Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(4):864-873. doi:10.7150/jca.27663 This issue Cite
Research Paper
Cadherin Related Family Member 2 Acts As A Tumor Suppressor By Inactivating AKT In Human Hepatocellular Carcinoma
Ziyuan Xia1*, Meijin Huang3*, Qiangqiang Zhu1*, Yinghua Li1, Qian Ma4, Yang Wang5, Xia Chen6, Jianzhong Li1, Lei Qiu1, Junping Zhang1, Jiaoyang Zheng2, Bin Lu1 ![]()
1. Department of Biochemical Pharmacy, School of Pharmacy, Second Military Medical University, Shanghai, China
2. Department of Endocrinology, Changzheng Hospital, Second Military Medical University, Shanghai, China
3. Department of Oncology, 920th Hospital of PLA Joint Logistics support Force, Yunnan, China
4. Department of Pathology, Changhai Hospital, Second Military Medical University, Shanghai, China
5. Department of Pathology, Shuguang Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China
6. Yangpu Hospital, Tongji University School of Medicine, Shanghai, China
* These authors contributed equally to this work.
Received 2018-6-5; Accepted 2019-1-2; Published 2019-1-29
Abstract
Cadherin related family member 2 (CDHR2) belongs to the protocadherin family and is abundant in normal liver, kidney, and colon tissues, but weakly expressed in cancers arising from these tissues. In this study, we demonstrated that CDHR2 was highly expressed in para-cancer tissues of human hepatocellular carcinoma (HCC), but significantly downregulated or silenced in 85.7% (6/7) of HCC cell lines by both semi-quantitative PCR and western blot, and 79.1% (19/24) and 80.2% (89/111) of tumor tissues from patients with HCC by semi-quantitative PCR, and immunohistochemistry, respectively. Interestingly, CpG islands in the promoter of CDHR2 gene were hypermethylated in HCC cell lines and tissues compared with the para-cancer tissues by methylation-specific PCR analysis, leading to transcriptional repression and silencing of CDHR2 in HCC. In addition, CDHR2 overexpression by lentiviral vectors had suppressive effects on HCC cell growth and proliferation, as evidenced by prolonged cell doubling time and reduced colony-forming ability in vitro, as well as by decreased tumorigenicity in vivo. Mechanistically, CDHR2 overexpression resulted in AKT dephosphorylation along with downregulation of cyclooxygenase-2 (COX2), a downstream target of AKT. This effect was reversed by myristoylated AKT, a constitutively active form of AKT, suggesting an involvement of CDHR2-AKT-COX2 axis in the suppression of HCC growth. Taken together, our study identified CDHR2 as a novel tumor suppressor in HCC and provided a new therapeutic target for HCC.
Keywords: CDHR2, methylation, hepatocarcinoma, proliferation, AKT, COX2
Introduction
Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide with a very poor prognostic outcome. Despite recent advances in treatment options, including surgery, percutaneous radiofrequency ablation, chemotherapy, radiotherapy, and traditional Chinese medicine therapy, the morbidity and mortality of HCC are still increasing over time [1-3]. Therefore, clarifying the pathogenesis of HCC and identifying potential diagnostic biomarkers and therapeutic targets are clinically urgent.
Protocadherin (PCDH) family, a subset of cadherin superfamily [4], is a group of transmembrane proteins characterized by repeated, ~110 amino acids-containing extracellular cadherin (EC) domains. PCDH family plays an important role in calcium-dependent cell adhesion [4, 5]. Loss of expression of some PCDH family members (e.g., PCDH8, PCDH9, PCDH10, PCDH17, and PCDH20) has been reported in a number of human cancers, including breast cancer, leukemia, glioblastoma, stomach cancer, colorectal cancer, esophageal cancer, and lung cancer, suggesting that PCDH may serve as tumor suppressors in human cancers [6-12]. PCDH family member cadherin related family member 2 (CDHR2), also named PCDH24, has a featured tissue-specific expression pattern with a dominant expression in normal liver, kidney, and colon tissues. In contrast, the expression of CDHR2 is markedly reduced in the cancers derived from these tissues, which suggests CDHR2 as a candidate biomarker and potential tumor suppressor in these cancers [13]. Consistently, CDHR2 can inhibit cell-cell interaction mediated by submembranous retention of β-catenin, resulting in cell proliferation inhibition and tumor growth arrest in colorectal cancer [14, 15]. However, little is known about the role of CDHR2 in HCC and the underlying mechanism.
In this study, we found that CDHR2 was generally downregulated in HCC cell lines and tissues, which was at least partially attributable to hypermethylation in the promoter of CDHR2 gene. By introducing the lentiviral vectors expressing CDHR2 into CDHR2-deficient HCC cell lines, we examined the role of CDHR2 in HCC growth both in vitro and in vivo, providing the evidence for the first time that CDHR2 may act as a tumor suppressor in HCC.
Material and Methods
Patients and tissue samples
This study was approved by the Ethics Committee at Second Military Medical University (SMMU), Shanghai, China. HCC tissues and para-cancer tissues were obtained from 135 patients enrolled in Eastern Hepatobiliary Surgery Hospital (Shanghai, China) from 2012 to 2013 with written informed consents. Twenty-four pairs of HCC and para-cancer specimens were used for semi-quantitative reverse transcription polymerase chain reaction (RT-PCR) assay (Supplementary table 1) and 111 pairs were used for immunohistochemical (IHC) analysis of tissue microarrays (Supplementary table 2).
Cell lines and transfection
SMMC-7721, HepG2, Hep3B, PLC/PRF/5, Huh7, HCC-LM3, MHCC-97L, and 293T cell lines were used and grown in DMEM (Gibco, Thermo Fisher Scientific, Waltham, MA, USA) or RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) in a humidified atmosphere with 5% CO2 at 37°C. HepG2, Hep3B, PLC/PRF/5, and Huh7 were provided by Shanghai Cell Collection, Chinese Academy of Sciences (Shanghai, China). HCC-LM3 and MHCC-97L were purchased from the Liver Cancer Institute, Zhongshan Hospital, Fudan University (Shanghai, China). SMMC-7721 and HEK293T (293T) were kept by SMMU (Shanghai, China). Lentiviral vectors expressing CDHR2 (LV-CDHR2) or myristoylated AKT (myr-AKT) were prepared as previously described [16] using primers shown in Table 1. Lentiviral vector expressing luciferase (LV-Luc vector) was used as a negative control. Cells were infected with lentivirus at multiplicities of infection of 20 and selected with puromycin (Sigma-Aldrich, St. Louis, MO, USA) to prepare stable cell lines.
Primer sets used for PCR
| Target | Forward sequences (5' to 3') | Reverse sequences (5' to 3') |
|---|---|---|
| CDHR2 | AAACCGGTCGCCACCATGGCCCAGCTATGGCTGTCC | AAATCGATTCACAGGTCCGTGGTGTCCAG |
| myr-AKT | AACCGGTCACCATGGGGAGCAGCAAGAGCAAGCCCAAGATGAGCGACGTGGCTATTGTG | AAATCGATTCACTTATCGTCGTCATCCTTGTAATCGGCCGTGCCGCTGGC |
| CDHR2 (for RT-PCR†) | ATCATAGGATTGGGAGTGGC | GTCTTTGTTGGGGAGGTTCA |
| β-Actin (for RT-PCR†) | CCAGCACAATGAAGATCAAG | GAAAGGGTGTAACGCAACTAA |
| Cox2 (for RT-PCR†) | GAAACTCTGGCTAGACAGCGTAA | AAACCGTAGATGCTCAGGGACT |
| Methylated CDHR2 promoter (for MSP‡) | ATAGAGAAAAGAGTTTAGGGGTACG | AAACAAAACACAAATAAAAACCGA |
| Unmethylated CDHR2 promoter (for MSP‡) | TAGAGAAAAGAGTTTAGGGGTATGG | AAACAAAACACAAATAAAAACCAAA |
†RT-PCR, reverse transcription polymerase chain reaction.
‡MSP, methylation-specific PCR.
RT-PCR analysis
Total RNA was isolated from cells or tissues using an NucleoSpin RNA II Kit (Macherey-Nagel, Düren, Germany) with removal of genomic DNA by RNase-free DNase. First-strand cDNA was generated with a PrimeScript RT reagent Kit (Takara, Dalian, China). Semi-quantitative and real-time quantitative PCR were performed using a S1000 thermal cycler (Bio-Rad, Hercules, CA, USA) and an ABI 7500 system (Applied Biosystems, Foster, CA, USA), respectively. The primers used were listed in Table 1. Actin was used as an endogenous control. For semi-quantitative PCR, PCR products were loaded onto 2% agarose gel and visualized under UV light. For real-time quantitative PCR, relative expression of genes was calculated and expressed as 2-△△CT [17].
Methylation-Specific PCR (MSP)
Genomic DNA was isolated from the cells and tissues using genomic DNA extraction kit (Tiangen, Beijing, China). Bisulfite modification of genomic DNA was performed using a CpGenome Turbo Bisulfite Modification Kit (Merck Millipore, Darmstadt, Germany) followed by PCR analysis with MSP system and primer sets specific for methylated or unmethylated CDHR2 (Table 1). PCR products were loaded onto 2% agarose gel and visualized under UV light.
Western blot analysis
Cells were lysed with radio-immuno precipitation assay (RIPA) buffer on ice. Cell lysates were collected by centrifugation at 12,000 rpm for 10 min, followed by heating at 95°C for 5 min in loading buffer. Proteins were separated by SDS-PAGE and transferred onto PVDF membranes which were then blotted with primary antibody (Ab) against β-actin (Sigma-Aldrich) or CDHR2 (rabbit polyclonal antibody produced in our laboratory). After washing three times with TBST, membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary Ab (Santa Cruz Biotechnology, Dallas, TX, USA) for 1 h at room temperature and washed with TBST. The chemiluminescence signals were visualized with a SuperSignal West Pico chemiluminescent substrate kit (Pierce Biotechnology, Rockford, IL, USA) and imaged with ChemiDoc™ System (Bio-rad).
IHC staining and tissue microarray analysis
Paraffin-embedded tissue sections of 5 μm thickness were deparaffinized and rehydrated followed by incubation with the CDHR2 polyclonal antibody. Normal IgG was used as a control. CDHR2 expression was evaluated by two pathologists in a blind manner. Five randomly selected fields were scored in each section. The staining intensity was quantified as follows: 0, no staining; 1, yellow; 2, brown; 3, dark brown. Grade 0 denoted positive immunostaining observed in < 5% cells; 1: 5-20% cells; 2: 20-50% cells; 3: 50-75%; and 4: > 75%. The final IHC score, the product of staining gradeintensity and staining intensity grade, was described as negative (0-3) and positive (> 3).
Cell proliferation assay
Cells were plated in 6-well plates at a density of 5.0 × 105 cells per well and grown at 37°C. Cell numbers were counted with a hemocytometer at 24, 48, and 72 h, respectively, and uploaded onto http://www.doubling-time.com/compute.php for doubling time computing.
EdU cell proliferation assay
Cells were seeded in 96-well plates at a density of 3.0 × 105 cells per well. Edu incorporation assay was performed with Cell-Light EdU Apollo 567 in vitro imaging kit (RiboBio, Guangzhou, China) according to the manufacturer's instruction. Images were obtained using an Olympus IX71 microscope (Olympus, Tokyo, Japan) and data were analyzed with Image-Pro software (Media Cybernetics, Silver Spring, USA).
Plate colony formation assay
For anchorage-dependent monolayer colony formation assay, cells were seeded into 100-mm petri dishes at a density of 1 × 103 cells per dish and grown at 37°C for 14 days. Cells were then fixed with methanol/acetone (1:1) and stained with crystal violet (Shenggong, Shanghai, China). Colonies containing more than 50 cells were counted and photographed with an Olympus IX71 microscope. Experiments were performed in triplicate and repeated at least three times.
Soft agar colony formation assay
For anchorage-independent soft agar colony formation assay, cells were seeded in 0.3% top agar over a layer of 0.6% agar in a 24-well plate at a density of 2.5 × 103 cells per well and grown at 37°C. After 15 days of incubation, colonies more than 100 µm in diameter were counted and photographed using an Olympus IX71 microscope. Experiments were performed in triplicate and repeated at least three times.
Animal Studies
Six-week-old male BALB/c nude mice were purchased from Shanghai Silaike Experiment Animal Co., Ltd (Shanghai, China). All mice were maintained under pathogen-free conditions in laminar flow boxes in accordance with established institutional guidance and approved protocols of SMMU. Mice were randomly divided into 4 groups (n = 6/group). Five million of SMMC-7721 or PLC/PRF/5 cells stably transfected with LV-CDHR2 or LV-Luc were injected subcutaneously into the right flank of each mouse. The tumor dimensions were measured every 3 to 4 days using a digital caliper. The tumor volumes were calculated as length × width2 × 0.5. After 5 weeks of tumor formation, mice were sacrificed and tumors were collected. Growth curves were plotted based on mean tumor volume within each group at the indicated time points.
High-throughput protein microarray assay
Cells were lysed with RIPA buffer and the lysates were collected by centrifugation at 12,000 rpm for 10 min. Proteins were labelled with biotin and loaded on protein microarray slides (Full Moon BioSystems, Sunnyvale, CA, USA) with 6 replicates according to the manufacturer's instruction. Array scanning was performed using an Axon GenePix 4000B scanner (Molecular Devices, Sunnyvale, CA, USA). Data were analyzed with GenePix Pro 6.1 software (Molecular Devices).
Statistical analysis
Statistical analysis was performed using the Analysis ToolPack of Microsoft Excel (Microsoft, Redmond, WA, USA). A two-sample Student t test assuming unequal variances was used to determine the equality of means of two samples. Results were considered statistically significant at P < 0.05.
Results
CDHR2 is significantly downregulated in HCC cell lines and tissues
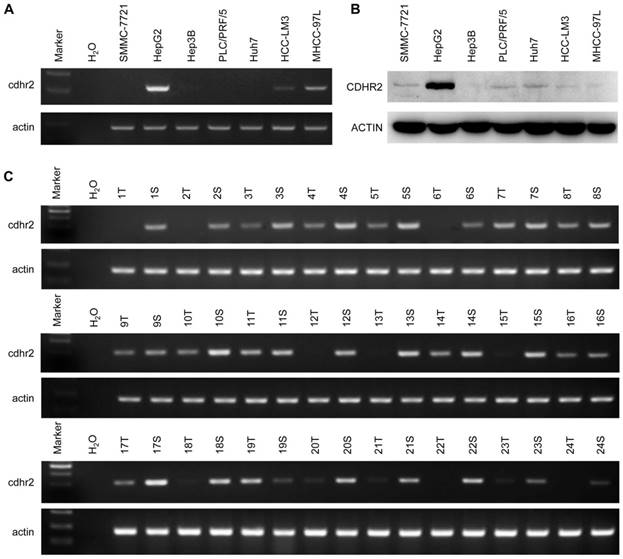
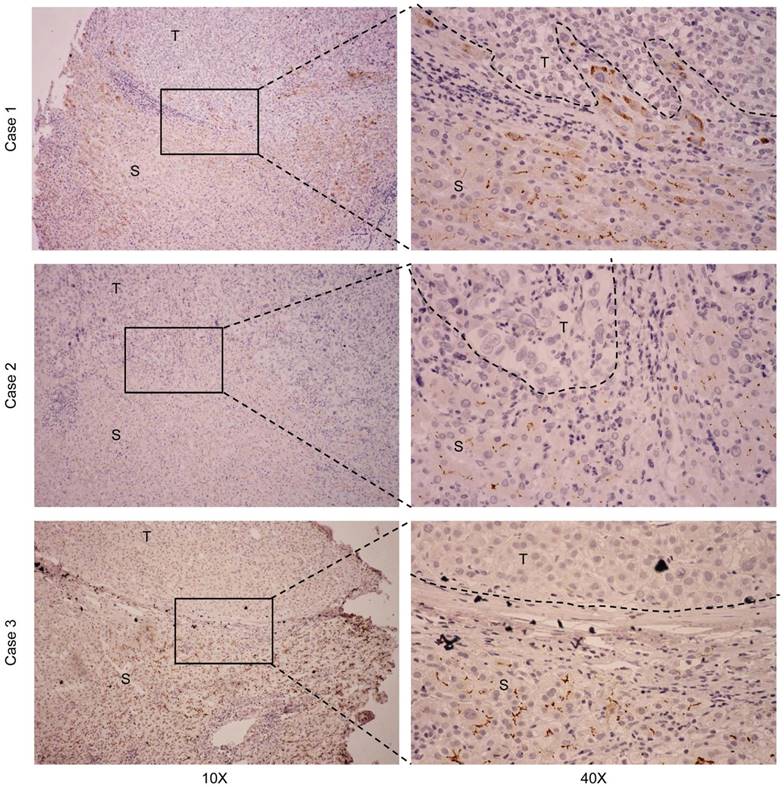
To detect endogenous CDHR2 protein, we produced and purified a polyclonal antibody against a synthetic peptide derived from C-terminal of CDHR2 protein by immunizing rabbits. The specificity of the antibody was validated by Western blot assay and a single band above 170 kDa corresponding to CDHR2 was detected (Supplementary Fig. 1). We next sought to determine the expression of CDHR2 in HCC cell lines and tissues. As shown in Figure 1A and 1B, both mRNA and protein expression of CDHR2 were significantly downregulated in 6 HCC cell lines. Similarly, CDHR2 mRNA is weakly or not expressed in 79.2% (19/24) of HCC tissues compared with adjacent para-tumor tissues (Fig. 1C). We further examined CDHR2 protein expression in 111 HCC tissues by immunohistochemistry, and found that CDHR2 was expressed mainly in the cell membrane of normal liver tissue or para cancerous tissue. However, 89 cases (80.2%) have reduced expression or deletion phenomenon of CDHR2 expression in HCC tissue, suggesting that CDHR2 may play an inhibitory role in HCC development (Fig. 2).
Expression of CDHR2 in HCC cell lines and tissues. Semi-quantitative RT-PCR was performed to examine mRNA expression of CDHR2 in 7 HCC cell lines (A) and 24 pairs of primary HCC tissues and adjacent normal tissues (B). (C) Western blot was performed to detect the protein expression levels of CDHR2 in 7 HCC cell lines. T, HCC tissue; S, Tumor-surrounding tissue.
Representative IHC staining for CDHR2 in 3 pairs of primary HCC tissues and adjacent non-neoplastic liver tissues. T, HCC tissue; S, Tumor-surrounding tissue.
CDHR2 inhibits HCC growth and proliferation in vitro
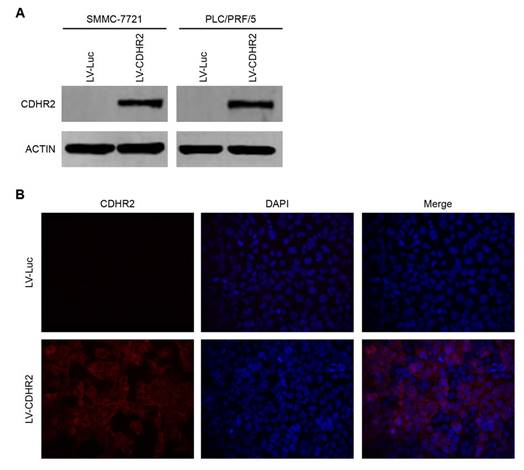
To determine the potential effects of CDHR2 on HCC growth, we established the cell lines stably expressing CDHR2 by introducing lentivirus carrying CDHR2 gene into SMMC-7721 and PLC/PRF/5 cells (named SMMC-7721-CDHR2 and PLC/PRF/5-CDHR2, respectively). CDHR2 expression was confirmed by Western blot (Fig. 3A). Cell immunofluorescent staining showed that CDHR2 is mainly located on the cell membrane (Fig. 3B).
Validation of ectopic expression and subcellular localization of CDHR2 protein in HCC cell lines. (A) Western blot detection of ectopic CDHR2 expression in SMMC-7721 (left panel) cells and PLC/PRF/5 cells (right panel) infected with LV-CDHR2 or LV-Luc. (B) Immunofluorescence staining shows the over-expression and localization of CDHR2 (red) in SMMC-7721 cells infected with LV-CDHR2, compared with LV-Luc group.
Then, prolonged doubling time and decreased DNA synthesis were observed in SMMC-7221-CDHR2 or PLC/PRF/5-CDHR2 cells (Fig. 4A and 4B, respectively), indicating that CDHR2 inhibits HCC cell proliferation in vitro. Interestingly, we found that overexpression of CDHR2 inhibited the cell proliferation more significantly in the confluent cultures than in the sparse cultures (Fig. 4B and Supplementary Fig. 2), especially for PLC/PRF/5 cells, suggesting that CDHR2 plays a role on cell contact inhibition.
The effect of CDHR2 on HCC cell growth and proliferation. (A) The effect of CDHR2 over-expression on doubling time of HCC cell lines, SMMC-7721 and PLC/PRF/5 cells, respectively. (B) Edu incorporation assay was performed on LV-CDHR2-infected SMMC-7721 and PLC/PRF/5 cells to evaluate the proliferation rate of sparse and confluent cultures. (C) Cells were plated in 10-cm Petri dishes at densities varying from 1,000 to 5,000 cells per dish in fresh medium for colony formation. Colonies were stained with crystal violet after 14 days, and counted as indicated. (D) Cells were plated in semisolid soft agar medium to monitor anchorage-independent growth. The numbers represent the mean±SD of 3 independent experiments. Photographs are representative images from each group as indicated under the microscope. The experiments were repeated 3 times. *p<0.05 vs. control lentivirus group.
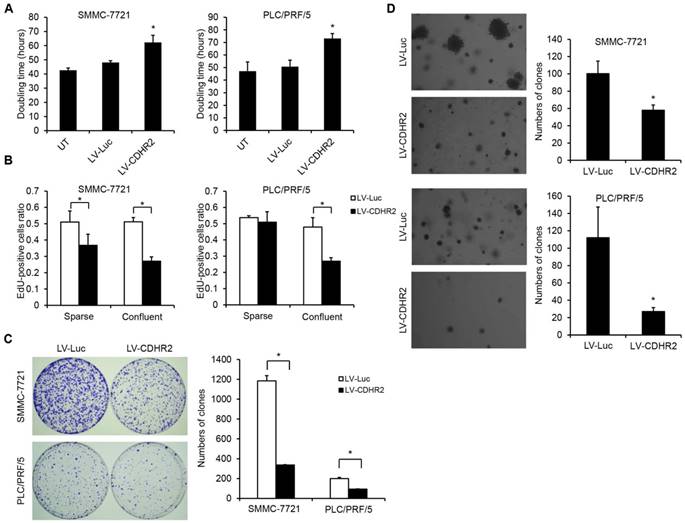
In addition, as shown in Figure 4C and 4D, SMMC-7221-CDHR2 or PLC/PRF/5-CDHR2 cells exhibited a significant reduction in both number and size of colonies compared with corresponding control cells, suggesting that CDHR2 inhibits anchorage-dependent and -independent growth of HCC cells.
CDHR2 suppresses HCC tumorigenicity in vivo
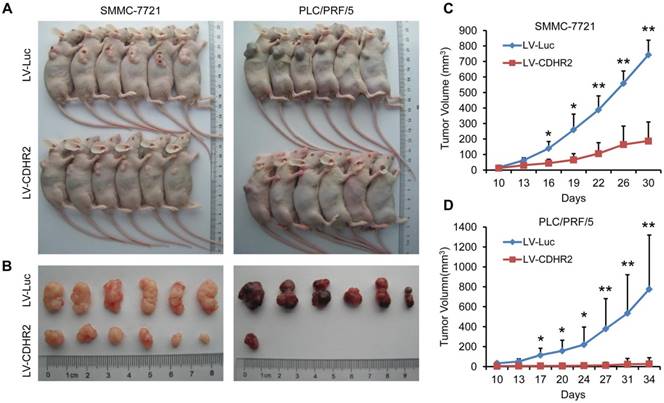
To further examine the in vivo role of CDHR2 in HCC cells, SMMC-7721-CDHR2 or PLC/PRF/5-CDHR2 cells and their respective control cells were subcutaneously inoculated into Balb/c nude mice. As shown in Figure 5, mice inoculated with CDHR2-expressing HCC cells exhibited a marked reduction in both tumor growth and tumor incidence compared with those inoculate with control HCC cells. The average volumes of tumors derived from SMMC-7721-CDHR2 or PLC/PRF/5-CDHR2 cells compared to their controls are 743.445±94.12 vs 187.21±122.65 mm3 and 776.57±542.20 vs 20.53±2.15 mm3, respectively (p<0.01). These data suggest the suppressive effect of CDHR2 on HCC tumorigenicity in vivo.
Overexpression of CDHR2 inhibited tumorigenicity HCC xenografts in nude mice. (A and B) Photographs of mice with primary tumors derived from CDHR2-overexpressing SMMC-7721 (left panel) and PLC/PRF/5 (right panel) cells. (C and D) Tumor volumes. *p<0.05, **p<0.01, n=6.
CDHR2 inhibits tumor growth via downregulation of AKT activity and cytochrome c oxidase subunit 2 (COX2)
To investigate the mechanism underlying the inhibitory effect of CDHR2 on HCC growth, we performed high-throughput mRNA and protein microarray analyses to examine a variety of cancer-associated signaling pathways between SMMC-7721-CDHR2 and control cells. As shown in Figure 6A, the protein expression of phosphorylated AKT (p-AKT) was considerably decreased in SMMC-7721-CDHR2 cells compared with that in control cells (0.8 vs 0.5), which was confirmed by Western blot assay (Fig. 6B). This result suggests the possible involvement of CDHR2 in inactivation of AKT pathway. We next sought to examine the role of AKT inactivation in CDHR2-mediated inhibition of HCC growth. As shown in Fig. 6C, the doubling time of SMMC-7721-CDHR2 cells was significantly decreased when the activity of AKT was restored by infection of lentiviral vectors expressing myristoylated AKT (myr-AKT), an active form of AKT, suggesting that AKT dephosphorylation or inactivation is essential for CDHR2-mediated inhibition of HCC proliferation.
Involvement of CDHR2 in dephosphorylation of AKT and COX2 downregulation of downstream. High-throughput protein microarray analysis (A) and Western blot detection (B) of p-AKT and total AKT in LV-CDHR2 or LV-luc infected SMMC-7721 cells. (C) The effect of myr-Akt on doubling time of SMMC-7721 cells stably expressing CDHR2. (D) Real-time PCR analysis of mRNA expression of COX2 in SMMC-7721 cells stably expressing CDHR2. (E) The effect of myr-Akt on mRNA expression of COX2 in CDHR2-expressing SMMC-7721 cells.
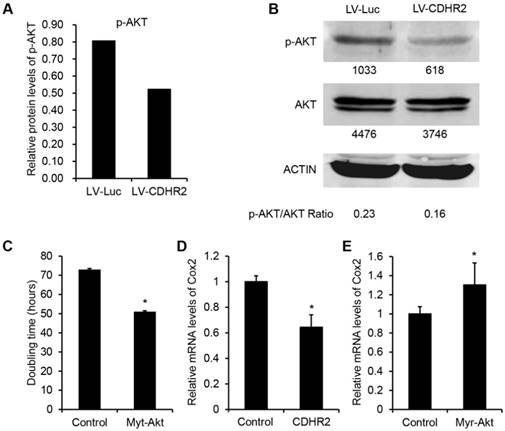
In addition, in the mRNA profile microarray analysis, we observed a significant downregulation of COX2, a critically important regulator of tumor growth [18], in SMMC-7721-CDHR2 cells compared with that in the control cells (data not shown), which was verified by the result of real-time RCR (Fig. 6D). Moreover, AKT activation by myr-AKT reversed the suppressive effect of CDHR2 on expression of COX2 (Fig. 6E). Taken together, these data suggest that inhibition of AKT-COX2 axis is required for CDHR2-induced suppression of HCC cell growth and proliferation.
CDHR2 silence in HCC is associated with CpG island methylation in its promoter
A lot of tumor suppressor genes were shown to be often inactivated by hypermethylation of their promoter regions. We supposed that the downregulated expression or absence of CDHR2 in HCC was due to the hypermethylation of CDHR2 promoter. In silico analysis reveals that there are four CpG islands around transcription start site of CDHR2 gene (data not shown).
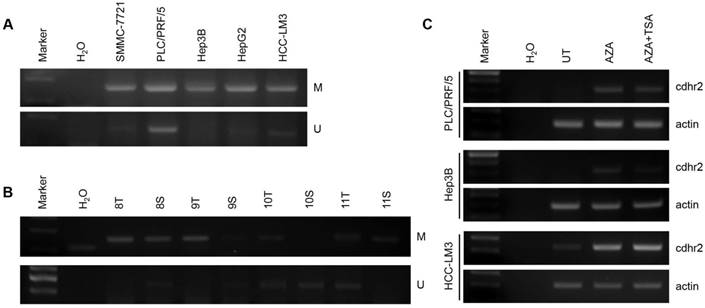
Using primer pairs located on the first CpG island, we found that the CpG island of CDHR2 promoter was methylated in 5 HCC cell lines detected by MS-PCR analysis (Fig. 7A). The mRNA expression levels of CDHR2 were significantly restored in PLC/PRF/5, Hep3B, and HepG2 cells, after treated with demethylating agent 5-azacytidine and histone deacetylase-inhibitor Trichostatin A individually or in combination (Fig. 7C). Furthermore, the methylation of CpG island was also found in some HCC tissues compared with adjacent noncancerous tissues (Fig. 7B). These data suggest that downregulation of CDHR2 in HCC may be at least partially due to the hypermethylation in its promoter region.
CpG island methylation of CDHR2 gene promoter. Methylation-Specific PCR analysis in 5 HCC cell lines (A) and some primary HCC tissues (B). (C) RT-PCR detection of CDHR2 expression in HCC cell lines treated with DNA and TSA methylation inhibitor AZA individually or in combination. UT: untreated; AZA: 5-Aza-2'-deoxycytidine; TSA: Trichostatin A.
Discussion
CDHR2, lowly expressed in tumor tissues, is being considered as a potential tumor suppressor because of its important role in cell-cell adhesion and contact inhibition in epithelial cells [13, 14]. However, the role of CDHR2 and the regulatory mechanism underlying downregulation of CDHR2 in HCC have never been investigated. In this study, we examined the expression pattern of CDHR2 in 7 HCC cell lines and totally 135 pairs of matched HCC tissues and para-cancer tissues. The mRNA and protein expression of CDHR2 were found generally downregulated or undetectable in most HCC cell lines and tumor tissues (Fig. 1), suggesting a possible inhibitory role of CDHR2 in HCC development.
Epigenetic modification has been reported to be an important mechanism for silencing tumor suppressor genes in cancer [19-22]. During hepatocarcinogenesis, a number of tumor suppressor genes can be ubiquitously silenced by hypermethylation in the CpG island of the promoter by DNA methyltransferases [19, 20]. Consistent with these findings, a ubiquitous hypermethylation also occurred in the promoter of CDHR2 in 5 HCC cell lines (Fig. 7). However, it appears that the degree of hypermethylation in the promoter of CDHR2 in HCC tissues was not as high as that in HCC cell lines, which is likely due to the complex microenvironment of HCC tissues. This is a possible explanation for the relatively high mRNA expression of CDHR2 in HCC tissues (Fig. 1C).
We also examined the role of CDHR2 in HCC by CDHR2 overexpression in HCC cells or inoculation of CDHR2-overexpressing HCC cells into nude mice. As expected, CDHR2 was able to inhibit HCC cell growth and proliferation in vitro, and reduce tumor formation and growth in vivo (Fig. 4, and Fig. 5). To our knowledge, this is the first study to show the protective role of CDHR2 against HCC growth both in vitro and in vivo. CDHR2 is a type of transmembrane protein, as confirmed by immunostaining analysis in our study (Fig. 3), containing 7 EC domains, a structural hallmark of cadherin superfamily [4, 5]. Cadherins interact with β-catenin via their cytoplasmic tails [23, 24], playing critical roles in contact inhibition, which is a key feature of normal proliferative cells and contributes to proper cell growth [25]. Loss of contact inhibition is thought to be a characteristic of cancer cells [26]. Previous study has revealed that CDHR2 can retain β-catenin in the cytoplasm from translocating into the nuclei which is a key process in Wnt/β-catenin signaling pathway leading to cell proliferation, thus restores contact inhibition in HCC cells [14]. Consistently, CDHR2 also abolished HCC cell anchorage-independent growth (Fig. 4D), another hallmark of carcinogenesis [26], suggesting that CDHR2 may suppress the metastatic potential of HCC cells.
Microarray analysis showed that AKT signaling pathway was inactivated in CDHR2-overexpressing HCC cells. Furthermore, Myr-AKT appeared to reverse the effect of CDHR2 on HCC growth and proliferation (Fig. 6), suggesting a pivotal role of AKT signaling in HCC development. This is in similar to a previous study on another tumor suppressor CYP3A5 which can suppress HCC migration and invasion through the inhibition of ROS/mTORC2/p-AKT signaling [27]. Consistent with the fact that COX-2 is a downstream target of AKT signaling in cancer [18, 28], our results also showed that Myr-AKT was able to restore CDHR2-suppressed COX-2 expression (Fig. 6). It appears that the imbalance between CDHR2 expression and AKT/Cox-2 signaling is correlated with the risk of HCC. The effect of CDHR2 on HCC migration and invasion requires further investigation.
In conclusion, our study demonstrates that CDHR2 is downregulated in HCC cell lines and tissues, and acts as a novel tumor suppressor in HCC growth both in vitro and in vivo. The protective role of CDHR2 in HCC is possibly through inactivation of AKT along with downregulation of COX-2 expression. Meanwhile, the silencing of CDHR2 in HCC is partially due to hypermethylation in its promoter. Better understanding of mechanisms of CDHR2 function may provide valuable clues for tumorigenesis and development of HCC.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
The authors thank National Nature Science Foundation of China (Grant No: 81472283, 81172306) for financial support.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen W, Zheng R, Baade PD. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-132
2. El-Fattah MA, Aboelmagd M, Elhamouly M. Prognostic factors of hepatocellular carcinoma survival after radiofrequency ablation: A US population-based study. United European Gastroenterol J. 2017;5:227-235
3. Dhanasekaran R, Limaye A, Cabrera R. Hepatocellular carcinoma: current trends in worldwide epidemiology, risk factors, diagnosis, and therapeutics. Hepat Med. 2012;4:19-37
4. Takeichi M. Cadherins: a molecular family important in selective cell-cell adhesion. Annu Rev Biochem. 1990;59:237-252
5. Morishita H, Yagi T. Protocadherin family: diversity, structure, and function. Curr Opin Cell Biol. 2007;19:584-592
6. Yu JS, Koujak S, Nagase S. et al. PCDH8, the human homolog of PAPC, is a candidate tumor suppressor of breast cancer. Oncogene. 2008;27:4657-4665
7. Harada H, Miyamoto K, Yamashita Y. et al. Prognostic signature of protocadherin 10 methylation in curatively resected pathological stage I non-small-cell lung cancer. Cancer Med. 2015;4:1536-1546
8. Haruki S, Imoto I, Kozaki K. et al. Frequent silencing of protocadherin 17, a candidate tumour suppressor for esophageal squamous cell carcinoma. Carcinogenesis. 2010;31:1027-1036
9. Imoto I, Izumi H, Yokoi S. et al. Frequent silencing of the candidate tumor suppressor PCDH20 by epigenetic mechanism in non-small-cell lung cancers. Cancer Res. 2006;66:4617-4626
10. Wang C, Yu G, Liu J. et al. Downregulation of PCDH9 predicts prognosis for patients with glioma. J Clin Neurosci. 2012;19:541-545
11. Fang S, Huang SF, Cao J. et al. Silencing of PCDH10 in hepatocellular carcinoma via de novo DNA methylation independent of HBV infection or HBX expression. Clin Exp Med. 2013;13:127-134
12. Narayan G, Scotto L, Neelakantan V. et al. Protocadherin PCDH10, involved in tumor progression, is a frequent and early target of promoter hypermethylation in cervical cancer. Genes Chromosomes Cancer. 2009;48:983-992
13. Okazaki N, Takahashi N, Kojima S. et al. Protocadherin LKC, a new candidate for a tumor suppressor of colon and liver cancers, its association with contact inhibition of cell proliferation. Carcinogenesis. 2002;23:1139-1148
14. Ose R, Yanagawa T, Ikeda S. et al. PCDH24-induced contact inhibition involves downregulation of beta-catenin signaling. Mol Oncol. 2009;3:54-66
15. Ose R, Oharaa O, Nagase T. Galectin-1 and Galectin-3 Mediate Protocadherin-24-Dependent Membrane Localization of beta-catenin in Colon Cancer Cell Line HCT116. Curr Chem Genomics. 2012;6:18-26
16. Lu B, Guo H, Zhao J. et al. Increased expression of iASPP, regulated by hepatitis B virus X protein-mediated NF-kappaB activation, in hepatocellular carcinoma. Gastroenterology. 2010;139:2183-2194 e2185
17. Zhao J, Lu B, Xu H. et al. Thirty-kilodalton Tat-interacting protein suppresses tumor metastasis by inhibition of osteopontin transcription in human hepatocellular carcinoma. Hepatology. 2008;48:265-275
18. Sobolewski C, Cerella C, Dicato M. et al. The role of cyclooxygenase-2 in cell proliferation and cell death in human malignancies. Int J Cell Biol. 2010;2010:215158
19. Dong Y, Wang A. Aberrant DNA methylation in hepatocellular carcinoma tumor suppression (Review). Oncol Lett. 2014;8:963-968
20. Jurkowska RZ, Jeltsch A. Silencing of gene expression by targeted DNA methylation: concepts and approaches. Methods Mol Biol. 2010;649:149-161
21. Leshchenko VV, Kuo PY, Shaknovich R. et al. Genomewide DNA methylation analysis reveals novel targets for drug development in mantle cell lymphoma. Blood. 2010;116:1025-1034
22. Miyamoto K, Fukutomi T, Akashi-Tanaka S. et al. Identification of 20 genes aberrantly methylated in human breast cancers. Int J Cancer. 2005;116:407-414
23. Shapiro L, Weis WI. Structure and biochemistry of cadherins and catenins. Cold Spring Harb Perspect Biol. 2009;1:a003053
24. Brasch J, Harrison OJ, Honig B. et al. Thinking outside the cell: how cadherins drive adhesion. Trends Cell Biol. 2012;22:299-310
25. Kim NG, Koh E, Chen X. et al. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc Natl Acad Sci U S A. 2011;108:11930-11935
26. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-674
27. Jiang F, Chen L, Yang YC. et al. CYP3A5 Functions as a Tumor Suppressor in Hepatocellular Carcinoma by Regulating mTORC2/Akt Signaling. Cancer Res. 2015;75:1470-1481
28. Xia S, Zhao Y, Yu S. et al. Activated PI3K/Akt/COX-2 pathway induces resistance to radiation in human cervical cancer HeLa cells. Cancer Biother Radiopharm. 2010;25:317-323
Author contact
![]() Corresponding author: Bin Lu, Department of Biochemical Pharmacy, School of Pharmacy, Second Military Medical University, Shanghai 200433, China. Tel: +86 21 81871327, Email: binluedu.cn; Jiaoyang Zheng, Department of Endocrinology, Changzheng Hospital, Second Military Medical University, Shanghai 200003, China. Tel: +86 21 81885373, Email: jyzhengedu.cn.
Corresponding author: Bin Lu, Department of Biochemical Pharmacy, School of Pharmacy, Second Military Medical University, Shanghai 200433, China. Tel: +86 21 81871327, Email: binluedu.cn; Jiaoyang Zheng, Department of Endocrinology, Changzheng Hospital, Second Military Medical University, Shanghai 200003, China. Tel: +86 21 81885373, Email: jyzhengedu.cn.