Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(4):853-863. doi:10.7150/jca.27803 This issue Cite
Research Paper
Comprehensive Analysis of Aberrantly Expressed ceRNA network in gastric cancer with and without H.pylori infection
Aining Chu, Jingwei Liu, Yuan Yuan ![]() , Yuehua Gong
, Yuehua Gong ![]()
1. Tumor Etiology and Screening Department of Cancer Institute and General Surgery, the First Hospital of China Medical University, Shenyang 110001, China
2. Key Laboratory of Cancer Etiology and Prevention in Liaoning Education Department, the First Hospital of China Medical University, Shenyang 110001, China
3. Key Laboratory of GI Cancer Etiology and Prevention in Liaoning Province, the First Hospital of China Medical University, Shenyang 110001, China
Received 2018-6-11; Accepted 2018-12-14; Published 2019-1-29
Abstract
Objective: This study mainly focused on revealing ceRNA network in gastric cancer (GC) with Hp infection after comparing with GC without Hp infection and exploring the biological function and prognostic relevance of related molecules.
Methods: The RNA expression profile data of GC patients with or without Hp infection were extracted from TCGA GDC data portal, including 20 GC cases with Hp infection and 168 GC cases without Hp infection. Differentially expressed lncRNAs, miRNAs and mRNAs were unveiled by package edgeR of R, and lncRNA-miRNA-mRNA ceRNA network was constructed by integrating the miRNA target information and the expression data of lncRNAs, miRNAs and mRNAs. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of aberrantly expressed mRNAs were performed to identify the related biological functions and pathologic pathways, and protein-protein interaction (PPI) network was constructed by STRING database. The overall survival (OS) of aberrantly expression lncRNAs and miRNAs were analyzed by package survival of R. A total of 30 gastric cancer tissues were used to validate the bioinformatics analysis results by real-time PCR.
Results: Among the 32 differentially expressed miRNAs, 27 differentially expressed lncRNAs and 257 differentially expressed mRNAs were identified by comparing GC patients with and without Hp infection. Totally 10 miRNA, 11 lncRNA, 219 mRNA were included to build ceRNA network. GO and KEGG analysis revealed that differentially expressed mRNAs involved in the ceRNA network were mainly involved in extracellular exosomes, structural molecular activities, proteolysis and P13K-Akt signaling pathways. And PPI analysis obtained six hub genes of NTS, APOC3, OTX2, KRT13, CALCA, GNG4. Survival analysis showed that four lncRNAs (LINC01254, LINC01287, LINC01524, U95743.1) and four miRNAs (miR-302a, miR-302b, miR-1286, miR-378g) were associated with overall survival of GC with Hp infection. The real-time PCR results showed that, the levels of LINCO1254, LINCO1287, LINCO1524, U95743.1 were significantly higher in Hp positive GC patients than Hp negative patients (P=0.02, 0.048, 0.04, 0.036, respectively).
Conclusion: Using TCGA database for data mining, we have successfully constructed a ceRNA regulatory network of GC with Hp infection, consisting of 10 lncRNAs, 11 miRNAs and 219 mRNAs. These findings might provide critical clues for the regulatory role of ceRNA network in the development of GC with Hp infection.
Keywords: H. pylori, gastric cancer, ceRNA network, miRNA, lncRNA
Introduction
Gastric cancer (GC) is the fourth most common cancer nowadays as well as the second leading cause of cancer-deaths worldwide [1, 2]. Helicobacter pylori (H. pylori,Hp) infection is closely associated with GC [3]. However, the pathogenesis of GC with Hp infection is still not fully understood.
Post-transcriptional regulation of non-coding RNA plays an important role in the development of cancer, which has become a research focus involved in the pathogenesis of GC, including Hp-related GC [4]. A large number of studies have explored miRNA and lncRNA expression in Hp positive GC, suggesting that different pathogenesis mechanisms could exist between Hp-positive and Hp-negative GC [5, 6]. Recently, Salmena et al. (2011) put forward the hypothesis of competing endogenous RNA (ceRNA), which triggered a hot topic in the research field [7]. At present, the regulation of ceRNA network in tumor development has been found in colon cancer, ovarian cancer, and also GC [8-10]. However, the Hp related non-coding RNA regulation in GC is still at elementary stage and relevant ceRNA regulation in GC with and without Hp infection remains largely unclear.
Online public databases provide an available resource platform for data mining and research exploration. In this study, using online bioinformatics resources, we explored ceRNA regulatory network of GC with and without Hp infection and further detected relevant functional pathway and potential prognostic value of differential expressed genes based on ceRNA network. Meanwhile, we used real time-PCR validation for some of these bioinformatics analysis results in gastric cancer tissue with or without Hp infection. Our research might expend the understanding of the molecular mechanism of Hp positive GC.
Materials and methods
Acquisition and selection of datasets
A total of 443 patients with GC were collected from the Cancer Genome Atlas (TCGA) database (https://gdc-portal.nci.nih.gov/). According to the clinical data, patients without Hp information were excluded. A total of 188 patients were extracted from 443 data, including 20 Hp positive patients and 168 Hp negative patients. And the clinical parameters of these 188 cases were listed in supplementary file Table S1. The RNA expression profile data of the corresponding patients were downloaded from TCGA GDC data portal. Our study was in accordance with the publication guidelines provided by TCGA.
Identification of DEGs, DE-lncRNAs, DE-miRNAs
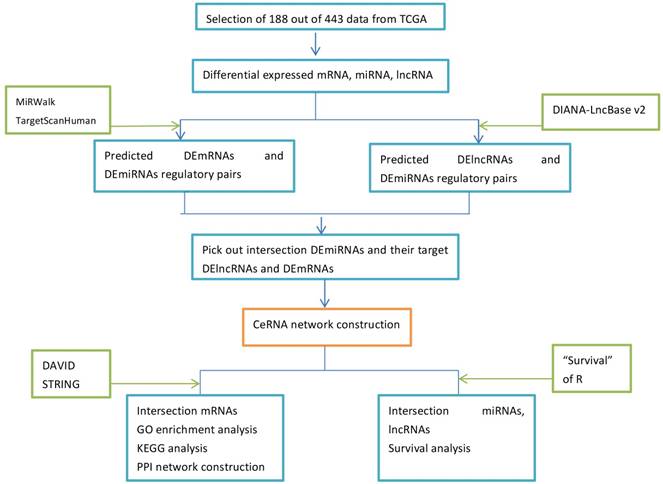
Empirical Analysis of Digital Gene Expression Data in R (edger) package (http://bioconductor.org/packages/release/bioc/html/edgeR.html) were applied to screen out differentially expressed lncRNAs, mRNAs and miRNAs between GC with Hp infection and GC without Hp infection, using P<0.01, |fold change|>1 (including up-regulation and down-regulation) for miRNAs and P<0.01, |fold change|>2.5 for mRNA as cut-offs. Then we used BIOMART (www.ensembl.org/biomart) to define and annotate the differentially expressed lncRNAs (DElncRNAs) from DERNAs. Fig. 1 depicted the flow diagram of bioinformatics analysis.
The flow of the bioinformatics analysis.
Construction of ceRNA network
We predicted DEmRNAs and DEmiRNA regulatory relationship through miRWalk 3.0 (http://mirwalk.umm.uni-heidelberg.de/) and TargetScanHuman 7.1 (http://www.targetscan.org/vert_71/), DEmiRNAs and DElncRNAs regulatory relationship through DIANA-LncBase v2 (http://www.microrna.gr/LncBase). Then we combined the results with differentially expressed data of TCGA to pick out the intersection mRNAs and lncRNAs. Next, we selected the intersectant DElncRNAs and DEmiRNAs which possess opposite expression trends, as well as DEmiRNAs and DEmRNAs. By this means, DElncRNAs and DEmRNAs regulated by the same DEmiRNAs were filtered out. The selected genes were chosen to construct the ceRNA network and visualized by Cytoscape software (http://cytoscape.org/) [11].
Functional enrichment analysis
We used the overlapping differentially expressed RNA to conduct the functional enrichment analysis on Database for Annotation, Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/), including GO annotation and KEGG pathway analysis [12]. And GO annotation contained biological process, molecular function and cellular component.
Protein-protein interaction (PPI) network construction
The Retrieval of Interacting Genes (STRING) database tool (string-db.org) was used to figure out the interactive relationships of DEGs. The interacting pairs with confidence score more than 0.4 were considered as significant and retained. Based on STRING results, PPI network was visualized utilizing Cytoscape software.
Survival analysis
To identity the possible relationship of DElncRNAs and DEmiRNAs with GC prognosis, package “survival” of R was conducted to test the prognostic value, with clinical data of TCGA combined. This test is based on the Kaplan-Meier method and P<0.05 would be considered as significant.
Real-time PCR validation of DElncRNAs
A total of 30 patients, including 23 men and 7 women, mean age of 62± 9 years old, with gastric cancer were enrolled for the validation, from whom cancerous gastric tissues were collected under gastrectomy at the First Affiliated Hospital of China Medical University. Total RNA was extracted using RNAiso Plus (Takara, Japan) and converted to cDNA using a PrimeScript RT reagent kit (Takara, Japan). The amplification was used with SYBR Green (SYBR Premix Ex Taq II, Takara, USA) in a fluorescence reader ABI Prism 7500. Each reaction was normalized to β-actin. All the primers are shown in supplementary table 2. Fasting venous blood was collected from each participant and the status of Hp infection was determined by ASSURE H.Pylori Rapid Test (MP bio, USA). Among the 30 GC patients, 11 cases were without Hp infection and 19 ones with Hp infection. This study was approved by the Ethics Committee of the First Affiliated Hospital of China Medical University Shenyang, China. Written informed consent was obtained from the participants.
Statistical analysis
All data were shown as mean ± SD. The fold change value and P-value were used to evaluate the differences of lncRNAs, miRNAs and mRNAs expression between GC tissues with and without Hp infection. False discovery rate (FDR) was calculated to correct the P-value. Student's t tests were used to analyze the differences of real-time PCR results. P<0.05 was considered as statistically significant.
Results
Identification of differentially expressed lncRNAs, miRNAs, mRNAs
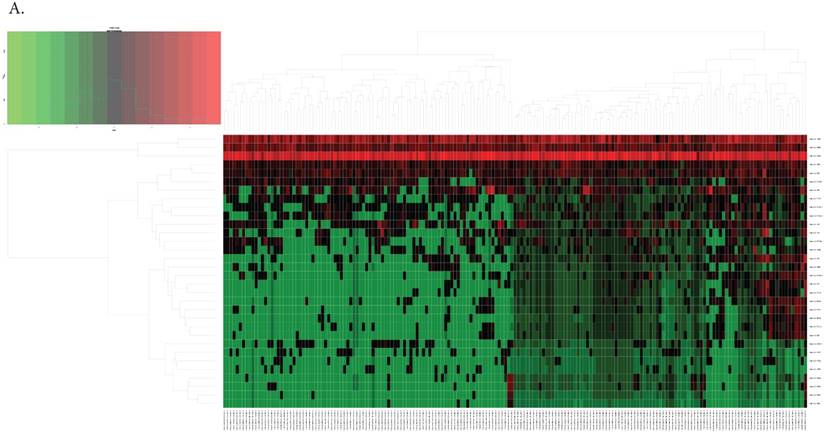
By differential analysis, we identified 32 DEmiRNAs and 462 DERNAs from miRNA-seq and RNA-seq of TCGA (The heat maps of them were shown in Fig. 2A and Fig. 2B). Twenty-one out of 32 DEmiRNAs were down-regulated and 11 were up-regulated. Excluding those RNAs that could not be defined or annotated by BIOMART, there were 27 DElncRNAs (24 DElncRNAs down-regulated and 3 up-regulated) and 257 DEmRNAs screened out. Top ten of DEmiRNAs and DElncRNAs were listed in Table 1 and 2, respectively.
The heat maps of the expression of DEmiRNAs(A) and DEmRNAs, DElncRNAs(B).

Top 10 differential expressed miRNAs
| Gene (miRNA) | Log FC | P Value | FDR |
|---|---|---|---|
| hsa-mir-302b | 4.800417487 | 2.11E-12 | 3.15E-09 |
| hsa-mir-302a | 4.73040372 | 4.65E-12 | 3.47E-09 |
| hsa-mir-302d | 4.153060037 | 1.52E-09 | 7.59E-07 |
| hsa-mir-302c | 2.544240507 | 1.66E-05 | 0.006186252 |
| hsa-mir-6715a | 2.059531155 | 6.76E-05 | 0.016809501 |
| hsa-mir-372 | -7.359051955 | 0.000100736 | 0.021485649 |
| hsa-mir-122 | -5.867951592 | 0.000367743 | 0.065727522 |
| hsa-mir-203a | -1.296017557 | 0.000420684 | 0.065727522 |
| hsa-mir-184 | -3.183847141 | 0.001012429 | 0.116273608 |
| hsa-mir-373 | -6.306192354 | 0.00114065 | 0.120854295 |
Top 10 differential expressed lncRNAs
| Gene (lncRNA) | Transcript ID | Log FC | P Value | FDR |
|---|---|---|---|---|
| OVAAL | ENST00000442621 | 7.016856432 | 1.29E-28 | 1.36E-24 |
| LINC00922 | ENST00000569736 | 4.112562259 | 6.05E-26 | 5.13E-22 |
| MYCNUT | ENST00000433810 | 4.475819305 | 3.08E-11 | 3.96E-08 |
| LINC01287 | ENST00000454441 | -5.141133564 | 2.65E-05 | 0.006442655 |
| LINC01254 | ENST00000567609 | -4.212536271 | 0.000106153 | 0.017152222 |
| LINC00645 | ENST00000557359 | -4.448490725 | 0.000310739 | 0.03564988 |
| U95743.1 | ENST00000573369 | -3.964689472 | 0.000395919 | 0.040290509 |
| LINC01361 | ENST00000452834 | -3.079983301 | 0.000474201 | 0.044370481 |
| LINC01524 | ENST00000454600 | -3.257166236 | 0.000892848 | 0.062906349 |
| AC005307.1 | ENST00000567877 | -3.505146705 | 0.001012023 | 0.067363162 |
Establishment of ceRNAs regulatory network
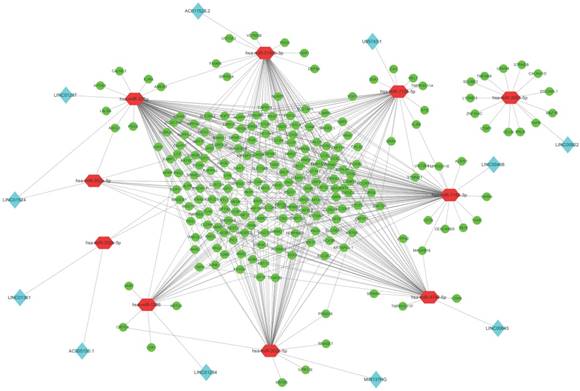
Based on the theory of ceRNA network, we built the regulation loops with lncRNAs-miRNAs and miRNAs-mRNAs regulation pairs. In order to establish lncRNA-miRNA-mRNA network, we focused on whether those DEmiRNAs had interaction with DElncRNAs and DEmRNAs respectively, which means DEmiRNAs could target on them. After prediction by DIANA-LncBase v2, we found 11 DElncRNAs which might be targeted by 10 DEmiRNAs. Then, through TargetScanHuman and miRWalk, 219 DEmRNAs were predicted to be targets of those 10 common DEmiRNAs.
According to the above results, we chose intersectant 11 DElncRNAs (LINC01361, AC005150.1, LINC01524, MIR137HG, LINC01287, LINC01524, LINC01254, AC011525.2, LINC00408, LINC00645, U95743.1, LINC00922), 10 DEmiRNAs (miR-1286, miR-302b-5p, miR-3150b-3p, miR-7156-3p, miR-302a-5p, miR-378g, miR-3065-5p, miR-302c-5p, miR-7156-5p, miR-4740-5p) and 219 DEmRNAs to build lncRNAs-miRNAs-mRNAs regulatory ceRNA network. And the Cytosacpe was used to draw the network for visualization (Fig. 3).
The differentially expressed lncRNA-miRNA-mRNA regulatory network in gastric cancer with and without Hp infection. Red octagon represents miRNA. Blue diamond represents lncRNA and green ellipse represents mRNA.
The cellular functions and pathway analysis of DEmRNAs involved in ceRNA network
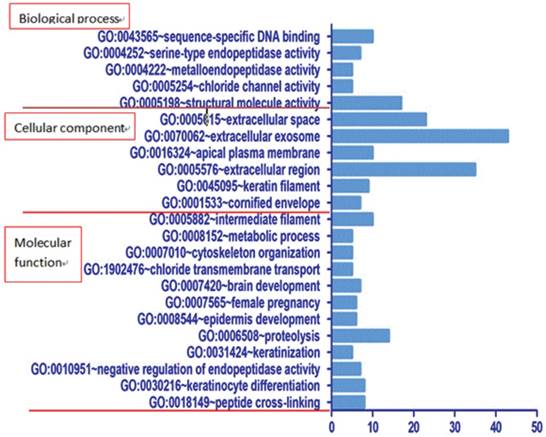
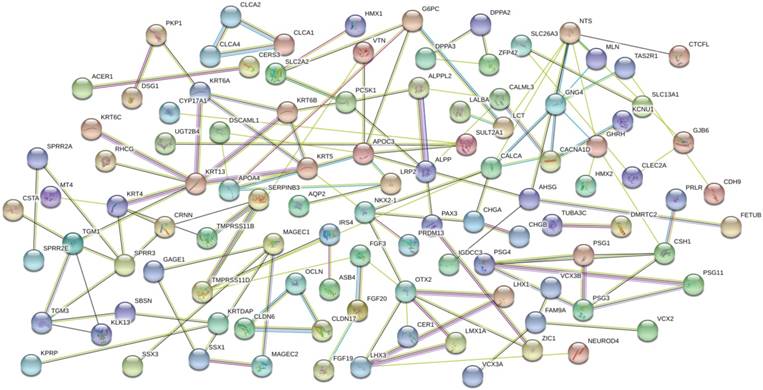
The GO analysis indicated that the 219 differentially expressed mRNAs involved in ceRNA network parcitipated in biological process of sequence-specific DNA binding, serine-type endopeptidase, metalloendopeptidase, chloride channel, and structural molecular. Molecular function enriched these genes in intermediate filament, metabolic process, cytoskeleton organization, chloride transmembrane transport, brain development, female pregnancy, epidemis development, proteolysis, keratinization, negative regulation of endopeptidase activity, keratinocyte differentiation, and peptide cross-linking. In addition, cellular component suggested enrichment mainly at extracellular space, extracellular exosome, apical plasma membrane, extracellular region, keratin filament, and comified envelope (Fig. 4). KEGG pathway enrichment analysis revealed that the most significant pathways were P13K-Akt signaling pathway and renin secretion (shown in Table 3). The PPI network was achieved, containing 109 nodes and 35 edges. According to the network, there were 6 hub genes (NTS, APOC3, OTX2, KRT13, CALCA, GNG4) and the gene with the highest degree (degree=8) was NTS (neurotensin, NTS), which could be seen in Fig. 5.
GO functional enrichment analyse for DEGs involved in the ceRNA network (the bar plot shows the enrichment scores of the significant enrichment pathways), mainly including three components, which are biological process, cellular component and molecular function.
Significantly KEGG pathways for DEGs
| Pathway ID | Description | Genes | P Value | FDR |
|---|---|---|---|---|
| hsa04924 | Renin secretion | CLCA2, CLCA1, CLCA4, CALML3, CACNA1D | 6.30E-04 | 0.70462716 |
| hsa04973 | Carbohydrate digestion and absorption | G6PC, SLC2A2, CACNA1D, LCT | 0.00217633 | 2.41508175 |
| hsa00052 | Galactose metabolism | LALBA, G6PC, LCT | 0.01473533 | 15.3441269 |
| hsa04911 | Insulin secretion | KCNU1, GPR119, SLC2A2, CACNA1D | 0.01556108 | 16.1368598 |
| hsa04151 | PI3K-Akt signaling pathway | FGF19, CSH1, G6PC, PRLR, VTN, GNG4, FGF20 | 0.01894243 | 19.3129631 |
| hsa04972 | Pancreatic secretion | SLC26A3, CLCA2, CLCA1, CLCA4 | 0.01975617 | 20.0607696 |
| hsa04930 | Type II diabetes mellitus | IRS4, SLC2A2, CACNA1D | 0.03561557 | 33.4311427 |
| hsa04917 | Prolactin signaling pathway | CYP17A1, PRLR, SLC2A2 | 0.07177503 | 56.6452718 |
| hsa04022 | cGMP-PKG signaling pathway | IRS4, KCNU1, CALML3, CACNA1D | 0.08363079 | 62.4688182 |
Protein-protein interaction (PPI) network for DEGs involved in the ceRNA network. Colored nodes: query proteins and first of interactors. Empty nodes: proteins of unknown 3D structure. Filled nodes: some 3D structure is known or predicted. Edges represent protein-protein associations. Pink represents experimentally determined, green represents gene neighborhood, wathet-blue represents from curated database, dark blue represents gene co-occurrence, red represents gene fusions, and black represents co-expression.
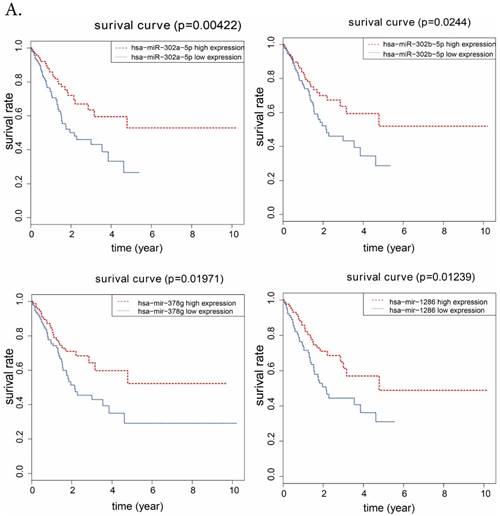
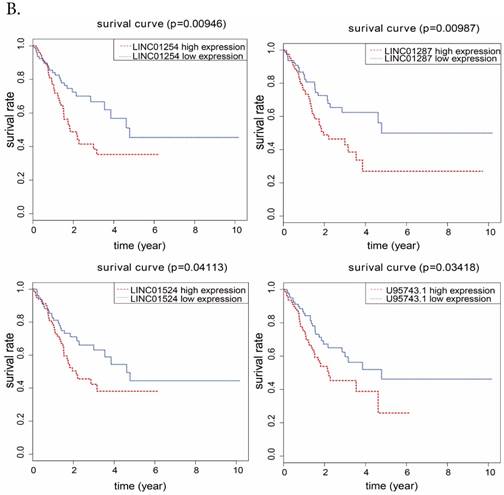
The Prognostic analysis of lncRNAs, miRNAs involved in ceRNA network
To further analyze the prognostic characteristics of 11 DElncRNAs and 10 DEmiRNAs, the data set and the overall survival information were profiled by univariate Cox regression. We found that 4 DElncRNAs and 4 DEmiRNAs were significantly associated with the survival of Hp positive GC patients (P<0.05), which were shown in Fig.6. It showed that LINCO1254, LINCO1287, LINCO1524, U95743.1 were negatively associated with overall survival, and the associations of miR-302a, miR-302b, miR-1286, miR-378g were positively.
A. Kaplan-Meier survival curvers for 4 DEmiRNAs associated with overall survival. Vertical axis represents survival rate, and horizontal axis represents survival time (year). B. for 4 DElncRNAs.
The verification of DElncRNAs
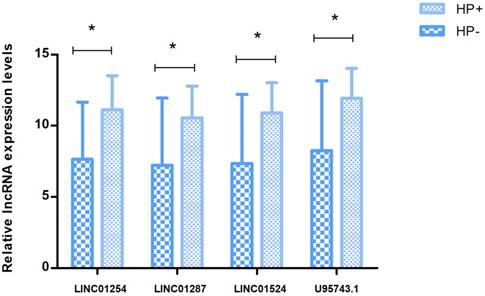
To further validate above results, we chose fresh tissues of 30 gastric cancer patients, detecting and analyzing the expression levels of 4 prognostic related lncRNAs that were involved in Hp positive ceRNA network. According to the results, the levels of LINCO1254, LINCO1287, LINCO1524, U95743.1 were significantly higher in Hp positive GC patients than Hp negative patients (P=0.02, 0.048, 0.04, 0.036, respectively). And the results from qRT-PCR validation were completely consistent with bioinformatics analysis ones (Fig. 7).
Quantitative RT-PCR validation of four differentially expressed lncRNAs. Relative expression levels of 4DElncRNAs are compared between Hp+ and Hp- group by △CT value (shown as mean+SD). P<0.05 is represented by *.
Discussion
In the present study, we identified cancer specific lncRNA and miRNA in GC with and without Hp infection by bioinformatics analysis using TCGA database, then built related ceRNA network. Besides, we predicted cellular functions and pathways of DEmRNAs involved in ceRNA network by GO and Pathway analysis. Moreover, the correlation between those DElncRNAs, DEmiRNA and overall survival were also explored. The ceRNA network we built provided a new thought of exploring the pathogenic mechanism and the survival analysis of GC with Hp infection.
In recent years, post-transcriptional regulation of non-coding RNAs has become a hot issue in the field of oncology [13]. Recent studies about miRNA and lncRNA have expanded our insights into Hp pathogenesis. Chang et al. suggested that miR-99b-3p, miR-564, and miR-638 were significantly increased in Hp-positive cancer tissues compared to the Hp-negative cancer tissues, while miR-204-5p, miR-338-5p, miR-375, and miR-548c-3p were significantly increased in Hp-negative cancer tissues [14]. Also, Zhu et al. reported they identified a number of differentially expressed lncRNAs in Hp-infected gastric mucosa tissues, and their data suggested that these lncRNAs might be involved in the pathological responses and development of Hp-related disorders and diseases.[6] In our study, based on the RNA sequence of TCGA database, we detected miRNA, lncRNA, and mRNA expression of GC with and without Hp infection. We found 27 differentially expressed lncRNAs, 32 differentially expressed miRNAs, and 257 differentially expressed mRNAs after comparing 168 patients with Hp negative GC patients and 20 patients with Hp positive GC. We newly found that these genes might be involved in regulating of the development of GC with Hp infection.
Competitive endogenous RNA (ceRNA) represents a novel layer of gene regulation that contribute to the physiology and development of diseases such as cancer, the dysregulation of which could lead to carcinogenesis [7]. Addressing this, in the present study we predicted DEmRNAs and DEmiRNA regulatory through miRWalk 3.0, TargetScanHuman 7.1, DEmiRNAs and DElncRNAs regulation through DIANA-LncBase v2. Next, we selected the intersectant DElncRNAs and DEmiRNAs with opposite expression trends, as well as DEmiRNAs and DEmRNAs. In the end, we constructed the ceRNA network of Hp positive GC by bioinformatics prediction and co-relation analysis of above differentially expressed RNAs, which eventually included intersectant 11 DElncRNAs, 10 DEmiRNAs and 219 DEmRNAs in co-expression network. Some researchers infer that Hp might regulate and control the expression of lncRNA through its toxin or other virulence factor, as what could also be found in the report of Zhu et al. that lncRNA might play key roles in the development of Hp-associated disorders and diseases through interaction with many other transcripts[6]. In addition, Yang et al. reported that immune response was confirmed to be dominant in Hp-positive GC, which was linked to lncRNA was overexpression [15]. However, until now, there is not any report about ceRNA network of Hp positive GC. Our analyses revealed that ceRNA regulation based on lncRNA-microRNA-mRNA is a complex and sophisticated mechanism, and might demonstrate essential regulatory role in the development of GC with Hp infection. Future biological studies might provide more detailed information and carry out essential clues for complex ceRNA regulation.
Then we predicted cellular functions and pathway analysis of DEmRNAs, which were involved in the ceRNA network. GO analysis indicated enrichment in biological process of sequence-specific DNA binding, serine-type endopeptidase, metalloendopeptidase, chloride channel, and structural molecular, which reminded us that Hp might contribute to the process of DNA binding and peptide cleavage. Cellular component suggested main enrichment at extracellular space, extracellular exosome, apical plasma membrane, extracellular region, keratin filament, and comified envelope. The obvious enrichment at extracellular exosome indicated that exosomes might be involved in the development of gastric and extragastric disorders associated with Hp infection, which should be focused on in the future [16]. In addition, intermediate filament, brain development, proteolysis, negative regulation of endopeptidase activity, keratinocyte differentiation, and peptide cross-linking were predominantly dysfunctional molecular function. As the most significant difference was proteolysis, it is reasonable to infer that Hp actively interferes with host cells by activating cellular proteases and the secretion of bacterial proteases, which has also been reported before [17]. As for the pathway, the results suggested that cancer-related pathway 'PI3K-Akt signaling pathway' has the most significant difference. And this pathway has already been reported to be associated with Hp infection that virulent factor of Hp, like CagA could inhibit the autophagy through PI3K-Akt signaling pathway to promote gastric inflammation, in turn, promote gastric carcinogenesis. Liu et al and Ren et al, also found 'PI3K-Akt signaling pathway' was related with GC cell functions.[18-20] About other pathways, like carbohydrate digestion and absorption, galactose metabolism, suggested that these metabolic processes might exert antimicrobial activity and have effect on Hp infection. From PPI network we built, we selected 6 hub genes, and the gene with the highest degree was NTS (neurotensin), regulated by miR-7156-5p. There is evidence that NTS contributes to the pathogenesis of Hp infection. The presence of Hp tends to alter the gastrointestinal hormone pattern, inducing neurotensin. And neurotensin is known to inhibit postprandial activity, effecting on some symtoms, like postprandial fullness [21].
At last, we explored the association between overall survival time and DElncRNAs, DEmiRNAs involved in the ceRNA network we built, respectively. We found from results that 4 DElncRNAs (LINC01254, LINC01287, LINC01524, U95743.1) and 4 DEmiRNAs (miR-302b, miR-302a, miR-378g, miR-1286) were related to Hp positive GC patients' survival. Among them, miR-302b and miR-302a have been reported to show associations with the survival of GC [22]. In addition, miR-302a may also be involved in endometrial cancer and hepatocellular carcinoma [23, 24]. MiR-378g has been reported to be involved in bladder urothelial carcinoma [25], while miR-1286 could associate with breast cancer and cervical cancer [26, 27]. There were few reports about the relationship between lncRNA and the survival of GC or other kinds of cancer, and lncRNAs (LINC01254, LINC01287, LINC01524, U95743.1) have not been reported yet. The results of lncRNAs, miRNA and overall survival revealed potential indictors of prognosis in Hp positive GC. In addition, we chose fresh tissues of 30 gastric cancer patients, detecting and analysing the expression level of 4 prognostic related lncRNAs that were involved in Hp positive ceRNA network. Results revealed that LINC01254, LINC01287, LINC01524, U95743.1 were expressed higher in Hp positive GC tissue than in Hp negative GC tissue which was in agreement with the results from TCGA, and showed that our bioinformatics analysis was incredible.
However, the limitation should be acknowledged in this study. The sample size of gastric cancer with Hp infection was relatively small for the pooled analysis of comparing with gastric cancer without Hp infection. So the results still need future large-scale researches to confirm.
Conclusions
Here we carried out a comprehensive analysis of aberrantly expressed ceRNA network in GC with and without Hp infection by bioinformatics analysis using TCGA database, and built the ceRNA regulatory network, which included 11lncRNAs, 10RNAs and 218mRNAs. We suggested the ceRNA regulatory network is associated with diseases pathways and essential cellular functions. Moreover, the prognostic analysis revealed that lncRNAs (LINC01254, LINC01287, LINC01524, U95743.1) and miRNAs (miR-302b, miR-302a, miR-378g, miR-1286) may be considered as potential specific biomarkers in diagnosis and prognosis of Hp positive GC, which could be continually validated and explored in our future study. Furthermore, the real-time PCR verification revealed that LINC01254, LINC01287, LINC01524, U95743.1 might be considered as potential biomarkers in the diagnosis and prognosis of GC with Hp infection. As far as I concerned, there are seldom reports that studied functional lncRNA induced by Hp infection. Besides, it has few reports about ceRNA network related to Hp infection until now. All together, we expect the study presented here brings biological insights into the essential roles of ceRNA regulation in GC with Hp infection.
Supplementary Material
Supplementary tables.
Acknowledgements
This work was supported by the National Key R&D Program of China (Grant No. 2017YFC0908300).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Crew KD, Neugut AI. Epidemiology of gastric cancer. World journal of gastroenterology. 2006;12:354-62
2. Bilici A. Treatment options in patients with metastatic gastric cancer: current status and future perspectives. World journal of gastroenterology. 2014;20:3905-15
3. Houghton J, Wang TC. Helicobacter pylori and gastric cancer: a new paradigm for inflammation-associated epithelial cancers. Gastroenterology. 2005;128:1567-78
4. Li PF, Chen SC, Xia T, Jiang XM, Shao YF, Xiao BX. et al. Non-coding RNAs and gastric cancer. World journal of gastroenterology. 2014;20:5411-9
5. Zou M, Wang F, Jiang A, Xia A, Kong S, Gong C. et al. MicroRNA-3178 ameliorates inflammation and gastric carcinogenesis promoted by Helicobacter pylori new toxin, Tip-alpha, by targeting TRAF3. Helicobacter. 2017:22
6. Zhu H, Wang Q, Yao Y, Fang J, Sun F, Ni Y. et al. Microarray analysis of Long non-coding RNA expression profiles in human gastric cells and tissues with Helicobacter pylori Infection. BMC medical genomics. 2015;8:84
7. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353-8
8. Zhou M, Wang X, Shi H, Cheng L, Wang Z, Zhao H. et al. Characterization of long non-coding RNA-associated ceRNA network to reveal potential prognostic lncRNA biomarkers in human ovarian cancer. Oncotarget. 2016;7:12598-611
9. Zhang G, Li S, Lu J, Ge Y, Wang Q, Ma G. et al. LncRNA MT1JP functions as a ceRNA in regulating FBXW7 through competitively binding to miR-92a-3p in gastric cancer. Molecular cancer. 2018;17:87
10. Liu ML, Zhang Q, Yuan X, Jin L, Wang LL, Fang TT. et al. Long noncoding RNA RP4 functions as a competing endogenous RNA through miR-7-5p sponge activity in colorectal cancer. World journal of gastroenterology. 2018;24:1004-12
11. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13:2498-504
12. Zhang T, Jiang M, Chen L, Niu B, Cai Y. Prediction of gene phenotypes based on GO and KEGG pathway enrichment scores. BioMed research international. 2013;2013:870795
13. Chan JJ, Tay Y. Noncoding RNA:RNA Regulatory Networks in Cancer. International journal of molecular sciences. 2018:19
14. Chang H, Kim N, Park JH, Nam RH, Choi YJ, Lee HS. et al. Different microRNA expression levels in gastric cancer depending on Helicobacter pylori infection. Gut and liver. 2015;9:188-96
15. Yang L, Long Y, Li C, Cao L, Gan H, Huang K. et al. Genome-wide analysis of long noncoding RNA profile in human gastric epithelial cell response to Helicobacter pylori. Japanese journal of infectious diseases. 2015;68:63-6
16. Shimoda A, Ueda K, Nishiumi S, Murata-Kamiya N, Mukai SA, Sawada S. et al. Exosomes as nanocarriers for systemic delivery of the Helicobacter pylori virulence factor CagA. Scientific reports. 2016;6:18346
17. Posselt G, Crabtree JE, Wessler S. Proteolysis in Helicobacter pylori-Induced Gastric Cancer. Toxins. 2017:9
18. Li N, Tang B, Jia YP, Zhu P, Zhuang Y, Fang Y. et al. Helicobacter pylori CagA Protein Negatively Regulates Autophagy and Promotes Inflammatory Response via c-Met-PI3K/Akt-mTOR Signaling Pathway. Frontiers in cellular and infection microbiology. 2017;7:417
19. Liu Y, Shao C, Zhu L, Jiang S, Li G, Zhang W. et al. High Expression of ABL2 Suppresses Apoptosis in Gastric Cancer. Digestive diseases and sciences. 2018
20. Ren X, Zheng D, Guo F, Liu J, Zhang B, Li H. et al. PPARgamma suppressed Wnt/beta-catenin signaling pathway and its downstream effector SOX9 expression in gastric cancer cells. Medical oncology (Northwood, London, England). 2015;32:91
21. Wu Z, Galmiche A, Liu J, Stadler N, Wendum D, Segal-Bendirdjian E. et al. Neurotensin regulation induces overexpression and activation of EGFR in HCC and restores response to erlotinib and sorafenib. Cancer letters. 2017;388:73-84
22. Kotachi T, Ito M, Yoshihara M, Boda T, Kiso M, Masuda K. et al. Serological Evaluation of Gastric Cancer Risk Based on Pepsinogen and Helicobacter pylori Antibody: Relationship to Endoscopic Findings. Digestion. 2017;95:314-8
23. Ma J, Li D, Kong FF, Yang D, Yang H, Ma XX. miR-302a-5p/367-3p-HMGA2 axis regulates malignant processes during endometrial cancer development. Journal of experimental & clinical cancer research: CR. 2018;37:19
24. Qin C, Zha W, Fan R, Ding H, Xu Y, Wang C. MicroRNA302a inhibits cell proliferation and invasion, and induces cell apoptosis in hepatocellular carcinoma by directly targeting VEGFA. Molecular medicine reports. 2017;16:6360-7
25. Zhang ZC, Huang Y, Wang XJ, Wang M, Ma LL. [Expression of circulating microRNAs in patients with bladder urothelial carcinoma]. Beijing da xue xue bao Yi xue ban = Journal of Peking University Health sciences. 2013;45:532-6
26. Yao T, Rao Q, Liu L, Zheng C, Xie Q, Liang J. et al. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in cervical cancer. Virology journal. 2013;10:175
27. Cordero F, Ferrero G, Polidoro S, Fiorito G, Campanella G, Sacerdote C. et al. Differentially methylated microRNAs in prediagnostic samples of subjects who developed breast cancer in the European Prospective Investigation into Nutrition and Cancer (EPIC-Italy) cohort. Carcinogenesis. 2015;36:1144-53
Author contact
![]() Corresponding authors: Dr. Gong Yuehua, Tumor Etiology and Screening Department of Cancer Institute and General Surgery, The First Hospital of China Medical University, No.155 NanjingBei Street, Heping District, Shenyang, Liaoning Province, P.R. China 110001, Telephone: +86-024-83282153; fax: +86-024-83282383. Email: yhgongedu.cn. Dr. Yuan Yuan, Tumor Etiology and Screening Department of Cancer Institute and General Surgery, The First Hospital of China Medical University, No.155 NanjingBei Street, Heping District, Shenyang, Liaoning Province, P.R. China 110001, Telephone:+86-024-83282153;fax:+86-024-83282292.Email: yuanyuanedu.cn
Corresponding authors: Dr. Gong Yuehua, Tumor Etiology and Screening Department of Cancer Institute and General Surgery, The First Hospital of China Medical University, No.155 NanjingBei Street, Heping District, Shenyang, Liaoning Province, P.R. China 110001, Telephone: +86-024-83282153; fax: +86-024-83282383. Email: yhgongedu.cn. Dr. Yuan Yuan, Tumor Etiology and Screening Department of Cancer Institute and General Surgery, The First Hospital of China Medical University, No.155 NanjingBei Street, Heping District, Shenyang, Liaoning Province, P.R. China 110001, Telephone:+86-024-83282153;fax:+86-024-83282292.Email: yuanyuanedu.cn