Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(2):323-331. doi:10.7150/jca.27615 This issue Cite
Research Paper
Mutation Profiling of Malignant Lymphoma by Next-Generation Sequencing of Circulating Cell-Free DNA
Peng Sun1,2*, Cui Chen3*, Yi Xia1,2*, Yu Wang1,2, Pan-Pan Liu1,2, Xi-Wen Bi1,2, Yang W. Shao4, Qiu-Xiang Ou4, Xue Wu4, Hang Yang1,2, Man Nie1,2, Xue-Wen Zhang1,2, Zhi-Ming Li1,2 ![]() , Wen-Qi Jiang1,2
, Wen-Qi Jiang1,2 ![]()
1. State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, 651 Dong Feng RD East, Guangzhou, Guangdong Province 510060, P. R. China.
2. Department of Medical Oncology, Sun Yat-Sen University Cancer Center, 651 Dong Feng RD East, Guangzhou, Guangdong Province 510060, P. R. China.
3. Department of Oncology, the First Affiliated Hospital, Sun Yat-Sen University, 58 Zhongshan Road II, Guangzhou, Guangdong Province 510080, P. R. China.
4. Geneseeq Technology Inc., 101 College Street, Suite 300, Toronto, Ontario, Canada.
*These authors contributed equally to this work.
Received 2018-6-2; Accepted 2018-10-21; Published 2019-1-1
Abstract
Background: Malignant lymphomas are a group of distinct lymphoid neoplasms, exhibiting marked diversity in biological behaviors and clinical outcomes. Liquid biopsy, such as circulating cell-free DNA (cfDNA), has recently been attempted to be used for mutation profiling of lymphomas using next-generation sequencing (NGS). However, only limited data about cfDNA are restricted in Hodgkin's lymphoma and B cell lymphoma, and there is no report in the T cell lymphoma so far.
Patient and Methods: Medical records of a total of 50 lymphoma patients were retrospectively reviewed, and cfDNA samples were analyzed by capture-based NGS targeting 390 lymphoma- and cancer- relevant genes. We sought to explore the clinical utility of cfDNA in establishing the mutation profiles of different lymphoma subtypes and analyze the correlation between cfDNA concentration and other clinical indices such as serum LDH and IPI.
Results: Somatic alterations were identified in cfDNA samples with a median of 64 variants per sample. The concentration of cfDNA in the plasma was found to be significantly correlated with the clinical indices in diffuse large B cell lymphoma (DLBCL). The genetic heterogeneity of different lymphoma subtypes was clearly observed in cfDNAs from germinal center B-cell (GCB) DLBCL, non-GCB DLBCL and natural killer/T-cell lymphoma (NKTCL), confirming that distinct molecular mechanisms are involved in the pathogenesis of different lymphomas.
Conclusion: Our findings demonstrate that NGS-based cfDNA mutation profiling reveals genetic heterogeneity across lymphoma subtypes, with potential implications for the discovery of therapeutic targets, the exploration of genome evolution and the development of risk-adapted treatment.
Keywords: malignant lymphoma, circulating cell-free DNA, tumor heterogeneity, next-generation sequencing (NGS), China
Background
Malignant lymphomas including non-Hodgkin's lymphoma (NHL) and Hodgkin's lymphoma (HL) are the most common hematologic malignancies worldwide, with over 80,000 new cases being diagnosed in the United States per year[1]. Either NHL or HL could be further classified into plenty of distinct subtypes, which exhibit marked diversity in biological behavior and clinical outcomes[2]. Recently, assessment of the molecular heterogeneity has been revealed as a useful tool for diagnosis, evaluation and personalized treatment of lymphoma patients[3, 4]. For instance, most diffuse large B cell lymphoma (DLBCL) could be divided into two subtypes, germinal center B cell-like (GCB) type and activated B cell-like (ABC) type, through their different gene expression profiling (GEP)[4]. The molecular classification of DLBCL showed distinct correlation with the risk stratification and could influence treatment decision-making for DLBCL patients.
Tissue biopsy is a traditional method to detect the molecular features of a tumor, however, it is fundamentally limited by its invasiveness and difficulty to obtain serially samples in clinical practice. By contrast, utility of circulating cell-free DNA (cfDNA) as a liquid biopsy has become a more feasible and noninvasive tool. Since cfDNA contains the DNA fragment released by the tumor cells, it harbors the tumor-derived genetic alterations and could reflect the molecular heterogeneity across multiple disease sites compared to the conventional single tissue biopsy[5]. Currently, next-generation sequencing (NGS) technology has emerged as a promising approach for mutation profiling of cfDNA due to its high-throughput, better sensitivity and specificity[6, 7]. cfDNA has been recognized as a tumor-specific biomarker and showed encouraging results in heralding a revolution for cancer diagnosis, prognosis and treatment[8-12] in multiple solid cancers as well as in malignant lymphomas[13].Recently, the tumor-specific mutations has been successfully detected in the peripheral blood of HL[14-16] and DLBCL[17, 18].NGS-based cfDNA detection could reflect tumor burden[19], demonstrate prognostic genetic variation and actionable targets[15, 17-20], and elucidate genome evolution[20] for lymphoma patients.
However, these studies are only limited to HL and B-NHL, and there are no available data in T-NHL. Herein, we sought to analyze the mutation profiles of different lymphoma subtypes, including HL, B-NHL and T-NHL, using patients' cfDNA targeting 390 lymphoma- and cancer- related genes by NGS. We aimed to explore the clinical utility of cfDNA mutation profiling in lymphomas and reveal the genetic heterogeneity in different subtypes of lymphoma to facilitate prognosis prediction and treatment decision-making.
Patients and methods
Patient
A total of 50 patients who were diagnosed of lymphoma at the Cancer Center of the Sun Yat-Sen University, Guangzhou, China, between April 1st and November 30th 2015 were enrolled in this study. Patient's demographic data and clinical characteristics were retrospectively reviewed and summarized in Table 1. Written informed consent was collected from each patient according to the Ethics Committee of the Cancer Center of the Sun Yat-Sen University. We have uploaded the essential raw data onto the Research Data Deposit (RDD) public platform (https://www.researchdata.org.cn), with the RDD approval number as RDDA2018000401.
Baseline characteristics of all patients
| Variables | No. | % |
|---|---|---|
| Age (years) | ||
| Median (Range) | 49 (18-80) | |
| Mean±SD | 47.4±16.9 | |
| Gender | ||
| Male | 35 | 70 |
| Female | 15 | 30 |
| ECOG score | ||
| 0 | 10 | 20 |
| 1 | 29 | 58 |
| 2 | 11 | 22 |
| Serum LDH ≤245U/L | 22 | 44 |
| B symptoms | 23 | 46 |
| BM involment | 4 | 8 |
| Bulky disease | 5 | 10 |
| Bone involvement | 7 | 14 |
| Liver involvement | 3 | 6 |
| Lung involvement | 4 | 8 |
| Spleen involvement | 10 | 20 |
| CNS involvement | 2 | 4 |
| Ann Arbor Stage | ||
| I-II | 24 | 48 |
| III-IV | 26 | 52 |
| Pathological diagnosis | ||
| DLBCL | 24 | 48 |
| NKTCL | 8 | 16 |
| PTCL, NOS | 2 | 4 |
| FL | 5 | 10 |
| ALCL | 1 | 2 |
| MALT-L | 2 | 4 |
| MCL | 3 | 6 |
| LPL | 1 | 2 |
| HL | 3 | 6 |
| ATCL | 1 | 2 |
SD=standard deviation, ECOG=Eastern Cooperative Oncology Group, LDH=lactate dehydrogenase, BM=Bone marrow, CNS=central nervous system, DLBCL=diffuse large B cell lymphoma, NKTCL= NK/T cell lymphoma, PTCL,NOS=Peripheral T cell lymphoma, non-origin specified, FL= follicular lymphoma, ALCL=anaplastic large cell lymphoma, MALT-L=mucosa-associated lymphoid tissue lymphoma, LPL=lymphoplasmacytic lymphoma, HL=Hodgkin's lymphoma, ATCL=angioimmunoblastic T-cell lymphoma
Sample collection and cfDNA extraction
All the blood samples (5-10ml peripheral blood) were collected within 24 hours into EDTA-coated tubes (BD Biosciences) before the start of the treatment. Plasma was extracted within 2 hours of blood collection and shipped to the central testing laboratory (Nanjing Geneseeq Technology Inc.) within 48 hours. cfDNA was extracted using the QIAamp Circulating Nucleic Acid Kit (QIAGEN). Genomic DNA of the whole blood sample was prepared by DNeasy Blood & Tissue kit (QIAGEN) as control for germline mutations. DNA was quantified by dsDNA HS Assay Kit (Life Technologies) on Qubit 2.0 and qualified by Nanodrop2000 (Thermo Fisher Scientific).
Library preparation and sequencing
cfDNA or fragmented genomic DNA (300~350bp with Covaris M220 instrument) underwent sequencing library preparation using KAPA Hyper Prep kit (KAPA Biosystems) with optimized protocols. In summary, DNA was experienced with end-repairing, A-tailing, adapter ligation, size selection using AgencourtAMPure XP beads (Beckman Coulter), and then PCR amplification and purification before targeted enrichment.
DNA libraries with unique indices were pooled up to 2µg together with Human cot-1 DNA (Life Technologies) and xGen Universal blocking oligos (Integrated DNA Technologies) as blocking reagents. Customized xGen lockdown probes panel (Integrated DNA Technologies) were used to targeted-enrich for 390 predefined genes. NimbleGen SeqCap EZ Hybridization and Wash Kit (Roche) was used to perform the hybridization reaction. Dynabeads M-270 (Life Technologies) was used to capture probe-bind fragments, followed by enriched library amplification with Illumina p5 (5' AAT GAT ACG GCG ACC ACC GA 3') and p7 primers (5' CAA GCA GAA GAC GGC ATA CGA GAT 3') in KAPA HiFiHotStartReadyMix (KAPA Biosystems), and purification by AgencourtAMPure XP beads. Library quantification was analyzed by KAPA Library Quantification kit (KAPA Biosystems). The size distribution of libraries was measured by Agilent Technologies 2100 Bioanalyzer (Agilent Technologies). Enriched libraries were sequenced on Hiseq 4000 NGS platforms (Illumina) to targeted mean coverage depths of at least 100x for whole blood control samples, and 3000x for cfDNAs.
Data processing and analysis
Trimmomatic[21] was applied for sequencing data quality control. The reads with a quality below the threshold of 15, as well as N bases were removed before mapping to reference sequence hg19 (Human Genome version 19) using Burrows-Wheeler Aligner (BWA) with optimized parameters. Genome Analysis Toolkit (GATK)[22] was used for indels local realignment and base quality score recalibration. SNPs/ indels were identified using VarScan2 when mutant allele frequency (MAF) is below 10% (http://dkoboldt.github.io/varscan/) or HaplotypeCaller/UnifiedGenotyper in GATK (MAF>10%). Common SNPs were filtered out with dbSNP and 1000 Genome data sets. Germline mutations were identified in the matched whole blood DNA. A mutation was called when at least 3 mutated reads were found in the sample on different strands with good quality scores and manually inspected in Integrative Genomics Viewer Software (IGV, Broad Institute). Genomic fusions were identified by FACTERA[23] with default parameters. ADTEx (http://adtex.sourceforge.net) was used to detect copy number variations (CNVs) with default parameters, while proposed discrete wavelet transform (DWT) was used to reduce intrinsic noise.
Statistics
Comparisons of the cfDNA load were analyzed by Student's t-test. Comparisons between the proportions of samples with certain genetic alterations were performed by the Chi-Square or Fisher's exact test.The relationships between different molecular parameters were determined using a non-parametric test (Mann-Whitney). An alpha value of p<0.05 was considered statistically significant. The statistical analyses were performed using the Statistical Package for the Social Sciences (SPSS, version 19.0).
Results
Targeted NGS-based mutation profiling of cfDNA from lymphoma patients
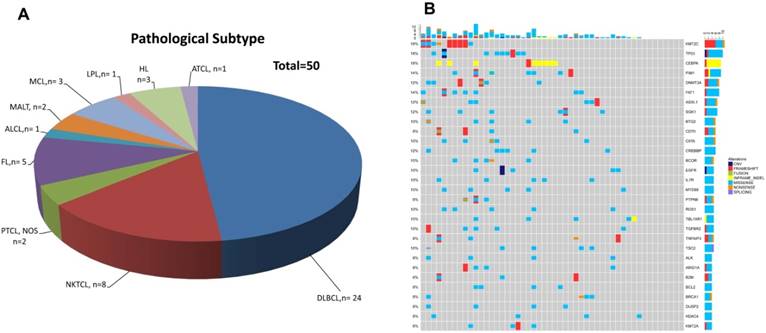
Patients' demographic and clinical characteristics were summarized in Table 1, including 3 HL patients, 35 B-NHL patients and 12 T-NHL patients (Table 2 and Figure 1A). DLBCL was the most common subtype of B-NHL (24/35, 68.6%) in this study, while natural killer/T-cell lymphoma (NKTCL) was the most common subtype of T-NHL (8/12, 66.7%). Although none of our patients harboured detectable circulating lymphoma cells by routine blood smear examination, all the blood samples had detectable cfDNAs, with a median concentration of 5.31 ng/ml plasma. Furthermore, somatic genetic alterations were identified in all the cfDNA samples, with a median of 64 variants in each sample. The median number of single nucleotide variant (SNV) was 8 (range 1-48). Copy number variants (CNVs) of 14 genes were identified in 9 patients. 30 recurrent gene mutations were identified in at least three patients (6%), as shown in Figure 1B. KMT2C, TP53, and CEBPA were the three most frequently mutated genes found in the whole cohort (9/50, 18% of the patients, Figure 1B).
cfDNA concentration in patients of different pathological subtypes
| Subtype | cfDNA concentration (ng/ml) | Mutation (median) |
|---|---|---|
| DLBCL | 11.70±12.05 | 13 |
| NKTCL | 19.6±26.04 | 4 |
| PTCL, NOS | 5.04±0.42 | 9 |
| FL | 4.21±3.10 | 7 |
| ALCL | 6.36 | 12 |
| MALT-L | 4.22±0.42 | 8 |
| MCL | 9.22±10.88 | 3 |
| LPL | 10.26 | 10 |
| HL | 5.23±3.06 | 5 |
| ATCL | 19.98 | 4 |
DLBCL=diffuse large B cell lymphoma, NKTCL= NK/T cell lymphoma, PTCL,NOS=Peripheral T cell lymphoma, non-origin specified, FL= follicular lymphoma, ALCL=anaplastic large cell lymphoma, MALT-L=mucosa-associated lymphoid tissue lymphoma, LPL=lymphoplasmacytic lymphoma, HL=Hodgkin's lymphoma, ATCL=angioimmunoblastic T-cell lymphoma
Distribution of pathological subtypes and genetic alterations of cfDNA in the total cohort.(A) Detailed distribution of pathological subtypes of 50 malignant lymphomas. (B) Genetic alterations of cfDNA in the total cohort. DLBCL=diffuse large B cell lymphoma, NKTCL= NK/T cell lymphoma, PTCL,NOS=Peripheral T cell lymphoma, non-origin specified, FL= follicular lymphoma, ALCL=anaplastic large cell lymphoma, MALT=mucosa-associated lymphoid tissue lymphoma, LPL=lymphoplasmacytic lymphoma, HL=Hodgkin's lymphoma, ATCL=angioimmunoblastic T-cell lymphoma
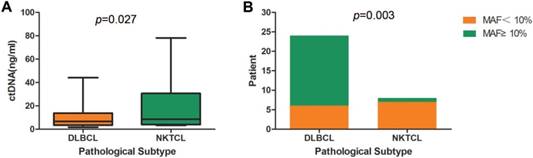
Interestingly, DLBCL and NKTCL showed a significant difference in the concentrations of cfDNA, as well as in the mutant allele frequencies (MAFs) of identified mutations (Figure 2). NKTCL patients exhibited a higher mean plasma concentration of cfDNA compared to that of DLBCL patients (19.6 ng/ml vs. 11.7ng/ml, p=0.027, Table 3 and Figure 2A). Since other factors including pathologic stage could contribute to the difference of cfDNA, we assessed baseline characteristics, including age (p=0.584), gender (p=0.313), IPI (p=0.813), ECOG (p=0.564), stage (p=0.915), between DLBCL and NKTCL patients. However, no significant difference was observed. Furthermore, 18 out of 24 DLBCL patients exhibited a maximal MAF over 10% in cfDNA, whereas only one NKTCL patient presenting a maximal MAF over 10% (75% vs. 12.5%, p=0.003, Figure 2B).
Comparison of cfDNA detection between DLBCL and NKTCL. (A) Comparison of cfDNA concentration between DLBCL and NKTCL. (B) Comparison of mutant allele frequency (MAF) between DLBCL and NKTCL.
Relationship between cfDNA concentration and clinical characteristics of DLBCL patients
| Characteristics | cfDNA concentration (ng/ml) | P value | Mutation (median) |
|---|---|---|---|
| Subtype | |||
| GCB | 4.72±3.63 | 0.007* | 11 |
| non-GCB | 15.95±13.42 | 13 | |
| Bulky disease | |||
| No | 10.67±10.58 | 0.358 | 13 |
| Yes | 16.88±18.99 | 25 | |
| Ann Arbor Stage | |||
| I-II | 10.81±12.27 | 0.726 | 7 |
| III-IV | 12.59±12.29 | 18 | |
| Serum LDH | |||
| ≤245U/L | 6.59±4.07 | 0.045* | 9 |
| >245U/L | 16.02±14.84 | 14 | |
| IPI | |||
| 0-2 | 7.16±5.97 | 0.044* | 12 |
| 3-4 | 22.73±16.16 | 17 | |
| Ki-67 | |||
| <90% | 12.49±12.78 | 0.591 | 13 |
| ≥90% | 9.34±10.17 | 13 | |
* p<0.05, DLBCL=diffuse large B cell lymphoma, GCB=germinal center B cell-like, LDH=lactate dehydrogenase, IPI=International prognostic index.
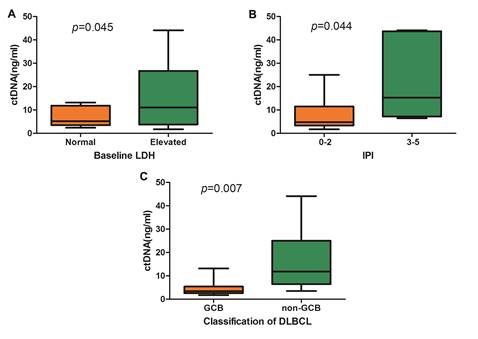
Among DLBCL patients, it was observed that the cfDNA concentration was significantly correlated with the baseline lactate dehydrogenase (LDH) level and the International Prognostic Index (IPI) (Figure 3A and Figure 3B). According to the Hams classification system, 8 DLBCL patients were further classified as germinal center B-cell (GCB) and 15 patients as non-GCB. The mean concentration of cfDNA was significantly higher in the non-GCB patients than in the GCB patients (15.95 ng/ml vs. 4.72ng/ml, p=0.015, Figure 3C).
Comparison of cfDNA concentration in DLBCL. (A) cfDNA concentration according to baseline LDH level, (B) cfDNA concentration according to IPI, and (C) Comparison of cfDNA concentration between GCB subtype and non-GCB subtype.
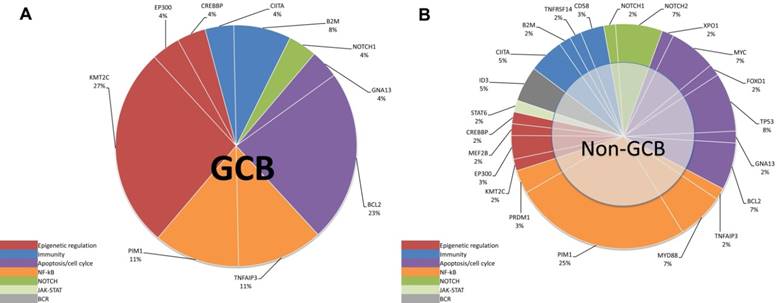
According to the method previously reported by Dubois et al. [7], we divided the top affected genes of DLBCL into eight specific pathways. We then demonstrated that the non-GCB subtype was mostly characterized by mutations in NF-κB pathway (Figure 4B and 5B), whereas the GCB subtype is mostly characterized by mutations in the epigenetic modulation pathway and the apoptosis/cell-cycle pathway (Figure 4A and 5A).
NGS detection of cfDNA in DLBCL. (A) Genetic alterations of cfDNA in the GCB subtype. (B) Genetic alterations of cfDNA in the non-GCB subtype.
Mutation pathway heterogeneity among DLBCL patients. Mutated genes of cfDNA were grouped into 8 specific pathways. Pie chart representations of mutation frequencies represented by pathway are represented for GCB (A) subtype and non-GCB (B) subtype. Mutation frequencies per gene and per subtype are shown here as the percentage of the total number of variants.
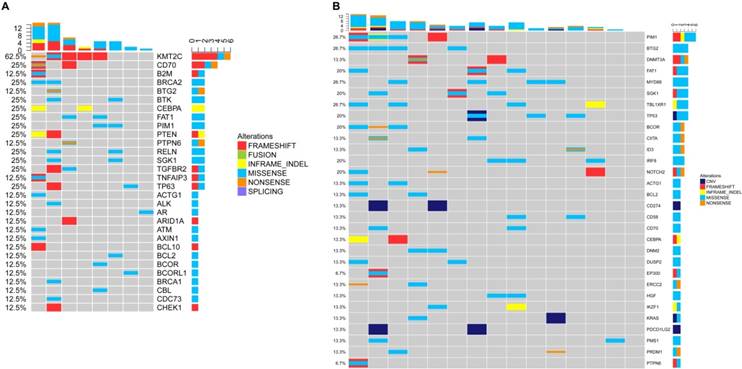
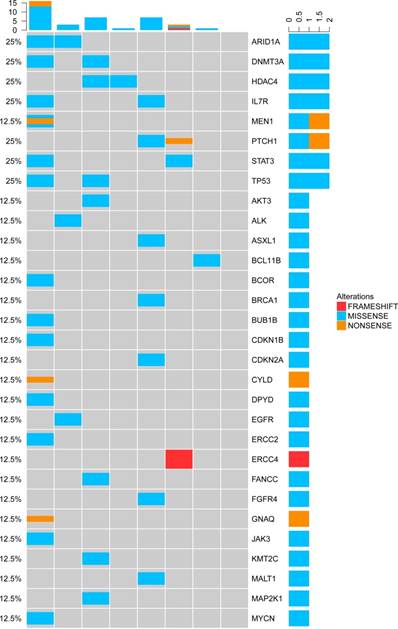
TP53 mutations appeared most frequently in the cfDNA of T-NHL (3/12, 25%). In total, there were 30 mutated genes detected in the cfDNA of NKTCL. Mutations of ARID1A, DNMT3A, HDAC4, IL7R, TP53, STAT3 and PTCH1 were the most frequent genetic alterations identified in NKTCL patients (2/8, 25% for all, Figure 6), implying that the mutated genes in cfDNA were most involved in the tumor suppressing and epigenetic modulation pathways.
Genetic alterations of cfDNA in the NKTCL.
Potential biomarkers identified in cfDNA with clinical implications
Our customized gene panel included several genes, which could have diagnostic implications or facilitate to predict the prognosis of lymphoma. MYD88 mutation which was revealed as a key diagnostic parameter for lymphoplasmacytic lymphoma (LPL)[2] was also detected in the cfDNA of one LPL patient in this study. Meanwhile, MYC mutations were detected in DLBCL patients with confirmed MYC translocation in the corresponding tumor tissue.
Besides, we also identified several gene mutations, which can be targeted by agents already approved or currently in clinical trials. There were three DLBCL patients' cfDNA harboring gene amplifications in the programmed cell death-1 (PD-1) pathway. Mutated MYD88 was detected in four non-GCB DLBCL patients, mutated TNFAIP3 in the cfDNA of two DLBCL patients, mutated PIM1 in the cfDNA of six DLBCL patients and one follicular lymphoma (FL) patient. Furthermore, mutated epigenetic pathway-related genes in cfDNA, such as EZH2, CREBBP, KMT2D, and EP300 were also found in our patient cohort.
Discussion
With the advances in NGS technology, comprehensive exploration of somatic alterations within cfDNA has been increasingly accessible in various malignancies including malignant lymphomas. Our study applied a targeted panel sequencing of cfDNA in 50 lymphoma patients on 390 key genes, and analyzed their genetic alterations. The results demonstrated that our panel was informative enough to track genetic landscape as well as novel or rare mutations in lymphoma. In addition, NGS-based cfDNA analysis could reflect genetic heterogeneity between different lymphoma subtypes, demonstrating that cfDNA could be a noninvasive and feasible biomarker for lymphoma patients' diagnosis. Taken together, our study fully validates the feasibility of NGS-based cfDNA mutation profiling and highlights the utility of cfDNA as a noninvasive biomarker in lymphoma patients.
Previous studies showed that cfDNA load could strongly reflect tumor burden. Florian Scherer et al[20] found that the amount of cfDNA was significantly correlated with LDH, Ann Arbor stage and IPI in DLBCL patients. Mark Roschewski et al[19] also found strong association between cfDNA amount and tumor stage. Our study showed similar findings that plasma concentration of cfDNA was significantly correlated with tumor burden of DLBCL (LDH and IPI). Notably, we found that distinct subtypes of lymphomas exhibited significantly different concentration of cfDNA, with NKTCL having a significantly higher cfDNA concentration than DLBCL, which may be due to the high level of necrosis in NKTCLs[24]. Moreover, non-GCB-DLBCL had a much higher concentration of cfDNA than GCB-DLBCL.
Accurate molecular classification is extremely important for precision medicine in malignant lymphomas. In DLBCL patients, gene expression profiles (GEP) has been recognized as the gold standard for GCB/ABC classification, however, this method is limited by the requirement of invasive tumor biopsies[4]. Recently, Florian Scherer et al[20] established a method for DLBCL classification based on integrating somatic mutation profiles of circulating tumor DNA (ctDNA), and this noninvasive classification method showed a high tumor-plasma concordance rate. Our study further emphasized that GCB-DLBCLs exhibited a distinct mutation profile from non-GCB-DLBCLs, and the cfDNA genotyping of GCB and non-GCB patients was in line with the genotyping of tumor tissue[7]. Furthermore, the cfDNA of NKTCL showed a distinct genotyping from DLBCL. These findings implied that cfDNA might be a promising and noninvasive tool for molecular classification of lymphomas.
In addition, high-throughput NGS technology could detect plenty of mutations which could inform prognosis of lymphoma[6, 7, 25, 26]. MYC translocation is a well-known unfavorable prognostic factor for DLBCL[3]. In our study, MYC mutation of cfDNA has also been identified in patient with paired tumor tissue exhibiting FISH-confirmed MYC rearrangement. Alessandro Pastore et al[27] found that the m7-FLIPI risk model including mutation status of seven genes (EZH2, ARID1A, MEF2B, EP300, FOXO1, CREBBP, and CARD11) could identify a subset of patients with follicular lymphoma (FL) at high risk. Interestingly, EZH2 mutations could also be detected in the cfDNA of FL patients of our cohort, suggesting the feasibility of establishing cfDNA-based prognostic model in FL. Alteration of MYD88 is important for the diagnosis of LPL[28], which was identified in one of our LPL patient. Other potentially actionable mutations were also identified in this study. For example, DLBCL with MYD88 mutation might be sensitive to ibrutinib[29, 30], PIM1 mutation might predict sensitivity to PIM1 inhibitors[31, 32], and TNFAIP3 alteration might lead to decreased activity of both sotrastaurin and ibrutinib[33, 34]. These findings are promising in the precision medicine era and are especially encouraging for the subtypes lacking of targeted therapies.
This is the first study exploring the somatic mutations in cfDNA of NKTCL by NGS. We confirmed that cfDNA was detectable in NKTCL, whereas recurrent somatic mutations could also be identified, providing valuable experience and implications on future studies. Since malignant lymphoma is a systematic disease and always involves multiple organs at initial diagnosis. NGS-based cfDNA testing could comprehensively reveal the genetic heterogeneity of all the tumor lesions, especially when tumor tissue is unavailable. Fontanilles et al. found that somatic genomic alterations could be detected in the cfDNA of patients with primary central nervous system lymphoma[35].Moreover, cfDNA could be acquired and analyzed at multiple time points for response evaluation, disease surveillance, resistant mechanism discovery, and genomic evolution identification. Another superiority of our study is the broad coverage of our comprehensive gene panel, including far more genes than any previous reported panels[36, 37], which could provide new insights in the molecular mechanisms of lymphoma. However, our results were partly limited by the heterogeneous mixture of patients with different subtypes of lymphoma and relatively small sample size of certain subtypes.
In summary, NGS-based cfDNA mutation profiling is a promising approach for uncovering the biology and facilitating personalized therapy of malignant lymphomas. We anticipate that cfDNA has critical utility for dissecting genetic heterogeneity between different subtypes of lymphomas, with applications for the discovery of potential targets, the exploration of genomic evolution and the development of risk-adapted treatment. Future studies with a larger patient cohort are warranted to validate the discovery of our study in lymphoma diagnosis and prognosis.
Abbreviations
NHL: non-Hodgkin's lymphoma; HL: Hodgkin's lymphoma; DLBCL: diffuse large B cell lymphoma; GCB: germinal center B cell-like; ABC: activated B cell-like; GEP: gene expression profiling (GEP); cfDNA: circulating cell-free; ctDNA: circulating tumor DNA; NGS: next-generation sequencing; NKTCL: natural killer/T-cell lymphoma; LDH: lactate dehydrogenase; IPI: International Prognostic Index; FISH: Fluorescence in situ hybridization; LPL: lymphoplasmacytic lymphoma; PD-1: programmed cell death-1; FL: follicular lymphoma.
Acknowledgements
We thank all the research staff members and patients involved in this study.
Funding
This study was supported by the General Funds of the Natural Science Foundation of Guangdong Province, China (No. 2017A030313754).
Authors' contributions
PS, YX and CC collected clinical data and drafted the manuscript. PS, WQJ and ZML participated in the design of the study. XWB, PPL and YW performed the statistical analysis. MN, XWZ, HY and YWS conducted the experiments. QO and XW contributed to the revision of the manuscript. WQJ and ZML conceived of the study and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript. PS, CC and YX were co-first authors.
Ethics approval and consent to participate
All patients have provided written consent for their information to be stored in the Sun Yat-Sen University Cancer Center database and used for research. This study was conducted in accordance with the ethical standards of the World Medical Association's Declaration of Helsinki and approved by an independent ethics committee at the Cancer Center of Sun Yat-Sen University.
Competing Interests
Yang W. Shao, Qiuxiang Ou, and Xue Wu are the shareholders or employees of Geneseeq Technology Inc. Toronto, Canada. The remaining authors have no conflicts of interest to declare.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R. et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127:2375-90
3. Xu-Monette ZY, Deng Q, Manyam GC, Tzankov A, Li L, Xia Y. et al. Clinical and Biologic Significance of MYC Genetic Mutations in De Novo Diffuse Large B-cell Lymphoma. Clin Cancer Res. 2016;22:3593-605
4. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A. et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503-11
5. Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res. 1977;37:646-50
6. Dubois S, Jardin F. The role of next-generation sequencing in understanding the genomic basis of diffuse large B cell lymphoma and advancing targeted therapies. Expert Rev Hematol. 2016;9:255-69
7. Dubois S, Viailly PJ, Mareschal S, Bohers E, Bertrand P, Ruminy P. et al. Next-Generation Sequencing in Diffuse Large B-Cell Lymphoma Highlights Molecular Divergence and Therapeutic Opportunities: a LYSA Study. Clin Cancer Res. 2016;22:2919-28
8. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF. et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368:1199-209
9. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM. et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497:108-12
10. Leung F, Kulasingam V, Diamandis EP, Hoon DS, Kinzler K, Pantel K. et al. Circulating Tumor DNA as a Cancer Biomarker: Fact or Fiction. Clin Chem. 2016
11. Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I. et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8:346ra92
12. Cheng F, Su L, Qian C. Circulating tumor DNA: a promising biomarker in the liquid biopsy of cancer. Oncotarget. 2016
13. Gordon LI. Precision Monitoring by Next-Generation Sequencing in Lymphoma: Circulating Tumor DNA as a New Biomarker. Oncology (Williston Park). 2016:30
14. Oki Y, Neelapu SS, Fanale M, Kwak LW, Fayad L, Rodriguez MA. et al. Detection of classical Hodgkin lymphoma specific sequence in peripheral blood using a next-generation sequencing approach. Br J Haematol. 2015;169:689-93
15. Camus V, Stamatoullas A, Mareschal S, Viailly PJ, Sarafan-Vasseur N, Bohers E. et al. Detection and prognostic value of recurrent XPO1 mutations in tumor and cell-free circulating DNA of patients with classical Hodgkin Lymphoma. Haematologica. 2016
16. Vandenberghe P, Wlodarska I, Tousseyn T, Dehaspe L, Dierickx D, Verheecke M. et al. Non-invasive detection of genomic imbalances in Hodgkin/Reed-Sternberg cells in early and advanced stage Hodgkin's lymphoma by sequencing of circulating cell-free DNA: a technical proof-of-principle study. Lancet Haematol. 2015;2:e55-65
17. Camus V, Sarafan-Vasseur N, Bohers E, Dubois S, Mareschal S, Bertrand P. et al. Digital PCR for quantification of recurrent and potentially actionable somatic mutations in circulating free DNA from patients with diffuse large B-cell lymphoma. Leuk Lymphoma. 2016:1-9
18. Herrera AF, Kim HT, Kong KA, Faham M, Sun H, Sohani AR. et al. Next-generation sequencing-based detection of circulating tumour DNA After allogeneic stem cell transplantation for lymphoma. Br J Haematol. 2016;175:841-50
19. Roschewski M, Dunleavy K, Pittaluga S, Moorhead M, Pepin F, Kong K. et al. Circulating tumour DNA and CT monitoring in patients with untreated diffuse large B-cell lymphoma: a correlative biomarker study. Lancet Oncol. 2015;16:541-9
20. Scherer F, Kurtz DM, Newman AM, Stehr H, Craig AF, Esfahani MS. et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci Transl Med. 2016;8:364ra155
21. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114-20
22. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491-8
23. Newman AM, Bratman SV, Stehr H, Lee LJ, Liu CL, Diehn M. et al. FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution. Bioinformatics. 2014;30:3390-3
24. Au WY, Weisenburger DD, Intragumtornchai T, Nakamura S, Kim WS, Sng I. et al. Clinical differences between nasal and extranasal natural killer/T-cell lymphoma: a study of 136 cases from the International Peripheral T-Cell Lymphoma Project. Blood. 2009;113:3931-7
25. Todorovic BM, Jelicic J, Mihaljevic B, Kostic J, Stanic B, Balint B. et al. Gene Mutation Profiles in Primary Diffuse Large B Cell Lymphoma of Central Nervous System: Next Generation Sequencing Analyses. Int J Mol Sci. 2016:17
26. Jardin F. Next generation sequencing and the management of diffuse large B-cell lymphoma: from whole exome analysis to targeted therapy. Discov Med. 2014;18:51-65
27. Pastore A, Jurinovic V, Kridel R, Hoster E, Staiger AM, Szczepanowski M. et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015;16:1111-22
28. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le BMM. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391-405
29. Treon SP, Xu L, Hunter Z. MYD88 Mutations and Response to Ibrutinib in Waldenström's Macroglobulinemia. N Engl J Med. 2015;373:584-6
30. Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G. et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21:922-6
31. Wan X, Zhang W, Li L, Xie Y, Li W, Huang N. A new target for an old drug: identifying mitoxantrone as a nanomolar inhibitor of PIM1 kinase via kinome-wide selectivity modeling. J Med Chem. 2013;56:2619-29
32. Brault L, Menter T, Obermann EC, Knapp S, Thommen S, Schwaller J. et al. PIM kinases are progression markers and emerging therapeutic targets in diffuse large B-cell lymphoma. Br J Cancer. 2012;107:491-500
33. Zheng X, Ding N, Song Y, Feng L, Zhu J. Different sensitivity of germinal center B cell-like diffuse large B cell lymphoma cells towards ibrutinib treatment. Cancer Cell Int. 2014;14:32
34. Naylor TL, Tang H, Ratsch BA, Enns A, Loo A, Chen L. et al. Protein kinase C inhibitor sotrastaurin selectively inhibits the growth of CD79 mutant diffuse large B-cell lymphomas. Cancer Res. 2011;71:2643-53
35. Fontanilles M, Marguet F, Bohers É, Viailly PJ, Dubois S, Bertrand P. et al. Non-invasive detection of somatic mutations using next-generation sequencing in primary central nervous system lymphoma. Oncotarget. 2017;8:48157-68
36. Armand P, Oki Y, Neuberg DS, Faham M, Cummings C, Klinger M. et al. Detection of circulating tumour DNA in patients with aggressive B-cell non-Hodgkin lymphoma. Br J Haematol. 2013;163:123-6
37. Bohers E, Viailly PJ, Dubois S, Bertrand P, Maingonnat C, Mareschal S. et al. Somatic mutations of cell-free circulating DNA detected by next-generation sequencing reflect the genetic changes in both germinal center B-cell-like and activated B-cell-like diffuse large B-cell lymphomas at the time of diagnosis. Haematologica. 2015;100:e280-4
Author contact
![]() Corresponding authors: Wen-Qi Jiang, MD, PhD, 651 Dong Feng RD East, Guangzhou, Guangdong Province 510060, P. R. China; e-mail: jiangwqorg.cn, Phone/Fax: +86-020-87343352, and Zhi-Ming Li, MD, PhD, 651 Dong Feng RD East, Guangzhou, Guangdong Province 510060, P. R. China; e-mail: lizhmorg.cn, Phone/Fax: +86-020-87343765.
Corresponding authors: Wen-Qi Jiang, MD, PhD, 651 Dong Feng RD East, Guangzhou, Guangdong Province 510060, P. R. China; e-mail: jiangwqorg.cn, Phone/Fax: +86-020-87343352, and Zhi-Ming Li, MD, PhD, 651 Dong Feng RD East, Guangzhou, Guangdong Province 510060, P. R. China; e-mail: lizhmorg.cn, Phone/Fax: +86-020-87343765.