Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(2):355-366. doi:10.7150/jca.27832 This issue Cite
Research Paper
Aberrantly DNA Methylated-Differentially Expressed Genes and Pathways in Hepatocellular Carcinoma
Changzhou Cai1, Weilin Wang1,2, Zhenhua Tu1,2,3 ![]()
1. Key Laboratory of Precision Diagnosis and Treatment for Hepatobiliary and Pancreatic Tumor of Zhejiang Province, First Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou 310003, China.
2. Ward of Liver transplant, Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery. First Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou 310003, China.
3. Shenzhen Key Laboratory of Hepatobiliary Disease, Shenzhen Third People`s Hospital, Shenzhen 518112, China.
Received 2018-6-12; Accepted 2018-10-21; Published 2019-1-1
Abstract
Background: Methylation plays a significant role in the etiology and pathogenesis of hepatocellular carcinoma (HCC). The aim of the present study is to identify aberrantly methylated-diferentially expressed genes (DEGs) and dysregulated pathways associated with the development of HCC through integrated analysis of gene expression and methylation microarray.
Method: Aberrantly methylated-DEGs were identified from gene expression microarrays (GSE62232, GSE74656) and gene methylation microarrays (GSE44909, GSE57958). Functional enrichment and pathway enrichment analyses were performed through the database of DAVID. Protein-protein interaction (PPI) network was established by STRING and visualized in Cytoscape. Subsequently, overall survival (OS) analysis of hub genes was performed by OncoLnc. Finally, we validated the expression level of CDCA5 by quantitative real-time PCR (qRT-PCR) and western blotting, and performed Immunohistochemical experiments utilizing a tissue microarray. Cell growth assay and flow cytometry were behaved to explore the function of CDCA5.
Results: Aberrantly methylated-DEGs were enriched in biological process, molecular function, cellular component and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway. Among them, cell cycle was enriched most frequently, and some terms associated with cancer were enriched, such as p53 signaling pathway, pathways in cancers, PI3K-Akt signaling pathway and AMPK signaling pathway. After survival analysis and validation in TCGA database including methylation and gene expression status, 12 hub genes were identified. Furthermore, the expression level of new gene CDCA5 was validated in HCC cell lines and hepatic normal cell lines through qRT-PCR and western blotting. In additional, immunohistochemistry experiments revealed higher CDCA5 protein expression from HCC tumor tissues compared with paracancer tissues by tissue microarray. Finally, through loss of function, we demonstrated that CDCA5 promoted proliferation by regulating the cell cycle.
Conclusions: In summary, the present study implied possible aberrantly methylated-differentially expressed genes and dysregulated pathways in HCC by bioinformatics analysis and experiments, which could be helpful in understanding the molecular mechanisms underlying the development and progression of HCC. Hub genes including CDC20, AURKB, BIRC5, RRM2, MCM2, PTTG1, CDKN2A, NEK2, CENPF, RACGAP1, GNA14 and especially the new gene CDCA5 may serve as biomarkers for diagnosis, treatment and prognosis of HCC.
Keywords: methylation, expression, bioinformatics, hepatocellular carcinoma, CDCA5
Introduction
Liver cancer is the third leading cause of mortality worldwide. Hepatocellular carcinoma (HCC), the most common cancer in the liver, accounts for more than 80% of liver tumors [1]. Several studies indicate that chronic hepatitis B virus (HBV) or hepatitis C virus (HCV) infection, alcohol abuse, obesity and non-alcoholic fatty liver disease (NAFLD) are multiple risk factors inducing HCC [2]. It is generally accepted that the accumulation of various genetic and epigenetic changes in hepatocytes drives initiation and progression of HCC [3].
Epigenetics is known as heritable changes in gene expression that is not associated with an alteration in DNA sequence but plays a critical role in carcinogenesis [4]. Cancer epigenetics covers aspects of aberrant DNA methylation, dysregulated non- coding RNA, altered post-translational histone modification, of which aberrant DNA methylation is most widely investigated [5]. Aberrant DNA methylation could influence the key genes involved in the carcinogenesis and progression of HCC especially influence some tumor suppressor genes by altering their expression thus inhibiting their function [6]. Although effects of certain genes with aberrant DNA methylation on HCC have been reported extensively, the comprehensive profile of the interaction network still needs further elucidation.
During the last decades, the microarray technology and bioinformatics analysis have been widely performed to identify general genetic or epigenetic alterations in carcinogenesis and screen biomarkers for diagnosis and prognosis in cancer [7]. Lots of gene expression microarrays have been used for identifying differentially expressed genes (DEGs) and pathways in HCC [8]. Additionally, some researches concerning aberrant DNA methylation have been conducted to find various differentially methylated genes (DMGs) [9, 10]. However, false-positive rates in independent microarray analysis from individual investigation make it difficult to identify reliable key genes and pathways. Thus, it is credible to screen results via overlapping relevant datasets.
Up to now, no research has been performed to analyze gene expression profiling microarrays together with gene methylation profiling microarrays in the progression of HCC. In this work, two gene expression microarray datasets (GSE62232, GSE74656) and two gene methylation microarray datasets (GSE44909, GSE57958) were integrated and analyzed to identify aberrantly methylated-DEGs. Subsequently, function and pathway were enriched while protein-protein interaction network was established and hub genes were indicated. By this mean, it is hopeful to screen novel aberrantly methylated genes and pathways in HCC and improve understanding of underlying molecular mechanisms.
Materials and methods
Microarray data
The Gene Expression Omnibus (GEO) is a public repository for data storage. In the present study, two gene expression profiling data sets (GSE62232, GSE74656) and two gene methylation profiling data sets (GSE44909, GSE57958) were obtained from GEO database. The microarray data of GSE62232 included 81 HCC samples and 10 normal liver tissue samples [11]. GSE76427 consisted of 115 HCC samples and 52 adjacent non-tumor samples. The gene methylation profile of GSE44909 included a total of 24 paired normal and HCC samples from 12 patients while GSE57958 consisted of 61 HCC samples and 59 adjacent non-tumor samples [12, 13].
Data procession
GEO2R is an interactive online tool which allows users to compare two groups of samples under the same experimental conditions in a GEO series in order to screen genes differentially expressed [14]. GEO2R was applied to identify differentially expressed genes (DEGs) and differentially methylated genes (DMGs) between HCC and normal samples. P < 0.05 and |t| ≥ 2 were set as the cut-off criterion. Subsequently, Funrich was adopted to identify overlapping DEGs in the two gene expression profiles of GSE62232 and GSE74656 as well as overlapping DMGs in the two methylation gene expression profiles of GSE44909 and GSE57958. Finally, hypomethylation-high expression genes were identified by overlapping hypomethylation and up-regulated genes. Hypermethylation-low expression genes were identified by overlapping hypermethylation and down-regulated genes.
Functional and pathway enrichment analysis
The Database for Annotation, Visualization and Integrated Discovery (DAVID) is an online program offering systematic and integrative functional annotation tools for researchers to unravel biological meaning behind large list of genes [15]. Hypomethylation-high expression genes and hypermethylation- low expression genes were performed Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis in DAVID [16]. P < 0.05 was set as the cut-off criterion.
Protein-protein interaction (PPI) network establishment and module analysis
The functional interactions between proteins can provide context in molecular mechanisms of key cellular activities in carcinogenesis. In present study, protein-protein interaction (PPI) network of hypomethylation-high expression genes and hypermethylation-low expression genes were constructed using the Search Tool for the Retrieval of Interacting Genes (STRING) database, respectively. Interaction score ≥ 0.4 was set as the cut-off criterion and the PPI was visualized using Cytoscape [17]. Subsequently, the Molecular Complex Detection (MCODE) was conducted to screen modules within PPI network with degree cutoff = 2, k-core = 2, node score cutoff = 0.2, and max. depth = 100 [18]. Hub genes were selected with degrees ≥ 5 and included in the hub module in aberrantly methylated-DEGs.
Survival analysis of hub genes
OncoLnc is an online database in which TCGA survival data linking to mRNA, miRNA, or lncRNA expression levels are available. The patients with HCC were divided into two groups according to the expression of a particular gene (high vs. low expression). The overall survival of patients with HCC was analyzed using a Kaplan-Meier plot. Log rank P value was calculated and displayed on the web page.
Verification of the hub genes in TCGA database
The Cancer Genome Atlas (TCGA) database has stored comprehensive, multi-dimensional data of the key genomic and epigenetic alterations in various cancers. In order to confirm the results, hub genes of aberrantly methylated-DEGs were verified in database TCGA including gene expression and methylation status. 50 HCC samples and 50 adjacent normal liver samples from TCGA were adopted.
Cell culture and treatment
The cell lines used in this study include the human HCC cells (HepG2, Hep3B, Huh7 and LM3) and normal human hepatic cells (QSG-7701 and L-02). HCC cells (HepG2, Hep3B, Huh7 and LM3) were cultured in DMEM (Gibco, Shanghai, China), added with 10% fetal bovine serum (FBS, Gibco, Qualified, Australia Origin) and 1% penicillin/streptomycin in a humidified incubator with the atmosphere of 5% CO2 at temperature of 37°C. QSG-7701 and L-02 were cultured in RPMI 1640 (Gibco, Shanghai, China) with the same condition as above. Hep3B and Huh7 cells were transfected with small interfering RNA (siRNA) specific for CDCA5 (siCDCA5-1: GCAGGGAGCTTACTAAGGA, siCDCA5-2: CGAGCATCCTCCCTGAAAT, siCDCA5-3: GTGGTGTGCTCCAAACTCA) or corresponding scramble siRNA as negative control (Ribobio, China) using Lipofectamine 3000 (Invitrogen, Shanghai, China) following the manufacturer's instructions.
RNA extraction and qRT-PCR
Total RNAs was isolated from cells utilizing Trizol method according to the manufacturer's instructions (Invitrogen, Shanghai, China). The RNA were reversely transcribed into cDNA using a PrimeScript® RT reagent Kit (Takara, Japan). Quantitative real-time PCR (qRT-PCR) was conducted using an ABI 7500 real-time PCR System (Applied Biosystems, Carlsbad, CA) with SYBR Green (Takara, Japan). The sequences of the primers used were as follows: CDCA5-F (5'-GGC CAG AGA CTT GGA AAT GT-3') and CDCA5-R (5'- GGC CAG AGA CTT GGA AAT GT -3'); GAPDH-F (5'-TCA ACG ACC ACT TTG TCA AGC TCA-3') and GAPDH-R (5'-GCT GGT GGT CCA GGG GTC TTA CT-3'). The relative expression was calculated with formula 2-ΔΔCT, and using GAPDH as an internal control.
Western blotting
Total proteins were extracted from human HCC cells (HePG2, Hep3B, Huh7 and LM3) and 2 normal human hepatic cells (QSG-7701 and L-02) utilizing RIPA buffer (Applygen Technologies Inc.) added with protein and phosphatase inhibitor (Sigma), separated by SDS-PAGE, and electrophoretically transferred to PVDF membranes (Millipore). After blocking with 5% nonfat milk in TBST, the membranes were incubated overnight with antibodies. Primary antibodies specific to CDCA5 (Novus NBP1-89630), Tublin (Cell Signaling 2148S) were used. The membranes were incubated with the secondary antibodies against rabbit, mouse at a 1:5000 dilution for 1 h and visualized using chemiluminescence (Multisciences, Hangzhou, China).
Cell growth assay
Cells were seeded into 96-well plates, using 5 × 103/well for Huh7 and 3 × 103/well for Hep3B in complete medium with 50 nM synthetic siRNAs and 0.2% Lipofectamine 3000 in a final volume of 100 μl. After 72 h, viability of cells was evaluated by WST-8 assays (Cell counting Kit-8; China).
Flow cytometry
Huh7 and Hep3B cells were transfected with 50 nM synthetic siRNAs with Lipofectamine 3000 for 48 h. After incubation, cells were separated with trypsin at 37°C, washed with cold PBS, and resuspended in 2 ml cold 75% ethanol for overnight. The suspending cells were washed twice with PBS and then stained with DNA Staining Solution (Cat. No. 70-P1421; MultiSciences Biotech, Co., Ltd, Hangzhou, China) at 4°C. After 30 minutes, cells were analyzed using a BD FACSCantoTM II Flow Cytometer (Beckton, Dickinson and Company, BD Biosciences, 23350 Qume Drive, San Jose, CA 95131 USA).
Immunohistochemical analysis and statistical analysis
Immunohistochemical analysis of HCC tissues and adjacent normal liver tissues was performed utilizing a tissue microarray from National Human Genetic Resources Sharing Service Platform (Shanghai, China). The tissue microarray containing 81 tumor and 81 normal tissue specimens was used for immunostaining against anti-CDCA5 antibody (1:100). The expression of CDCA5 in liver specimens was blindly quantified by two pathologists using the histochemical score (H-score). We obtained the H-score (0-300) by multiplying the intensity of staining (0 = negative, 1 = weak, 2 = moderate and 3 = strong) and the percentage of positive cells (0%- 100%). The CDCA5 expression was finally divided into two groups: low (H-score ≤ 200) and high (H-score >200). Images were visualized and processed by Aperio ImageScope.
Statistical analyses were performed using GraphPad Prism software version 6 (GraphPad Software, San Diego, CA, USA) and SPSS version 19 (SPSS Inc.). Values were shown as mean ± SD. IHC data between groups were compared by Wilcoxon signed-rank test. Two-tailed Student's t-tests were used to evaluate the statistical differences. Statistically significant was displayed as *P<0.05, **P<0.01, ***P <0.001 and ****P <0.0001.
Results
Identification of aberrantly methylated-differentially expressed genes
Data from each microarray was analyzed by GEO2R to screen DEGs and DMGs, respectively. Among DEGs of gene expression microarrays, 3733 overlapping up-regulated genes (5946 in GSE62232, 6953 in GSE76427) as well as 2958 overlapping down- regulated genes (9243 in GSE62232, 6223 in GSE76427) were obtained. For DMGs of gene methylation microarrays, 2217 overlapping hypomethylation genes (3769 in GSE44909, 4868 in GSE57958) as well as 768 hypermethylation genes (2344 in GSE44909, 2651 in GSE57958) were obtained.
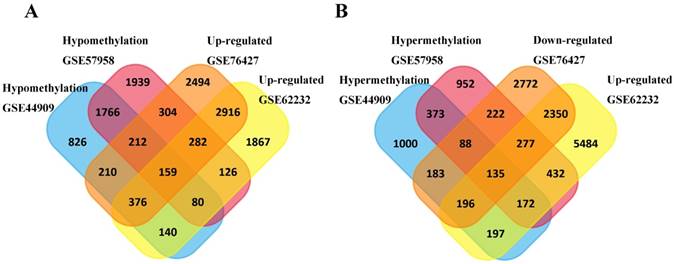
Subsequently, a total of 159 hypomethylation- high expression genes were screened out from overlapping 2217 hypomethylation and 3733 up-regulated genes while 135 hypermethylation-low expression genes were screened out from overlapping 768 hypermethylation genes and 2958 down-regulated genes (Figure 1).
Identification of aberrantly methylated-differentially expressed genes in mRNA expression profiling datasets (GSE62232, GSE74656) and gene methylation profiling datasets (GSE44909, GSE57958). (A) Hypomethylation and high expression gens; (B) Hypermethylation and low expression genes.
GO functional enrichment analysis and KEGG pathway enrichment analysis
The significant terms of GO enrichment analysis performed by DAVID were illustrated (Table 1). The hypomethylation-high expression genes were mainly involved in biological processes (BP) of mitotic nuclear division, sister chromatid cohesion, chromosome segregation, cell cycle and positive regulation of telomere maintenance via telomerase and apoptotic process. As for molecular functions (MF), these genes showed enrichment in protein binding, integrin binding, protein kinase activity, adiponectin binding and protein phosphatase regulatory activity. Besides, cell components (CC) indicated enrichment predominantly at nucleus and extracellular exosome, which indicated that hypomethylation-high expression genes might play a vital role in cell cycle and tumor microenvironment of HCC (Table 2).
Functional enrichment analysis of aberrantly methylated-DEGs in HCC.
| Category GO analysis | Terms | count | P value |
|---|---|---|---|
| Hypomethylation and high expression | |||
| GOTERM_BP_FAT | GO:0000278~ mitotic nuclear division | 11 | 7.9E-5 |
| GOTERM_BP_FAT | GO:0007062~ sister chromatid cohesion | 7 | 3.2E-4 |
| GOTERM_BP_FAT | GO:0007059~ chromosome segregation | 6 | 3.5E-4 |
| GOTERM_BP_FAT | GO:0032212~ positive regulation of telomere maintenance via telomerase and apoptotic process and cell cycle | 4 | 2.9E-3 |
| GOTERM_BP_FAT | GO:0007049~ cell cycle | 8 | 3.4E-3 |
| GOTERM_CC_FAT | GO:0005829~ cytosol | 58 | 3.1E-8 |
| GOTERM_CC_FAT | GO:0070062~ extrocellular | 50 | 3.1E-7 |
| GOTERM_CC_FAT | GO:0005737~ cytoplasm | 72 | 4.0E-6 |
| GOTERM_CC_FAT | GO:0005925~ focal adhesion | 14 | 3.1E-5 |
| GOTERM_CC_FAT | GO:0005654~ nucleuplasm | 40 | 8.9E-4 |
| GOTERM_MF_FAT | GO:0005515~ protein binding | 101 | 7.8E-4 |
| GOTERM_MF_FAT | GO:0005178~ integrin binding | 6 | 2.7E-3 |
| GOTERM_MF_FAT | GO:0004672~ protein kinase activity | 9 | 1.7E-2 |
| GOTERM_MF_FAT | GO:0055100~ adiponectin binding | 2 | 2.7E-2 |
| GOTERM_MF_FAT | GO:0019888~ protein phosphatase regulatory activity | 3 | 2.9E-2 |
| Hypermethylation and expression | |||
| GOTERM_BP_FAT | GO:0050885~ neuromuscular process controlling balance | 5 | 4.1E-4 |
| GOTERM_BP_FAT | GO:0032496~ response to lipopolysaccharide | 7 | 1.6E-3 |
| GOTERM_BP_FAT | GO:0001960~ negative regulation of cytokine-mediated signaling pathway | 3 | 2.0E-3 |
| GOTERM_BP_FAT | GO:0007165~ signal transduction | 19 | 3.1E-3 |
| GOTERM_BP_FAT | GO:0007204~ positive regulation of cytosolic calcium ion concentration | 6 | 3.6E-3 |
| GOTERM_CC_FAT | GO:0005887~ integral component of plasma membrane | 25 | 7.3E-5 |
| GOTERM_CC_FAT | GO:0005578~ proteinaceous extracellular matrix | 10 | 1.5E-4 |
| GOTERM_CC_FAT | GO:0005615~ extracellular space | 19 | 8.4E-3 |
| GOTERM_CC_FAT | GO:0005886~ plasma membrane | 43 | 9.2E-3 |
| GOTERM_CC_FAT | GO:0030426~ growth cone | 5 | 1.0E-2 |
| GOTERM_MF_FAT | GO:0005201~ extracellular matrix structural constituent | 5 | 1.7E-3 |
| GOTERM_MF_FAT | GO:0020037~ heme binding | 6 | 4.0E-3 |
| GOTERM_MF_FAT | GO:0019825~ oxygen binding | 4 | 5.5E-3 |
| GOTERM_MF_FAT | GO:0004601~ peroxidase activity | 3 | 1.2E-2 |
| GOTERM_MF_FAT | GO:0008236~ serine-type peptidase activity | 4 | 1.2E-2 |
Pathway enrichment analysis of aberrantly-DEGs in HCC
| Pathway ID | Term | Count | P value |
|---|---|---|---|
| Hypomethylation and high expression | |||
| hsa04110 | Cell cycle | 8 | 4.3E-4 |
| hsa00040 | Pentose and glucuronate interconversions | 4 | 7.3E-3 |
| hsa00240 | Pyrimidine metabolism | 5 | 2.8E-2 |
| hsa04114 | Oocyte meiosis | 5 | 3.3E-2 |
| hsa04115 | p53 signaling pathway | 4 | 3.8E-2 |
| Hypermethylation and high expression | |||
| hsa05204 | Chemical carcinogenesis | 5 | 4.1E-3 |
| hsa04022 | cGMP-PKG signaling pathway | 6 | 1.2E-2 |
| hsa04080 | Neuroactive ligand-receptor interaction | 7 | 2.6E-2 |
| hsa04916 | Melanogenesis | 4 | 4.9E-2 |
| hsa04930 | Type II diabetes mellitus | 3 | 5.9E-2 |
For hypermethylation-low expression genes, enriched biological processes included neuromuscular process controlling balance, response to lipopolysaccharide, negative regulation of cytokine-mediated signaling pathway, signal transduction and positive regulation of cytosolic calcium ion concentration. Molecular function enrichment indicated extracellular matrix structural constituent, heme binding, oxygen binding, peroxidase activity and serine-type peptidase activity. Furthermore, cell component displayed plasma membrane and extracellular space.
KEGG pathway enrichment analysis indicated that significant pathways of hypomethylation-high expression genes included cell cycle, pentose and glucuronate interconversions, and p53 signaling pathway, pathways in cancers and PI3K-Akt signaling pathway. Hypermethylation-low expression genes indicated enrichment in chemical carcinogenesis, cGMP-PKG signaling pathway, calcium signaling pathway, PI3K-Akt signaling pathway and AMPK signaling pathway.
PPI network construction, module analysis and hub genes selection
PPI network of hypomethylation-high expression genes was constructed by the database of STRING and visualized in Cytoscape software (Figure 2A). A significant module was obtained from PPI network by using MCODE in Cytoscape software, including 11 nodes and 55 edges (Figure 2B). The hub genes included CDC20, AURKB, BIRC5, RRM2, MCM2, PTTG1, CDKN2A, NEK2, CENPF, CDCA5 and RACGAP1.
Protein-protein interaction (PPI) networks and a core module of PPI network. (A) Hypomethylation and high expression genes; (B) Hypermethylation and low expression genes. Gene expression and methylation status of hub genes between HCC samples and adjacent normal samples were further validated in database TCGA, showed in Figure 5, Figure 6.
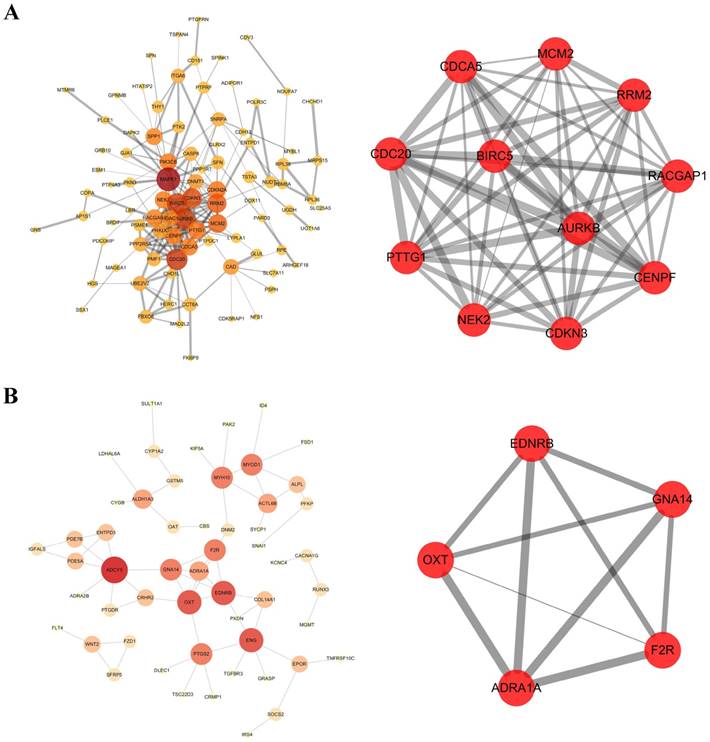
PPI network and the core module including 5 nodes and 10 edges of hypermethylation-low expression genes were displayed (Figure 2C, Figure 2D). The hub gene was GNA14.
The Kaplan-Meier analysis
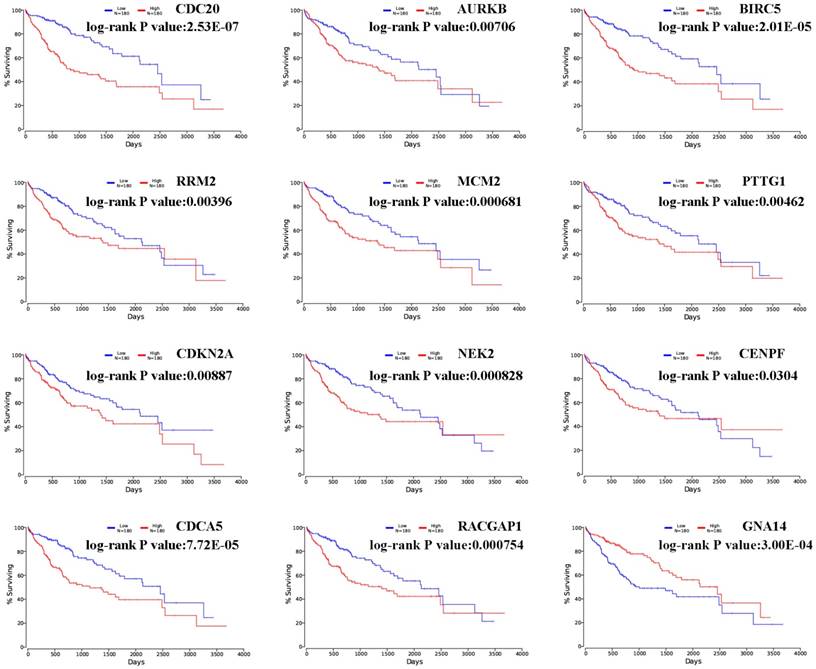
The prognostic values of hub genes in PPI network were obtained in OncoLnc and displayed in Figure 3 [19]. Overall survival for HCC patients was acquired in the accordance with the low or high expression of each gene. It was found that high mRNA expression of CDC20 (P= 2.53E-07) was associated with worse overall survival for HCC, as well as AURKB (P=0.00706), BIRC5 (P= 2.01E-05), RRM2 (P=0.00396), MCM2 (P=0.000681), PTTG1 (P=0.00462), CDKN2A (P=0.00887), NEK2 (P=0.000828), CENPF (P=0.0304), CDCA5 (P=7.72E-05), RACGAP1 (P= 0.000754). Meanwhile, it was found that low mRNA expression of GNA14 (P= 3.00E-04) was associated with worse overall survival for HCC.
Overall survivals of significant hub genes selected from aberrantly methylated-DEGs. Hub genes included CDC20, AURKB, BIRC5, RRM2, MCM2, PTTG1, CDKN2A, NEK2, CENPF, RACGAP1, CDCA5 and GNA14. P < 0.05 was considered statistically significant.
Verification of the hub genes in TCGA database
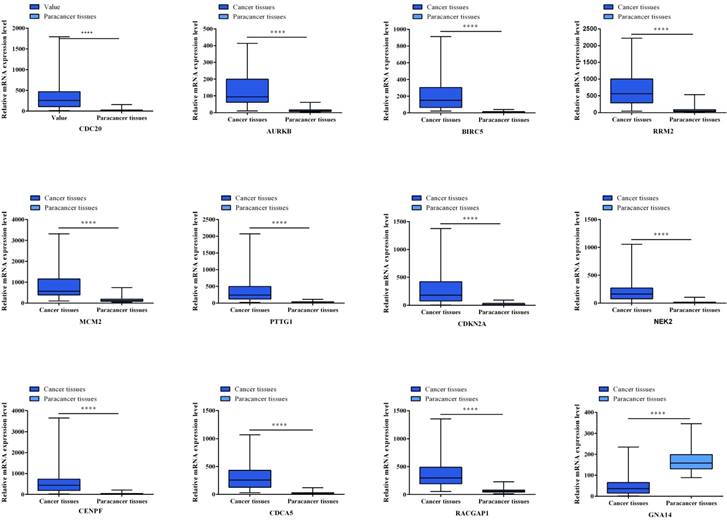
Twelve hub genes from aberrantly methylated- DMGs were validated in database TCGA to compare gene expression and methylation status between HCC samples and adjacent normal samples (Figure 5, Figure 6). Final significant genes consistent with the present study include CDC20, AURKB, RRM2, PTTG1, CDKN2A, NEK2, CENPF, CDCA5 and GNA14. Among them, Cell Division Cycle Associated 5 (CDCA5) was not described in HCC yet. Therefore, we further verified the expression of CDCA5 and study its function in the development of HCC.
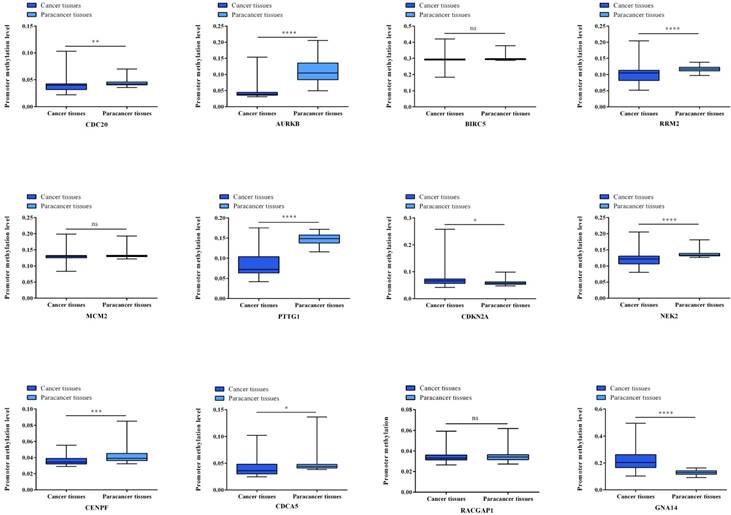
Gene expression status of 12 hub genes were validated in TCGA database. The results are expressed as the mean ± SD of 50 paired HCC samples and adjacent normal liver samples. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Gene methylation status of 12 hub genes were validated in TCGA database. The results are expressed as the mean ± SD of 50 paired HCC samples and adjacent normal liver samples. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001
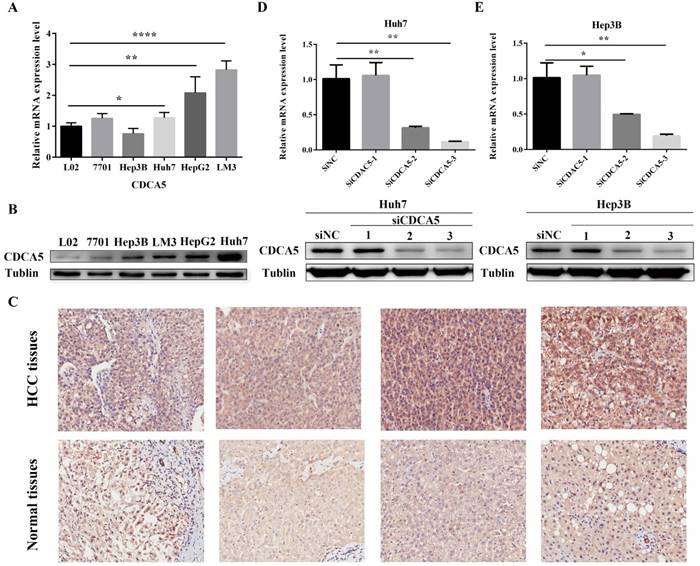
Upregulation of hepatic CDCA5 expression in HCC cell lines and patients with HCC and knockdown of CDCA5 in Huh7 and Hep3B. (A) Expression of CDCA5 mRNA in 4 human HCC cell lines and 2 normal human hepatic cells was evaluated by qRT-PCR. (B) Western blots show the CDCA5 protein level compared to Tubulin in HCC cell lines and normal hepatic cells. (C) Representative images of immunohistochemistry staining of liver sections of HCC tissues (n=81) and paired adjacent normal tissues (n=81). Original magnification, ×200. Upper panel, HCC tissues; Lower panel, adjacent normal tissues. (D and E) Western blots and qRT-PCR showing the effect of RNAi on CDCA5 expression. There different siCDCA5s at 50 nM were transfected into human HCC cell lines Huh7 and Hep3B using Lipofectamine 3000. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
CDCA5 is upregulated in human HCC cells and patients with HCC
We performed qRT-PCR to detect the expression of CDCA5 in 4 human HCC cells (HePG2, Hep3B, Huh7 and LM3) and 2 normal human hepatic cells (QSG-7701 and L-02). High levels of CDCA5 mRNA were observed in HCC cells compared with normal hepatic cells (Figure 6A). In addition, we confirmed high levels of CDAC5 protein expression by western blotting in HCC cells (Figure 6B). Finally, as is shown in Figure 6C, immunohistochemistry experiments revealed markedly higher CDCA5 protein expression from HCC tumor tissues compared with paracancer tissues , which was further semiquantitative analyzed by H-score (Z=-5.727, P<0.001).
CDCA5 promotes HCC cell proliferation by regulating the cell cycle
To investigate the role of CDAC5 in HCC, we transfected HCC cells (Huh7 and Hep3B) with siRNA specific for CDCA5 (siCDCA5) at a concentration of 50 nM. As illustrated in Figure 6D and Figure 6E, hepatic CDCA5 expression was remarkably downregulated in Huh7 and Hep3B cells by siCDCA5-2 or siCDAC5-3, but not siCDAC5-1. Moreover, we found that CDCA5 knockdown signifcantly inhibited cell growth compared to an untargeted siNC (Figure 7A).
CDCA5 promotes HCC cell proliferation by inducing G2 phase cell cycle arrest in HCC cells. (A) Influence of CDCA5 knockdown on vitality of Huh7 and Hep3B cells was measured by CCK8 assay. (B) Huh7 and Hep3B cells were transfected with siCDCA5s or siNC for 48h and analyzed by flow cytometry. Numbers of cell populations at different stages of cell cycles are listed within the panels. Histograms show the percentage (%) of cells in different stages of cell cycles from three independent experiments. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
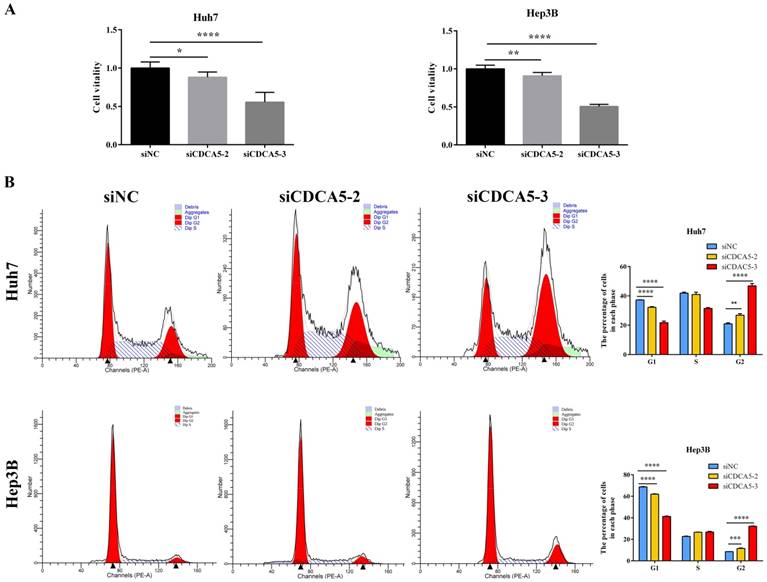
CDCA5 has been reported playing an important role in the cell cycle [20, 21]. Therefore, we performed flow cytometry to analyze the effect of CDCA5 on the cell cycle using Huh7 and Hep3B cells. CDCA5 silencing significantly reduced the cells in G0/G1 phase, whereas it increased the cells in G2 phase (Figure 7B). The data demonstrated that the anti-proliferative effect of siCDCA5 was on account of G2 phase cell cycle arrest in HCC.
Discussion
Despite advances in surgical and medicine treatment, the mortality of HCC has still increased in recent years [2]. Therefore, elucidating the etiological factors and mechanisms of the initiation and progression of HCC are essential for diagnosis, treatment and prognosis assessment. Recently, Microarray technology has been developing rapidly and it enables us to explore the genetic and epigenetic alterations, among which finding the new biomarkers in the development of HCC.
In the present study, a total of 159 hypomethylation-high expression genes and 135 hypermethylation-low expression genes were obtained by analyzing two mRNA microarray datasets (GSE62232, GSE74656) and two gene methylation microarrays (GSE44909, GSE57958). Functional and pathway enrichment analysis were performed to screen hub genes and pathways, which may provide novel insights for revealing pathogenesis of HCC.
As was demonstrated in DAVID, hypomethylation-high expression genes were involved in biological processes (BP) of mitotic nuclear division, sister chromatid cohesion, chromosome segregation, positive regulation of telomere maintenance via telomerase and apoptotic process and cell cycle. Previous studies have indicated that loss of chromatid cohesion and dysregulation of cell cycle progression including mitotic nuclear division, chromosome segregation and telomere regulation are obvious hallmarks of cancers [22-24]. KEGG pathway enrichment showed cell cycle, pentose and glucuronate interconversions, and p53 signaling pathway. It was believed that glucose metabolism was involved in anti-cancer drugs resistance and cells proliferation [25]. The dysregulation of P53 signaling pathway was already reported in many researches and consistent with the present work [26].
PPI network of hypomethylation-high expression genes displayed the functional interactions, in which hub genes were selected. Following, survival analysis of these genes demonstrated that all these eleven genes significantly worsen overall survival of patients with HCC, including CDC20, AURKB, BIRC5, RRM2, MCM2, PTTG1, CDKN2A, NEK2, CENPF, CDCA5 and RACGAP1. Finally, after validation in database TCGA to compare gene expression and methylation status between HCC samples and normal samples, CDC20, AURKB, RRM2, PTTG1, CDKN2A, NEK2, CENPF, CDCA5 were still significant. CDC20 has been proved to be over-expressed in various malignant tumors such as cancers of bladder, breast, colonic and liver [27]. Additionally, increased CDC20 expression was associated with development and progression of HCC [28]. It was reported that AURKB was overexpressed in HCC and may serve as biomarkers for diagnosis, treatment and prognosis of HCC [29]. High expression of RRM2 was significantly correlated with early recurrence and may be a biomarker for poor prognosis of HCC after surgery [30]. There was research demonstrating that the PTTG1 gene was up-regulated in the tissues from patients with HCC and involved in the progression of HCC [31]. CDKN2A was proved hypermythelated in the early stage of hepatitis B virus-associated hepatocarcinogenesis [32]. A study showed that NEK2 mediated drug resistance, liver cancer cell migration, angiogenesis via expression of an ABCC10 transporter, pAKT/NF-κB signaling and matrix metalloproteinase (MMP) activation, and IL-8 stimulation, respectively [33]. It was suggested that amplified CDK13, GMNN, and CENPF genes can function as common cancer-driver genes in HCC [34].
There was evidence that sister chromatids cohesion was mediated by cohesion complexes, consisting of the ATPases Smc1, Smc3, the kleisin Scc1/Rad21/Mcd1 and stromal antigen (SA) in vertebrates. Scc1 and SA proteins are further connected with a heterodimer of two proteins, named Wapl and Pds5, the latter of which exists in two subtypes invertebrates (Pds5A and Pds5B) [35]. It was proved that Sororin, encoded by CDCA5, is recruited to chromatin-bound cohesin complexes induced by DNA replication and Smc3 acetylation during S phage. Following, Sororin displaces Wapl from Pds5 within cohesin causing a conformational rearrangement, and that these molecular processes stabilize cohesin on DNA via antagonizing Wapl's function of dissociating cohesin from DNA [20]. Recently, more and more researches indicated pathogenic effect of CDCA5 in a variety of cancers, such as human lung carcinoma and oral squamous cell carcinoma [36, 37].
In this study, we have demonstrated the overexpression of CDCA5 in HCC cell lines. Furthermore, we have demonstrated that targeting CDCA5 using small interfering RNA (siRNA) inhibited the growth of human OSCC cells by inducing the G2 phase cell cycle arrest. These results raise the possibility that CDCA5 is a novel therapeutic target for HCC.
As for hypermethylation-low expression genes, functional analysis enriched in biological processes of neuromuscular process controlling balance, response to lipopolysaccharide, negative regulation of cytokine-mediated signaling pathway, signal transduction and positive regulation of cytosolic calcium ion concentration. A recent research showed that the lipopolysaccharide (LPS) response in healthy human PBMCs has been linked to cytokine response and implied that LPS negative responders may have unfavorable outcomes if afflicted with cancer [38]. Besides, dysregulation of cytokines correlated with invasiveness and metastasis of cancer cells [39]. Cytosolic calcium ion concentration has been widely reported in several types of cancer and played critical roles in cancer development by influencing proliferative signaling and invasive pathways [40]. Molecular function mainly enriched in extracellular matrix structural constituent, which was consistent with the theory that tumor phenotypes were influenced by molecular cross-talk between tumor cells and extracellular structures [41]. KEGG enriched in chemical carcinogenesis, cGMP-PKG signaling pathway, calcium signaling pathway, PI3K-Akt signaling pathway and AMPK signaling pathway. These results implied that cGMP-PKG signaling pathway might be a candidate study target for understanding the mechanism of HCC and chemical angiogenesis inhibitors were promising therapeutic methods for HCC [42].
After constructing PPI network, the hub gene was selected and followed with survival analysis, validation in database TCGA as above; GNA14 was still significant. There was evidence that GNA14 showed deregulated methylation in adjacent normal tissue but upregulated methylation in HCC [43].
In conclusion, the present study was designed to identify aberrantly methylated-DEGs that may be involved in the carcinogenesis or progression of HCC. A total of 12 hub genes were selected and CDCA5 may be regarded as new biomarkers for diagnosis, treatment and prognosis in HCC, so that targeting CDCA5 could be a potentially useful therapeutic approach for patients with HCC.
Abbreviations
HCC: hepatocellular carcinoma; DEGs: differentially expressed genes; DMGs: differentially methylated genes; GEO: gene expression omnibus; DAVID: database for annotation, visualization and integrated discovery; GO: gene ontology; BP: biological processses; MF: molecular functions; CC: cell components; KEGG: kyoto encyclopedia of genes and genomes; PPI: protein-protein interaction; STRING: search tool for the retrieval of interacting genes; MCODE: molecular complex detection; TCGA: the cancer genome atlas; siRNA: small interfering RNA; H-score: histochemical score; qRT-PCR: quantitative real-time PCR; CDC20: cell division cycle 20; AURKB: aurora kinase B; BIRC5: baculoviral IAP repeat containing 5; RRM2: ribonucleotide reductase regulatory subunit M2; MCM2: minichromosome maintenance complex component 2; PTTG1: pituitary tumor-transforming 1; CDKN2A: cyclin dependent kinase inhibitor 2A; NEK2: cyclin dependent kinase inhibitor 2A; CENPF: centromere protein F; CDCA5: cell division cycle associated 5; RACGAP1: RAC GTPase activating protein 1; GNA14: G protein subunit alpha 14.
Acknowledgements
This work was supported by the program of Shenzhen Science Technology an Innovation Committee (SYS201504301534057), the program of Shenzhen SanMing Project (SZSM201412006). The funders did not play any role in the study design, data collection and analysis, decisions regarding data release or manuscript preparation.
Author Contributions
CC designed and performed the study, analyzed the data and wrote the paper. WW and ZT designed the study and critically revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Berger E, Vega N, Vidal H, Geloen A. Gene network analysis leads to functional validation of pathways linked to cancer cell growth and survival. Biotechnology journal. 2012;7:1395-404
2. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557-76
3. Cancer Genome Atlas Research Network. Electronic address wbe, Cancer Genome Atlas Research N. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169:1327-41 e23
4. Momparler RL. Cancer epigenetics. Oncogene. 2003;22:6479-83
5. Wei L, Chiu DK, Tsang FH, Law CT, Cheng CL, Au SL. et al. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J Hepatol. 2017;67:758-69
6. Nishida N, Nagasaka T, Nishimura T, Ikai I, Boland CR, Goel A. Aberrant methylation of multiple tumor suppressor genes in aging liver, chronic hepatitis, and hepatocellular carcinoma. Hepatology. 2008;47:908-18
7. Iizuka N, Oka M, Yamada-Okabe H, Nishida M, Maeda Y, Mori N. et al. Oligonucleotide microarray for prediction of early intrahepatic recurrence of hepatocellular carcinoma after curative resection. The Lancet. 2003;361:923-9
8. Li L, Lei Q, Zhang S, Kong L, Qin B. Screening and identification of key biomarkers in hepatocellular carcinoma: Evidence from bioinformatic analysis. Oncol Rep. 2017
9. Lin YH, Wu MH, Huang YH, Yeh CT, Cheng ML, Chi HC. et al. Taurine upregulated gene 1 functions as a master regulator to coordinate glycolysis and metastasis in hepatocellular carcinoma. Hepatology. 2017
10. Villanueva A, Portela A, Sayols S, Battiston C, Hoshida Y, Mendez-Gonzalez J. et al. DNA methylation-based prognosis and epidrivers in hepatocellular carcinoma. Hepatology. 2015;61:1945-56
11. Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S. et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nature genetics. 2015;47:505-11
12. Mah WC, Thurnherr T, Chow PK, Chung AY, Ooi LL, Toh HC. et al. Methylation profiles reveal distinct subgroup of hepatocellular carcinoma patients with poor prognosis. PLoS One. 2014;9:e104158
13. Revill K, Wang T, Lachenmayer A, Kojima K, Harrington A, Li J. et al. Genome-wide methylation analysis and epigenetic unmasking identify tumor suppressor genes in hepatocellular carcinoma. Gastroenterology. 2013;145:1424-35 e1-25
14. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M. et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 2013;41:D991-5
15. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44-57
16. Du J, Yuan Z, Ma Z, Song J, Xie X, Chen Y. KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Molecular bioSystems. 2014;10:2441-7
17. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498-504
18. Li M, Wu X, Pan Y, Wang J. hF-measure: A new measurement for evaluating clusters in protein-protein interaction networks. Proteomics. 2013;13:291-300
19. Anaya J. OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Computer Science. 2016;2:e67
20. Nishiyama T, Ladurner R, Schmitz J, Kreidl E, Schleiffer A, Bhaskara V. et al. Sororin mediates sister chromatid cohesion by antagonizing Wapl. Cell. 2010;143:737-49
21. Schmitz J, Watrin E, Lenart P, Mechtler K, Peters JM. Sororin is required for stable binding of cohesin to chromatin and for sister chromatid cohesion in interphase. Current biology: CB. 2007;17:630-6
22. Marchese FP, Grossi E, Marin-Bejar O, Bharti SK, Raimondi I, Gonzalez J. et al. A Long Noncoding RNA Regulates Sister Chromatid Cohesion. Mol Cell. 2016;63:397-407
23. Hydbring P, Wang Y, Fassl A, Li X, Matia V, Otto T. et al. Cell-Cycle-Targeting MicroRNAs as Therapeutic Tools against Refractory Cancers. Cancer cell. 2017;31:576-90 e8
24. de Lange J, Faramarz A, Oostra AB, de Menezes RX, van der Meulen IH, Rooimans MA. et al. Defective sister chromatid cohesion is synthetically lethal with impaired APC/C function. Nat Commun. 2015;6:8399
25. Saito T, Ichimura Y, Taguchi K, Suzuki T, Mizushima T, Takagi K. et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun. 2016;7:12030
26. Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. Journal of Hepatology. 2016;64:S84-S101
27. Gayyed MF, El-Maqsoud NM, Tawfiek ER, El Gelany SA, Rahman MF. A comprehensive analysis of CDC20 overexpression in common malignant tumors from multiple organs: its correlation with tumor grade and stage. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37:749-62
28. Li J, Gao JZ, Du JL, Huang ZX, Wei LX. Increased CDC20 expression is associated with development and progression of hepatocellular carcinoma. International journal of oncology. 2014;45:1547-55
29. Lin ZZ, Jeng YM, Hu FC, Pan HW, Tsao HW, Lai PL. et al. Significance of Aurora B overexpression in hepatocellular carcinoma. Aurora B Overexpression in HCC. BMC cancer. 2010;10:461
30. Lee B, Ha SY, Song DH, Lee HW, Cho SY, Park CK. High expression of ribonucleotide reductase subunit M2 correlates with poor prognosis of hepatocellular carcinoma. Gut and liver. 2014;8:662-8
31. Liang M, Chen X, Liu W, Li S, Li C, Jiang L. et al. Role of the pituitary tumor transforming gene 1 in the progression of hepatocellular carcinoma. Cancer biology & therapy. 2011;11:337-45
32. Hu H, Li Z, Chen J, Wang D, Ma J, Wang W. et al. P16 reactivation induces anoikis and exhibits antitumour potency by downregulating Akt/survivin signalling in hepatocellular carcinoma cells. Gut. 2011;60:710-21
33. Wu SM, Lin SL, Lee KY, Chuang HC, Feng PH, Cheng WL. et al. Hepatoma cell functions modulated by NEK2 are associated with liver cancer progression. International journal of cancer. 2017;140:1581-96
34. Kim HE, Kim DG, Lee KJ, Son JG, Song MY, Park YM. et al. Frequent amplification of CENPF, GMNN and CDK13 genes in hepatocellular carcinomas. PLoS One. 2012;7:e43223
35. Gandhi R, Gillespie PJ, Hirano T. Human Wapl is a cohesin-binding protein that promotes sister-chromatid resolution in mitotic prophase. Current biology: CB. 2006;16:2406-17
36. Nguyen MH, Koinuma J, Ueda K, Ito T, Tsuchiya E, Nakamura Y. et al. Phosphorylation and activation of cell division cycle associated 5 by mitogen-activated protein kinase play a crucial role in human lung carcinogenesis. Cancer Res. 2010;70:5337-47
37. Tokuzen N, Nakashiro K, Tanaka H, Iwamoto K, Hamakawa H. Therapeutic potential of targeting cell division cycle associated 5 for oral squamous cell carcinoma. Oncotarget. 2016;7:2343-53
38. Buscher K, Ehinger E, Gupta P, Pramod AB, Wolf D, Tweet G. et al. Natural variation of macrophage activation as disease-relevant phenotype predictive of inflammation and cancer survival. Nat Commun. 2017;8:16041
39. Stender JD, Nwachukwu JC, Kastrati I, Kim Y, Strid T, Yakir M. et al. Structural and Molecular Mechanisms of Cytokine-Mediated Endocrine Resistance in Human Breast Cancer Cells. Mol Cell. 2017;65:1122-35 e5
40. Monteith GR, Prevarskaya N, Roberts-Thomson SJ. The calcium-cancer signalling nexus. Nat Rev Cancer. 2017;17:367-80
41. Matter MS, Marquardt JU, Andersen JB, Quintavalle C, Korokhov N, Stauffer JK. et al. Oncogenic driver genes and the inflammatory microenvironment dictate liver tumor phenotype. Hepatology. 2016;63:1888-99
42. Raoul JL, Gilabert M, Adhoute X, Edeline J. An in-depth review of chemical angiogenesis inhibitors for treating hepatocellular carcinoma. Expert opinion on pharmacotherapy. 2017;18:1467-76
43. Gao W, Kondo Y, Shen L, Shimizu Y, Sano T, Yamao K. et al. Variable DNA methylation patterns associated with progression of disease in hepatocellular carcinomas. Carcinogenesis. 2008;29:1901-10
Author contact
![]() Corresponding author: Associate Prof. Zhenhua Tu, Ward of Liver transplant, Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery. First Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou 310003, China. E-mail: richardtucom; Tel/Fax: +86-571-87236739
Corresponding author: Associate Prof. Zhenhua Tu, Ward of Liver transplant, Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery. First Affiliated Hospital of Zhejiang University School of Medicine, Hangzhou 310003, China. E-mail: richardtucom; Tel/Fax: +86-571-87236739