loss of BRG1 expression is occasionally present in some morphologically typical AT/RT tumors. In contrast, SCCOHT is predominantly associated with SMARCA4 (BRG1) mutations and rarely with SMARCB1 (INI1) [68, 95]. SMARCA4 (BRG1) mutations can be regarded as the hallmark of SCCOHT although deleterious germline SMARCA4 mutation has been reported in one high grade serous carcinoma to date [96]. A recent molecular study demonstrated that the genomic and epigenomic signature of SCCOHT are similar to those of AT/RT [97]. Both AT/RT and SCCOHT have a low mutational burden except deficient SMARCB1 and SMARCA4 [29, 71]. Moreover, both AT/RT and SCCOHT share high morphological and clinical similarities, such as early onset, aggressive clinical course and the presence of “rhabdoid cells”, etc. Witkowski L, et al [79] reported an ovarian immature teratoma arising in a young woman and a clonally distinct intracerebral immature teratoma developing in her daughter. They reclassified both tumors as familial rhabdoid tumors because of a germline SMARCA4 mutation (c.3533G>A; p.Trp1178*) and a somatic SMARCA4 mutation in the mother (c.2438+1G>T) and her daughter (c.3229C>T; p.Arg 1077*) after the histopathological and immunohistochemical re-assessment. Unequivocally, the ovarian malignant rhabdoid tumor should be regarded as SCCOHT according to the current opinion [10]. The unique familial cases indicated the common genetic rationale of SCCOHT and malignant rhabdoid tumors. Thereafter, Foulkes WD, et al [62] have suggested that SCCOHT should be renamed as malignant rhabdoid tumors of the ovary having SMARCA4 mutations. This conception is further supported by SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus), which showed rhabdoid morphology, SMARCA4 loss by immunohistochemistry and recurrent SMARCA4 mutations and few other molecular alterations [96]. However, Soslow RA and colleagues [36] think that the nomenclature change is premature currently due to the extensive accumulation of literatures under the traditional term SCCOHT. Moreover, a substantial number of SCCOHT tumors lack “rhabdoid cells” but have SMARCA4 mutations. Hence, they suggest that rather than the exchange of a histology-based term, it is preferable to create a molecular catalogue, for example, “SMARC-gene mutated tumors”, in the future when compelling molecular evidence accumulates. In line with their proposal, gene profiling analyses demonstrated that SMARCA4- deficient thoracic sarcomas (“SMARCA4-deficient thoracic sarcomas”) were related to malignant rhabdoid tumors and SCCOHTs [78]. SCCOHT and other tumors, such as thoracic sarcoma, undifferentiated/rhabdoid carcinomas of the gastrointestinal tract, and SMARCA4-deficient undifferentiated uterine sarcoma can be added to the list of this unique, highly lethal catalogue [43-45, 78, 91, 98, 99].
Diagnostic implications: discovery of the hallmark of SCCOHT
Several studies have demonstrated that loss of BRG1 is highly specific and sensitive for SCCOHT among ovarian cancers after the initial descriptions of SMARCA4 deficiency in SCCOHT [Figure 1D] [35-38]. Up to date, 121/129 (94%) SCCOHT cases show loss of BRG1 expression, as summarized by Clarke BA, et al [37]. However, SMARCA4 mutation is not always associated with loss of protein expression [Table 1]. The inconsistency between SMARCA4 mutation and loss of BRG1 may suggest the presence of inactivated protein products. Alternatively, intact BRG1 may indicate deficiency in other subunits of the SWI/SNF complex. Ramos P, et al. [6] noted that one SCCOHT did not display SMARCA4 mutations, but showed loss of SMARCB1/INI-1 instead. Karnezis et al. [95] demonstrated that INI-1 loss was present in 3 of 4 SCCOHT cases with retaining BRG1. These findings address the necessity of SMARCA4 and SMARCB1 mutation analysis in cases with SCCOHT morphology, but with equivocal BRG1 expression.
To test the specificity of BRG1 loss in the diagnosis of SCCOHT, several studies have investigated BRG1 expression in approximately 4,000 other tumors in the female tract including sex cord-stromal tumors, germ cell tumors, carcinomas, sarcomas, and metastatic cancers, etc [36-38, 95]. Most tumors showed intact BRG1 expression with rare exceptions. Absence of BRG1 expression was only found in ovarian clear cell carcinomas (17/447, 3.8%), ovarian undifferentiated carcinomas (1/22, 4.5%), uterine endometrioid carcinoma (1/360, 0.3%), uterine undifferentiated/dedifferentiated carcinoma (16/59, 27%), and melanoma (1/47, 2.1%) [37], which are not morphological mimics of SCCOHT. All granulosa cell tumors (273/273, 100%), the most challenging differential diagnosis of SCCOHT, have intact BRG1 expression. Heterogenous staining of BRG1 can be seen in a subset of SCCOHT mimics, such as adult granulosa cell tumor, high grade serous carcinoma, small round cell sarcoma and Ewing sarcoma; thereof, caution should be taken in the interpretation of BRG1 staining in small biopsy specimens. Taken together, loss of BRG1 expression is a specific and sensitive marker for the diagnosis of SCCOHT.
SMARCA2, also known as BRM, is one of two mutually-exclusively ATPases in the SWI/SNF complex. SMARCA2 is essential for the survival of SMARCA4-deficient non-small cell carcinoma [99]. However, minimal level of SMARCA2 expression was observed in BIN67, a SCCOHT cell line with SMARCA4-mutation [95]. Jelinic P, et al. [68] showed that dual loss of both BRM and BRG1 by immunohistochemistry was present in 10 of 11 SCCOHT with SMARCA4 mutations, and intact staining in 1 case. The concomitant expression pattern is suggestive of a signature for this rare tumor. The absence of germline or somatic mutations of SMARCA2 in SCCOHT implicates that posttranscriptional or epigenomic modifications are responsible for loss of gene expression as in other cancers [100]. Another study simultaneously found that SMARCA2 expression was lost in all 45 SCCOHT cases (43 lacking BRG1 and 2 lacking INI-1) [95]. Concomitant loss of both SMARCA2 and SMARCA4 expression was not seen in ovarian clear cell carcinoma, the only tumor lacking SMARCA4 expression in that study. They also confirmed that loss of SMARCA2 expression in SCCOHT was associated with (histone deacetylation) HDAC-mediated gene silencing. Collectively, both studies suggest that dual loss of BRG1 and BRM is the diagnostic hallmark of SCCOHT [68, 95].
Preventive implications: developing genetic test and counseling
Current data suggest that familial SCCOHT may be much more common than expected previously [26]. An analysis on the published SCCOHT cases with SMARCA4 mutations showed that 26 (43%) were carriers of germline mutations, and 21 had no reported familial history [10].The under-reported familial SCCOHT cases can be explained by germline mutations arising from unaffected father or de novo. Moreover, patients carrying germline mutations appear to be younger at the time of diagnosis than non-carriers. Patients diagnosed below 18 years have a significantly high risk for a germline mutation. Germline and somatic mutations of SMARCA4 should be detected in SCCOHT patients with loss of BRG1 expression. The next-generation sequencing based platform is recommended for mutation analysis because the hotspots of SMARCA4 mutations have not been identified yet. After the confirmation of deleterious germline mutations, the genetic test and counseling should be rendered for all her family members. Male carriers are also important since their daughters may inherit these mutations although the risk of other cancers in male carriers is unclear currently.
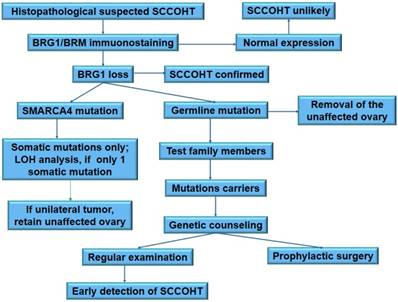
There is no census on the clinical management of SMARCA4 mutation carriers. Mutation carriers of hereditary breast and ovarian cancer have two main clinical options: increased surveillance for the detection of early (stage I) cancers, or risk-reducing salpingo-oophorectomy. SCCOHT lacks reliable biomarkers for cancer detection at present. The surveillance of SCCOHT largely depends on sonography. Nevertheless, sonography is not a good method for early detection. Therefore, taking the extremely progressive process of tumor growth and highly lethality of SCCOHT into account, SMARCA4 mutation carriers have to be counseled about prophylactic bilateral oophorectomy [26, 101]. The early onset of SCCOHT (median age: 21.5 years) indicates that prophylactic surgery should be performed at younger age in these carriers than in the hereditary breast and ovarian cancer carriers. Most carriers at this age are unmarried and will not consider for conception. Thereof, prophylactic bilateral oophorectomy is not acceptable for these carriers. However, great improvement on the oocyte cryopreservation and in vitro fertilization may facilitate the accomplishment of prophylactic surgery in women carrying SMARCA4 mutations. To prevent genetic transmission of the deleterious mutations, pre-implantation genetic diagnosis should be recommended for fertilization in vitro. The proposed clinical algorithm for the management of SMARCA4 mutation carriers is summarized in Figure 3.
A proposed schema for clinical and genetic management of SCCOHT
Therapeutic implications: mechanism-driven promising treatment
The survival is poor even for stage I SCCOHT patients. The current adjuvant treatment only achieves a modest improvement of survival underlying the critical requirement for novel therapies [24]. The recent advance on the molecular genetics can guide optimal management in addition to better understanding the pathogenesis although the rarity of SCCOHT limits prospective clinical trials. A study of gene expression profiling from a single case suggests a number of agents such as pazopanib, bortezomib, 5-azacytidine, and PARP inhibitors as possible treatment options against the disease [102]. The potential unification of AT/RT and SCCOHT implicates that chemotherapeutic regimens used to AT/RT, for example, high dose chemotherapy with autologous stem cell rescue, can be implemented in SCCOHT [9-11, 31, 103]. The activity of PD-1/PD-L1 blockage is associated with high tumor mutation load and tumor infiltration lymphocytes in cancers [104]. SCCOHT is not hypermutated [29, 71], but the immunogenic microenvironment resembling the landscape of the tumors with immune checkpoint blockade provides the rational for immunotherapy. Four recurrent SCCOHT patients have responded well to anti-PD-L1immunotherapy. One patient has a sustained partial response of 6 months, and the other three have remained disease free for at least 1.5 years [72].
SCCOHT is a monogenic disease, almost exclusively with SMARCA4 mutations and concurent loss of SMARCA2 expression [29, 71]; therefore, SCCOHT is an excellent prototype to develop novel therapy targeting the driver genes and related pathways. Moreover, novel therapies under development in cancers with similar genetic background (i.e., “SMARC-gene mutated tumors” or SWI/SNFoma”) might be encouraged for treating SCCOHT patients.
Previous studies reported an antagonistic relationship between SMARCB-1 and the histone trimethylase EZH2 [84]. SMARCB-1-defficient cancers are dependent on EZH2 oncogenic activities. Kim KH et al. [105] demonstrate that cancer cells harboring mutations of SMARCA4 and other subunits in the SWI/SNF complex are primarily dependent on a non-catalytic role of EZH2 in the stabilization of the polycomb repressive complex 2 (PRC2), and only partially dependent on EZH2 histone methyltransferase activity. Accordingly, EZH2 is overexpressed in SMARCB1-inactivated rhabdoid tumors and other tumors with deficient SWI/SNF complex [106]. Selective EZH2 inhibition appears to be a promising treatment in the SWI/SNF complex-deficient tumors [107]. In AT/RT, EZH2 inhibition can suppress cell proliferation, promote cell apoptosis and increase radiotherapy sensitivity in preclinical models [108]. A preclinical study demonstrated that EPZ-6438 (tazemetostat), a selective inhibitor of EZH2, had significant antitumor activity in preclinical rhabdoid tumor models [109]. Two recent studies have demonstrated that EZH2 inhibitors, such as (tazemetostat), can restore the SMARCA-2 expression in SCCOHT cells, and show significantly potent anti-proliferative and anti-tumoral effects in cell lines and xenografts [110, 111]. These findings suggest that pharmacological inhibition of EZH2 will be a promising therapeutic strategy.
Little attention has been paid on the effects of the overall oncogenes, such as RTKs in SMARC-deficient cancers. A recent study showed that RTKs were activated in SCCOHT cell lines by using high through-put siRNA screens [84]. Previous studies in SMARCB1-mutant AT/RT harbored elevated FGFR and PDGFR expression, and sensitivity to ponatinib, an FDA-approved, FGFR/PDGFR-selective inhibitor [112]. Similarly, ponatinib and other RTK inhibitors, significantly suppressed cell proliferation in SCCOHT cell lines whereas re-expression of SMARCA4 conferred a 1.7-fold increase in resistance to ponatinib. Ponatinib substantially prolonged the tumor doubling time and reduced final tumor volumes in xenografts and patient derived xenografts of SCCOHT [85]. Ponatinib provides a great hope to improve the SCCOHT patients' outcomes although further clinical investigation should be encouraged.
AURKA overexpression is essential in the survival of non-small cell lung carcinoma (NSCLC) cells lacking SMARCA4. Inhibition of AURKA expression induces cell apoptosis and death in vivo and in vitro [113]. A pilot clinical study on MLN8237 (Alisertib), an Aurora K inhibitor has achieved early success in the treatment of AT/RT patients [114]. AURKA inhibitor is an emerging biomarker-driven therapy to treat SCCOHT. HDAC inhibitor, trichostatin A, can restore SMARCA2 expression and suppress cell growth in SCCOHT cells [95]. Reversal of epigenetic silencing of SMARCA2 merits further exploration as a potential treatment option for SCCOHT.
Concluding Remarks
The finding of driver mutations of SMARCA4 and a relative simple genome may contribute significantly to our diagnosis and treatment in SCCOHT. Loss of BRG1 and BRM as well as mutation analysis has been recognized as the hallmark for the diagnosis of SCCOHT. Genetic tests enable the identification of the mutation carriers, and promote the development of genetic counseling, clinical screening and prophylactic surgery. SSCOHT can be put into the expanding catalogue of AT/RT due to their genetic and morphological similarities. The merging evidence supports the benefits of a multi-modality treatment approach including surgery, high dose multi-agent chemotherapy with possible stem cell transplantation and radiotherapy, for SCCOHT patients. Novel target therapies on SMARCA4 mutations and other related molecular events, such as tazemetostat and ponatinib, are under development for the treatment of this highly aggressive tumor. Future work should be focused on the development of novel target therapy directing to restore the function of inactive SMARCA4 gene. A worldwide collaboration is critically required to reach a census in standard screening, preventive surgery and treatment for this rare, lethal disease.
Abbreviations
AT/RT: atypical teratoid/rhabdoid tumor; EZH2: enhancer of zeste homolog 2; FIGO: International Federation of Gynecology and Obstetrics; GCIG: the Gynecological Cancer Intergroup; MRT: malignant teratoid/rhabdoid tumor; NCDB: national cancer database; PD-L1: Programmed Death Ligand- 1; PRC2: polycomb repressive complex 2; SCCOHT: small cell carcinoma of the ovary, hypercalcemic type; SCCOPT: small cell carcinoma of the ovary, pulmonary type; SWI/SNF: SWItch/Sucrose Non-Fermentable; WHO: World Health Organization.
Acknowledgements
We thank Dr. Brian Eyden from Manchester, UK for his kind help in the linguistic tuning of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Patibandla JR, Fehniger JE, Levine DA. et al. Small cell cancers of the female genital tract: Molecular and clinical aspects. Gynecol Oncol. 2018;149:420-7
2. Agarwal S, Pandey P, Ralli M. et al. Ovarian Small Cell Carcinoma: A Rare Case Report and Review of Literature. Iran J Pathol. 2018;13:99-102
3. Yin L, Li J, Wei Y. et al. Primary ovarian small cell carcinoma of pulmonary type with coexisting endometrial carcinoma in a breast cancer patient receiving tamoxifen: A case report and literature review. Medicine (Baltimore). 2018;97:e10900
4. Taylor-Weiner A, Zack T, O'Donnell E. et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature. 2016;540:114-8
5. Dong A, Lu Y, Lu B. Genomic/Epigenomic Alterations in Ovarian Carcinoma: Translational Insight into Clinical Practice. J Cancer. 2016;7:1441-51
6. Ramos P, Karnezis AN, Craig DW. et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet. 2014;46:427-9
7. Witkowski L, Carrot-Zhang J, Albrecht S. et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet. 2014;46:438-43
8. Jelinic P, Mueller JJ, Olvera N. et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nat Genet. 2014;46:424-6
9. Callegaro-Filho D, Gershenson DM, Nick AM. et al. Small cell carcinoma of the ovary-hypercalcemic type (SCCOHT): A review of 47 cases. Gynecol Oncol. 2016;140:53-7
10. Witkowski L, Goudie C, Ramos P. et al. The influence of clinical and genetic factors on patient outcome in small cell carcinoma of the ovary, hypercalcemic type. Gynecol Oncol. 2016;141:454-60
11. Harrison ML, Hoskins P, du Bois A. et al. Small cell of the ovary, hypercalcemic type - analysis of combined experience and recommendation for management. A GCIG study. Gynecol Oncol. 2006;100:233-8
12. Distelmaier F, Calaminus G, Harms D. et al. Ovarian small cell carcinoma of the hypercalcemic type in children and adolescents: a prognostically unfavorable but curable disease. Cancer. 2006;107:2298-306
13. Dickersin GR, Kline IW, Scully RE. Small cell carcinoma of the ovary with hypercalcemia, a report of eleven cases. Cancer. 1982;49:188-97
14. Bourgain A, Acker O, Lambaudie E. et al. Small cell carcinoma of the ovary of the hypercalcemic type revealed by a severe acute pancreatitis, about one case. Gynecol Obstet Fertil. 2005;33:35-8
15. Wynn D, Everett GD, Boothby RA. Small cell carcinoma of the ovary with hypercalcemia causes severe pancreatitis and altered mental status. Gynecol Oncol. 2004;95:716-8
16. Young RH, Oliva E, Scully RE. Small cell carcinoma of the ovary.; hypercalcemic type. A clinicopathological analysis of 150 cases. Am J Surg Pathol. 1994;18:1102-16
17. Lavrut PM, Le Loarer F, Normand C. et al. Small Cell Carcinoma of the Ovary, Hypercalcemic Type, Report of a Bilateral Case in a Teenager Associated with SMARCA4 Germline Mutation. Pediatr Dev Pathol. 2016;19:56-60
18. Witkowski L, Donini N, Byler-Dann R. et al. The hereditary nature of small cell carcinoma of the ovary, hypercalcemic type, two new familial cases. Fam Cancer. 2017;16:395-9
19. Popiolek DA, Kumar AR, Mittal K. Large cell variant of small cell carcinoma, hypercalcemic type, of primary peritoneal origin. Gynecol Oncol. 2005;96:249-53
20. Mansor S, Nagarajan S, Sumathi VP. et al. Borderline ovarian mucinous neoplasm recurring as small cell carcinoma of hypercalcemic type, evidence for an epithelial histogenesis and relationship with ovarian mucinous tumors for this enigmatic neoplasm. Int J Gynecol Pathol. 2011;30:380-5
21. Chen L, Dinh TA, Haque A. Small cell carcinoma of the ovary with hypercalcemia and ectopic parathyroid hormone production. Arch Pathol Lab Med. 2005;129:531-3
22. Matias-Guiu X, Prat J, Young RH. et al. Human parathyroid hormone-related protein in ovarian small cell carcinoma. An immunohistochemical study. Cancer. 1994;73:1878-81
23. Dickersin GR, Scully RE. An update on the electron microscopy of small cell carcinoma of the ovary with hypercalcemia. Ultrastruct Pathol. 1993;17:411-22
24. Nasioudis D, Chapman-Davis E, Frey MK. et al. Small Cell Carcinoma of the Ovary: A Rare Tumor With a Poor Prognosis. Int J Gynecol Cancer. 2018;28:932-8
25. Ghazi A, Ayaz A, Hamid T. et al. Small cell carcinoma of the ovary hypercalcemic type (SCCOHT): A rare case after in vitro fertilization (IVF). Pak J Med Sci. 2017;33:241-4
26. Ramos P, Karnezis AN, Hendricks WP. et al. Loss of the tumor suppressor SMARCA4 in small cell carcinoma of the ovary, hypercalcemic type (SCCOHT). Rare Dis. 2014;2:e967148
27. McCormick TC, Muffly T, Lu G. et al. Aggressive small cell carcinoma of the ovary, hypercalcemic type with hypercalcemia in pregnancy, treated with conservative surgery and chemotherapy. Int J Gynecol Cancer. 2009;19:1339-41
28. Dykgraaf RH, de Jong D, van Veen M. et al. Clinical management of ovarian small-cell carcinoma of the hypercalcemic type, a proposal for conservative surgery in an advanced stage of disease. Int J Gynecol Cancer. 2009;19:348-53
29. Gamwell LF, Gambaro K, Merziotis M. et al. Small cell ovarian carcinoma, genomic stability and responsiveness to therapeutics. Orphanet J Rare Dis. 2013;8:33
30. Otte A, Rauprich F, Hillemanns P. et al. In vitro and in vivo therapeutic approach for a small cell carcinoma of the ovary hypercalcaemic type using a SCCOHT-1 cellular model. Orphanet J Rare Dis. 2014;9:126
31. Pressey JG, Kelly DR, Hawthorne HT. Successful treatment of preadolescents with small cell carcinoma of the ovary hypercalcemic type. J Pediatr Hematol Oncol. 2013;35:566-9
32. Qin Q, Ajewole VB, Sheu TG. et al. Successful treatment of a stage IIIC small-cell carcinoma of the ovary hypercalcemic subtype using multi-modality therapeutic approach. Ecancermedicalscience. 2018;12:832
33. Ferlicot S, Bessoud B, Martin V. et al. Large cell variant of small cell carcinoma of the ovary with hypercalcemia. Ann Pathol. 1998;18:197-200
34. McCluggage WG, Oliva E, Connolly LE. et al. An immunohistochemical analysis of ovarian small cell carcinoma of hypercalcemic type. Int J Gynecol Pathol. 2004;23:330-6
35. Agaimy A, Thiel F, Hartmann A. et al. SMARCA4-deficient undifferentiated carcinoma of the ovary (small cell carcinoma, hypercalcemic type), clinicopathologic and immunohistochemical study of 3 cases. Ann Diagn Pathol. 2015;19:283-7
36. Conlon N, Silva A, Guerra E. et al. Loss of SMARCA4 Expression Is Both Sensitive and Specific for the Diagnosis of Small Cell Carcinoma of Ovary, Hypercalcemic Type. Am J Surg Pathol. 2016;40:395-403
37. Clarke BA, Witkowski L, Ton Nu TN. et al. Loss of SMARCA4 (BRG1) protein expression as determined by immunohistochemistry in small-cell carcinoma of the ovary, hypercalcaemic type distinguishes these tumours from their mimics. Histopathology. 2016;69:727-38
38. Karanian-Philippe M, Velasco V, Longy M. et al. SMARCA4 (BRG1) loss of expression is a useful marker for the diagnosis of ovarian small cell carcinoma of the hypercalcemic type (ovarian rhabdoid tumor), a comprehensive analysis of 116 rare gynecologic tumor, 9 soft tissue tumors, and 9 melanomas. Am J Surg Pathol. 2015;39:1197-205
39. Rod J, Renard C, Lacreuse I. et al. Hypercalcemia in a child with juvenile granulosa cell tumor of ovary, Report of an unusual paraneoplastic syndrome and review of the literature. Gynecol Oncol Case Rep. 2013;5:10-2
40. Piura B, Wiznitzer A, Shaco-Levy R. Juvenile granulosa cell tumor of the ovary associated with hypercalcemia. Arch Gynecol Obstet. 2008;277:257-62
41. Daubenton JD, Sinclair-Smith C. Severe hypercalcemia in association with a juvenile granulosa cell tumor of the ovary. Med Pediatr Oncol. 2000;34:301-3
42. McCluggag WG, Malpica A, Daya D. et al. Miscellaneous tomors. WHO classification of tumors of female reproductive organs, 4th Ed. Ryon: IARC. 2014;6:69-73
43. Geyer FC, Pareja F, Burke KA. et al. Genetic analysis of a morphologically heterogeneous ovarian endometrioid carcinoma. Histopathology. 2017;71:480-7
44. Yoshida A, Kobayashi E, Kubo T. et al. Clinicopathological and molecular characterization of SMARCA4-deficient thoracic sarcomas with comparison to potentially related entities. Mod Pathol. 2017;30:797-809
45. Araujo LH, Timmers C, Bell EH. et al. Genomic Characterization of Non-Small-Cell Lung Cancer in African Americans by Targeted Massively Parallel Sequencing. J Clin Oncol. 2015;33:1966-73
46. McMahon JT, Hart WR. Ultrastructural analysis of small cell carcinomas of the ovary. Am J Clin Pathol. 1988;90:523-9
47. Aguirre P, Thor AD, Scully RE. Ovarian small cell carcinoma. Histogenetic considerations based on immunohistochemical and other findings. Am J Clin Pathol. 1989;92:140-9
48. Förster C, Ostertag H, Schmitt J. et al. Small cell carcinoma of the ovary.; hypercalcemic type. A case report with immunohistochemical, ultrastructural and cytophotometric analysis and review of the literature. Gen Diagn Pathol. 1997;142:365-70
49. Eichhorn JH, Bell DA, Young RH. et al. DNA content and proliferative activity in ovarian small cell carcinomas of the hypercalcemic type. Implications for diagnosis, prognosis, and histogenesis. Am J Clin Pathol. 1992;98:579-86
50. Hauptmann S, Denkert C, Koch I. et al. Genetic alterations in epithelial ovarian tumors analyzed by comparative genomic hybridization. Hum Pathol. 2002;33:632-41
51. Zeimet AG, Fiegl H, Goebel G. et al. DNA ploidy, nuclear size, proliferation index and DNA-hypomethylation in ovarian cancer. Gynecol Oncol. 2011;121:24-31
52. Chien J, Sicotte H, Fan JB. et al. TP53 mutations, tetraploidy and homologous recombination repair defects in early stage high-grade serous ovarian cancer. Nucleic Acids Res. 2015;43:6945-58
53. Patch AM, Christie EL, Etemadmoghadam D. et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature. 2015;521:489-94
54. McConechy MK, Färkkilä A, Horlings HM. et al. Molecularly Defined Adult Granulosa Cell Tumor of the Ovary, The Clinical Phenotype. J Natl Cancer Inst. 2016:108 (11)
55. Conlon N, Schultheis AM, Piscuoglio S. et al. A survey of DICER1 hotspot mutations in ovarian and testicular sex cord-stromal tumors. Mod Pathol. 2015;28:1603-12
56. Goulvent T, Ray-Coquard I, Borel S. et al. DICER1 and FOXL2 mutations in ovarian sex cord-stromal tumours, a GINECO Group study. Histopathology. 2016;68:279-85
57. Ulbright TM, Roth LM, Stehman FB. et al. Poorly differentiated (small cell) carcinoma of the ovary in young women, evidence supporting a germ cell origin. Hum Pathol. 1987;18:175-84
58. Snir OL, Buza N, Hui P. Mucinous epithelial tumours arising from ovarian mature teratomas, a tissue genotyping study. Histopathology. 2016;69:383-92
59. Kupryjańczyk J, Dansonka-Mieszkowska A, Moes-Sosnowska J. et al. Ovarian small cell carcinoma of hypercalcemic type - evidence of germline origin and SMARCA4 gene inactivation: a pilot study. Pol J Pathol. 2013;64:238-46
60. McCluggage WG, Witkowski L, Clarke BA. et al. Clinical, morphological and immunohistochemical evidence that small-cell carcinoma of the ovary of hypercalcaemic type (SCCOHT) may be a primitive germ-cell neoplasm. Histopathology. 2017;70:1147-54
61. Kleinman GM, Young RH, Scully RE. Primary neuroectodermal tumors of the ovary. A report of 25 cases. Am J Surg Pathol. 1993;17:764-78
62. Foulkes WD, Clarke BA, Hasselblatt M. et al. No small surprise - small cell carcinoma of the ovary, hypercalcaemic type. is a malignant rhabdoid tumour. J Pathol. 2014;233:209-14
63. Longy M, Toulouse C, Mage P. et al. Familial cluster of ovarian small cell carcinoma, a new mendelian entity? J Med Genet. 1996;33:333-5
64. Lamovec J, Bracko M, Cerar O. Familial occurrence of small-cell carcinoma of the ovary. Arch Pathol Lab Med. 1995;119:523-7
65. Meganck C, Moerman PH, De Schrijver D. et al. A non-diploid.; small cell carcinoma of the ovary of the hypercalcemic type. Int J Gynecol Cancer. 1998;8:430-3
66. Seidman JD. Small cell carcinoma of the ovary of the hypercalcemic type, p53 protein accumulation and clinicopathologic features. Gynecol Oncol. 1995;59:283-7
67. Tandon B, Hagemann IS, Maluf HM. et al. Association of Li-Fraumeni Syndrome With Small Cell Carcinoma of the Ovary, Hypercalcemic Type and Concurrent Pleomorphic Liposarcoma of the Cervix. Int J Gynecol Pathol. 2017;36:593-9
68. Jelinic P, Schlappe BA, Conlon N. et al. Concomitant loss of SMARCA2 and SMARCA4 expression in small cell carcinoma of the ovary, hypercalcemic type. Mod Pathol. 2016;29:60-6
69. Schrader KA, Hurlburt J, Kalloger SE. et al. Germline BRCA1 and BRCA2 mutations in ovarian cancer: utility of a histology-based referral strategy. Obstet Gynecol. 2012;120:235-40
70. Herold N, Wappenschmidt B, Markiefka B. et al. Non-small cell neuroendocrine carcinoma of the ovary in a BRCA2-germline mutation carrier: A case report and brief review of the literature. Oncol Lett. 2018;15:4093-6
71. Lin DI, Chudnovsky Y, Duggan B. et al. Comprehensive genomic profiling reveals inactivating SMARCA4 mutations and low tumor mutational burden in small cell carcinoma of the ovary, hypercalcemic-type. Gynecol Oncol. 2017;147:626-33
72. Jelinic P, Ricca J, Van Oudenhove E. et al. Immune-Active Microenvironment in Small Cell Carcinoma of the Ovary, Hypercalcemic Type: Rationale for Immune Checkpoint Blockade. J Natl Cancer Inst. 2018;110:787-90
73. Kupryjańczyk J, Dansonka-Mieszkowska A, Moes-Sosnowska J. et al. Ovarian small cell carcinoma of hypercalcemic type - evidence of germline origin and SMARCA4 gene inactivation: a pilot study. Pol J Pathol. 2013;64:238-46
74. Lang JD, Hendricks WPD. Identification of Driver Mutations in Rare Cancers: The Role of SMARCA4 in Small Cell Carcinoma of the Ovary, Hypercalcemic Type (SCCOHT). Methods Mol Biol. 2018;1706:367-79
75. Bailey S, Murray MJ, Witkowski L. et al. Biallelic somatic SMARCA4 mutations in small cell carcinoma of the ovary, hypercalcemic type (SCCOHT). Pediatr Blood Cancer. 2015;62:728-30
76. Moes-Sosnowska J, Szafron L, Nowakowska D. et al. Germline SMARCA4 mutations in patients with ovarian small cell carcinoma of hypercalcemic type. Orphanet J Rare Dis. 2015;10:32
77. Laé M, Bourgoin R, Cornelis F. et al. Cytological features of small cell carcinoma of the ovary-hypercalcemic type/malignant ovarian rhabdoid tumor in ascitic fluid. Diagn Cytopathol. 2018;46:365-6
78. Le Loarer F, Watson S, Pierron G. et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat Genet. 2015;47:1200-5
79. Witkowski L, Lalonde E, Zhang J. et al. Familial rhabdoid tumour 'avant la lettre'-from pathology review to exome sequencing and back again. J Pathol. 2013;231:35-43
80. Pulice JL, Kadoch C. Composition and Function of Mammalian SWI/SNF Chromatin Remodeling Complexes in Human Disease. Cold Spring Harb Symp Quant Biol. 2016;81:53-60
81. Masliah-Planchon J, Bièche I, Guinebretière JM. et al. SWI/SNF chromatin remodeling and human malignancies. Annu Rev Pathol. 2015;10:145-71
82. Brownlee PM, Meisenberg C, Downs JA. The SWI/SNF chromatin remodelling complex: Its role in maintaining genome stability and preventing tumourigenesis. DNA Repair (Amst). 2015;32:127-33
83. Jubierre L, Soriano A, Planells-Ferrer L. et al. BRG1/SMARCA4 is essential for neuroblastoma cell viability through modulation of cell death and survival pathways. Oncogene. 2016;35:5179-90
84. Wilson BG, Wang X, Shen X. et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18:316-28
85. Lang JD, Hendricks WPD, Orlando KA. et al. Ponatinib Shows Potent Antitumor Activity in Small Cell Carcinoma of the Ovary Hypercalcemic Type (SCCOHT) through Multikinase Inhibition. Clin Cancer Res. 2018;24:1932-43
86. Pugh TJ, Weeraratne SD, Archer TC. et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106-10
87. Huether R, Dong L, Chen X. et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nat Commun. 2014;5:3630
88. Masliah-Planchon J, Machet MC, Fréneaux P. et al. SMARCA4-Mutated Atypical Teratoid/Rhabdoid Tumor with Retained BRG1 Expression. Pediatr Blood Cancer. 2016;63:568-9
89. Oike T, Ogiwara H, Nakano T. et al. Inactivating mutations in SWI/SNF chromatin remodeling genes in human cancer. Jpn J Clin Oncol. 2013;43:849-55
90. Love C, Sun Z, Jima D. et al. The genetic landscape of mutations in Burkitt lymphoma. Nat Genet. 2012;44:1321-5
91. Agaimy A, Daum O, Märkl B. et al. SWI/SNF Complex-deficient Undifferentiated/Rhabdoid Carcinomas of the Gastrointestinal Tract, A Series of 13 Cases Highlighting Mutually Exclusive Loss of SMARCA4 and SMARCA2 and Frequent Co-inactivation of SMARCB1 and SMARCA2. Am J Surg Pathol. 2016;40:544-53
92. Kohashi K, Oda Y. Oncogenic roles of SMARCB1/INI1 and its deficient tumors. Cancer Sci. 2017;108:547-52
93. Haberler C, Laggner U, Slavc I. et al. Reclassification of rhabdoid tumor and pediatric undifferentiated/unclassified sarcoma with complete loss of SMARCB1/INI1 protein expression, three subtypes of rhabdoid tumor according to their histological features. Mod Pathol. 2016;29:1232-42
94. Haberler C, Laggner U, Slavc I. et al. Immunohistochemical analysis of INI1 protein in malignant pediatric CNS tumors, Lack of INI1 in atypical teratoid/rhabdoid tumors and in a fraction of primitive neuroectodermal tumors without rhabdoid phenotype. Am J Surg Pathol. 2006;30:1462-8
95. Karnezis AN, Wang Y, Ramos P. et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J Pathol. 2016;238:389-400
96. Muppala R, Donenberg T, Huang MS. et al. SMARCA4 germline gene mutation in a patient with epithelial ovarian: A case report. Gynecol Oncol Rep. 2017;22:45-7
97. Fahiminiya S, Witkowski L, Nadaf J. et al. Molecular analyses reveal close similarities between small cell carcinoma of the ovary, hypercalcemic type and atypical teratoid/rhabdoid tumor. Oncotarget. 2016;7:1732-40
98. Kolin DL, Dong F, Baltay M. et al. SMARCA4-deficient undifferentiated uterine sarcoma (malignant rhabdoid tumor of the uterus): a clinicopathologic entity distinct from undifferentiated carcinoma. Mod Pathol. 2018 [Epub ahead of print]
99. Oike T, Ogiwara H, Tominaga Y. et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res. 2013;73:5508-18
100. Hoffman GR, Rahal R, Buxton F. et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc Natl Acad Sci U S A. 2014;111:3128-33
101. Berchuck A, Witkowski L, Hasselblatt M. et al. Prophylactic oophorectomy for hereditary small cell carcinoma of the ovary, hypercalcemic type. Gynecol Oncol Rep. 2015;12:20-2
102. Stephens B, Anthony SP, Han H. et al. Molecular Characterization of a Patient's Small Cell Carcinoma of the Ovary of the Hypercalcemic Type. J Cancer. 2012;3:58-66
103. Frühwald MC, Biegel JA, Bourdeaut F. et al. Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro Oncol. 2016;18:764-78
104. Rizvi NA, Hellmann MD, Snyder A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124-8
105. Kim KH, Kim W, Howard TP. et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med. 2015;21:1491-6
106. Venneti S, Le P, Martine D. et al. Malignant rhabdoid tumors express stem cell factors: which relate to the expression of EZH2 and Id proteins. Am J Surg Pathol. 2011;35:1463-72
107. Knutson SK, Warholic NM, Wigle TJ. et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110:7922-7
108. Alimova I, Birks DK, Harris PS. et al. Inhibition of EZH2 suppresses self-renewal and induces radiation sensitivity in atypical rhabdoid teratoid tumor cells. Neuro Oncol. 2013;15:149-60
109. Kurmasheva RT, Sammons M, Favours E. et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2017:64
110. Wang Y, Chen SY, Karnezis AN. et al. The histone methyltransferase EZH2 is a therapeutic target in small cell carcinoma of the ovary, hypercalcaemic type. J Pathol. 2017;242:371-83
111. Chan-Penebre E, Armstrong K, Drew A. et al. Selective Killing of SMARCA2- and SMARCA4-deficient Small Cell Carcinoma of the Ovary, Hypercalcemic Type Cells by Inhibition of EZH2, In Vitro and In Vivo Preclinical Models. Mol Cancer Ther. 2017;16:850-60
112. Wong JP, Todd JR, Finetti MA. et al. Dual Targeting of PDGFRα and FGFR1 Displays Synergistic Efficacy in Malignant Rhabdoid Tumors. Cell Rep. 2016;17:1265-75
113. Tagal V, Wei S, Zhang W. et al. SMARCA4-inactivating mutations increase sensitivity to Aurora kinase A inhibitor VX-680 in non-small cell lung cancers. Nat Commun. 2017;8:14098
114. Wetmore C, Boyett J, Li S. et al. Alisertib is active as single agent in recurrent atypical teratoid rhabdoid tumors in 4 children. Neuro Oncol. 2015;17:882-8
Author contact
![]() Corresponding author: Bingjian Lu, M.D., Department of Surgical Pathology, Women's Hospital, School of Medicine, Zhejiang University, 1 Xueshi Road, Hangzhou, Zhejiang Province, PR China, 310006. Email: lbjedu.cn; Tel: 86-571-89991702
Corresponding author: Bingjian Lu, M.D., Department of Surgical Pathology, Women's Hospital, School of Medicine, Zhejiang University, 1 Xueshi Road, Hangzhou, Zhejiang Province, PR China, 310006. Email: lbjedu.cn; Tel: 86-571-89991702