Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(4):893-902. doi:10.7150/jca.27491 This issue Cite
Research Paper
A network-based approach to identify DNA methylation and its involved molecular pathways in testicular germ cell tumors
Hao Bo1, Ke Cao4, Ruiling Tang1, Han Zhang1, Zhaojian Gong5, Zhizhong Liu6, Jianye Liu7, Jingjing Li3 ![]() , Liqing Fan1,2
, Liqing Fan1,2 ![]()
1. Institute of Reproductive and Stem Cell Engineering, School of Basic Medical Science, Central South University, Changsha, China
2. Reproductive & Genetic Hospital of CITIC-Xiangya, Changsha, China
3. Department of Plastic Surgery of Third Xiangya Hospital, Central South University, Changsha, China
4. Department of Oncology, Third Xiangya Hospital, Central South University, Changsha, Hunan, China
5. Department of Oral and Maxillofacial Surgery, The Second Xiangya Hospital, Central South University, Changsha, Hunan, China
6. Hunan Cancer Hospital and The Affliated Cancer Hospital of Xiangya School of Medicine, Central South University, Changsha, Hunan, China
7. Department of Urology, The Third Xiangya Hospital, Central South University, Changsha, Hunan, China
Received 2018-5-26; Accepted 2018-12-5; Published 2019-1-29
Abstract
Background: Testicular germ cell tumors (TGCT) is the most common testicular malignancy threaten young male reproductive health. This study aimed to identify aberrantly methylated-differentially expressed genes and pathways in TGCT by comprehensive bioinformatics analysis.
Methods: Data of gene expression microarrays (GSE3218, GSE18155) and gene methylation microarrays (GSE72444) were collected from GEO database. Integrated analysis acquired aberrantly methylated-genes. Functional and pathway enrichment analysis were performed using DAVID database. Protein-protein interaction (PPI) network was constructed by STRING and App Mcode was used for module analysis. GEPIA platform and DiseaseMeth database were used for confirming the expression and methylation levels of hub genes. Finally, Human Protein Atlas database was performed to evaluate the prognostic significance.
Results: Totally 604 hypomethylation-high expression and 147 hypermethylation-low genes were identified. The high expressed genes were enriched in biological processes of cell proliferation and migration. The top 8 hub genes of PPI network were GAPDH, VEGFA, PTPRC, RIPK4, MMP9, CSF1R, KRAS and FN1. After validation in GEPIA platform, all hub genes were elevated in TGCT tissues. Only MMP9, CSF1R and PTPRC showed hypomethylation-high expression status, which predicted the poor outcome of TGCT patients.
Conclusion: Our study indicated possible aberrantly methylated-differentially expressed genes and pathways in TGCT by bioinformatics analysis, which may provide novel insights for unraveling pathogenesis of TGCT.
Keywords: methylation, genetic etiology, bioinformatics analysis, testicular germ cell tumors
Introduction
Testicular germ cell tumors (TGCT) is the most common malignancy among young men between 17 and 45 years of age, which comprises around 98% of all testicular neoplasms [1]. The incidence rate of TGCT has been increasing over the last decades in large parts of the world [2]. TGCT are divided into seminomas (SE), non-seminomas (non-SE) and mixed tumors according histologic features. Currently, platinum chemotherapy and surgery are the main treatment for TGCT, but about 15% of the patients still result in poor prognosis [3].
And the embarrassing is that little has been known about the etiology of TGCT up to now. According to the statistics, approximately 25% of genetic factors is believed to contribute to TGCT susceptibility [4]. A gain of chromosome arm 12p is identified as highest effect and the most frequent event detected in TGCT [5]. But beyond that, only Kit and Ras gene family have been implicated repeatedly in different TGCT [6]. Therefore, identification of key differently expressed genes may provide a further understanding of the genetic etiology and molecular mechanism in TGCT occurrence and development.
DNA methylation is reported to lead to gene silencing via inhibiting gene transcription, which has become the most studied epigenetic mechanism [7]. Accumulated studies have showed that aberrant methylation of gene promoter regions is associated with occurrence, progression, and metastasis of several tumors, including ovarian carcinoma [8], gastric cancer [9] and so on. The profile of gene promoter methylation in TGCT was reported by several studies [10-12]. In this study, we focused on identifying the DNA methylation in TGCT from GEO online database. Integrated analysis and protein-protein interaction network were performed to explore the functions of abnormal expressed genes. Then the levels of mRNA and DNA methylation of top 8 hub genes were confirmed by the data from Cancer Genome Atlas (TCGA) or DiseaseMeth database, respectively. These identified genes may be used as potential biomarkers or therapeutic targets for TGCT.
Materials and methods
Microarray data information and gene identification
The gene express profile of GSE3218 (6 normal cases and 101 tumor cases) [13] and GSE18155 (7 normal cases and 26 tumor cases) [14], and gene methylation datasets of GSE72444 (9 normal cases and 3 tumor cases) were obtained from NCBI GEO database (GEO, http://www.ncbi.nlm.nih.gov/geo/). Limma package (http://www.bioconductor.org/packages/release/bioc/html/limma.html) in R (https://www.r-project.org/) was used to identify differentially expressed genes (DEGs) between normal testis cases and TGCT cases. Aberrantly methylated-DEGs were identified by GEO2R online software. The screening criteria of DEGs was defined with |t| > 2, P < 0.01, while which of differentially DNA methylation was defined with |t| > 8, P < 0.01. For integrated analysis, the gene microarray and DNA methylation data were processed in Bioinformatics & Evolutionary Genomics (Available online: http://bioinformatics.psb.ugent.be/webtools/Venn/), which were presented by Venn diagrams.
GO and KEGG pathway enrichment analysis
The annotation, visualization and integrated discovery (DAVID) database is a web-accessible program (https://david.ncifcrf.gov/), which provides an integrated analysis to reveal biological meaning behind given gene list. Candidate genes' functions and pathways enrichment were analyzed by using DAVID. The enrichment graphs were drawn by E Chart online software (http://www.ehbio.com/ImageGP/index.php/Home/Index/index.html). And the top 10 of the results were presented. The size of each circle indicates the counting number on each part, while the color represents the P-value of the enrichment analysis.
Integration of protein-protein interaction (PPI) network construction and modular analysis
The online database Retrieval of Interacting Genes (STRING, http://string-db.org) was used to evaluate proteins and protein-protein interaction information, the detail methods were used as previous report [15]. Interaction combined score ≥ 0.4 was regarded as significant. The results of significant interactions were imported into Cytoscape plugin to create network visualizations. After finishing PPI network, App Mcode was performed to make module analysis with the default parameters. (Degree cut off ≥ 8, Node score cut off ≥ 30 and Max depth = 100). Hub genes were defined as the genes with the top 8 degree.
Analysis of hub genes expression in TGCT tissue samples
The expression levels of top 8 hub genes in TGCT tissues and normal samples were validated by GEPIA (Gene Expression Profiling Interactive Analysis) platform, which is a free online database (http://gepia.cancer-pku.cn/). Search strategy was as previous report [16]. In brief, the difference in expression levels of each candidate gene between TGCT and normal testicular tissues from Genotype-Tissue Expression (GTEx) project were assessed by the linear model and the empirical Bayes method implemented by the R package limma, with adjusted P-value (Benjamini and Hochberg FDR). |log2FC| > 0.5 and P < 0.01 were considered to indicate a statistically significant difference.
Methylation and prognostic analysis of hub genes in TGCT tissue samples
The data of DNA methylation level of these top 8 hub genes were obtained from DiseaseMeth database (http://bio-bigdata.hrbmu.edu.cn/diseasemeth/), and analyzed as described previously [17]. The difference in DNA methylation between TGCT tissues and normal tissues were compared by Student's t-test. The survival curves of each hub gene were online drawn using the Human Protein Atlas database (http://www.proteinatlas.org/) [18].
Results
Identification of abnormal DNA methylated‑differentially expressed genes in TGCT
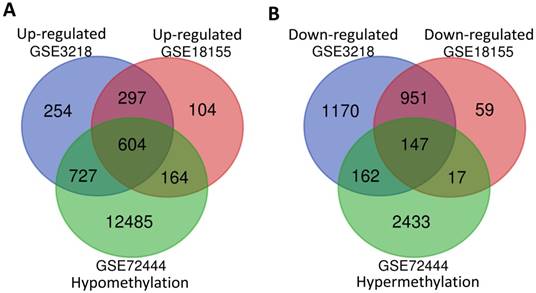
In order to screen the different expression of genes related to the development of TGCT, a total of two gene mRNA expression datasets (GSE3218 and GSE18155) and one DNA methylation dataset (GSE72444) were downloaded for integrated analysis from GEO database. As shown in Fig. 1A and B, 901 overlapping high-regulated genes and 1098 overlapping low-regulated genes were identified in the GSE3218, GSE18155 and GSE72444 datasets, respectively. For integrated analysis, totally 604 hypomethylation-up expression genes and 147 hypermethylation-down expression genes were screened from these three datasets.
The integrated analysis of abnormal DNA methylation genes in TGCT. The gene counting numbers were summarized in gene expression datasets (GSE3218 blue and GSE18155 red) and gene methylation dataset (GSE72444 green), respectively. Left panel (A) was represented the hypomethylation and up-regulated genes, while right panel (B) represented the hypermethylation and down-regulated genes. These two graphs were drawn by Bioinformatics & Evolutionary Genomics.
Gene function and pathways enrichment analysis
604 hypomethylation and up-regulated genes and 147 hypermethylation and down-regulated genes were respectively imported into online biological classification tool DAVID for functional analysis. GO term enrichment analysis was composed of three aspects, including biological process, cellular component, and molecular function. For biological process (Fig. 2A), the hypomethylation-upregulated genes were enriched in cell migration, osteoblast differentiation, cell adhesion, cell proliferation, ERK1 and ERK2 cascade, cell shape, innate immune response and apoptosis. The hypermethylation-downregulated genes were enriched in spermatid development, autophagy, cell cycle, starvation, fatty acid catabolic process, protein ubiquitination, membrane depolarization, phosphatidylethanolamine biosynthetic process and proteasome-mediated ubiquitin-dependent protein catabolic process. GO cell component analysis showed that the upregulated genes were significantly enriched in the extracellular exosome, focal adhesion, endoplasmic reticulum, cell surface, plasma membrane, while downregulated genes were enriched in nucleus, centriole, lysosome (Fig. 2B). Besides, the results showed that hypomethylation-upregulated genes were associated with the molecular function, including calcium ion binding, GTP binding, extracellular matrix structural constituent, GTPase activator activity (Fig. 2C). The hypermethylation-downregulated genes were mostly enriched in molecular function of Zinc ion binding and ubiquitin-protein transferase activity.
GO and KEGG pathway enrichment analysis. GO analysis covered three groups, including biological process (A), cellular component (B) and molecular function (C). (D) The most significantly enriched molecular pathways of the hypomethylation-upregulated and hypermethylation-downregulated genes were figured out by KEGG analysis. The size of each circle indicates the counting number on each part, while the color represents the P-value of the enrichment analysis.
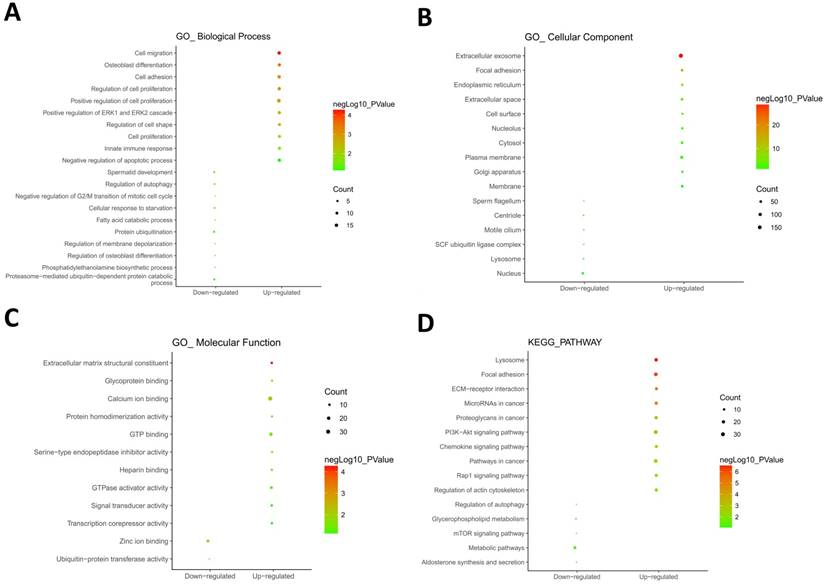
To further understand the functions of such identified genes, these overlapping genes were mapped into DAVID for KEGG pathway enrichment analysis (Fig. 2D). The most significantly molecular pathways of the hypomethylation-upregulated genes were enriched in lysosome, focal adhesion, ECM-receptor interaction, microRNA in cancer, proteoglycans in cancer, PI3K-Akt signaling pathway, chemokine signaling pathway, Rap 1 signaling pathway and regulation of actin cytoskeleton. In addition, the hypermethylation-downregulated genes were enriched in five KEGG pathways, which including autophagy, glycerophospholipid metabolism, mTOR signaling pathway, metabolic pathway, and aldosterone synthesis and secretion.
PPI networks and module analysis
The overlapping abnormal DNA methylated‑differentially expressed genes indicated a distinct set of interactions and networks, these identified genes were imported into STRING database to construct PPI networks. The analysis results were then put into Cytoscape software for creating network visualizations. The PPI network graph was shown in Fig. 3A, which included 638 nodes and 3382 edges. The most significant 8 node with higher degrees were identified as hub genes, including glyceraldehyde-3-phosphate dehydrogenase (GAPDH), vascular endothelial growth factor A (VEGFA), protein tyrosine phosphatase, receptor type C (PTPRC), receptor interacting serine/threonine kinase 4 (RIPK4), matrix metallopeptidase 9 (MMP9), colony stimulating factor 1 receptor (CSF1R), KRAS proto-oncogene, GTPase (KRAS), fibronectin 1 (FN1).
Protein-protein interaction (PPI) network complex and top three modules of the hypomethylation-upregulated and hypermethylation-downregulated genes. (A) The PPI networks. The size of node indicated the degree value. Yellow nodes represented hub genes. The thickness of edges represented correlation degree. (B-D) Module 1 consists of 30 nodes, module 2 consists of 35 nodes and module 3 consists of 58 nodes.
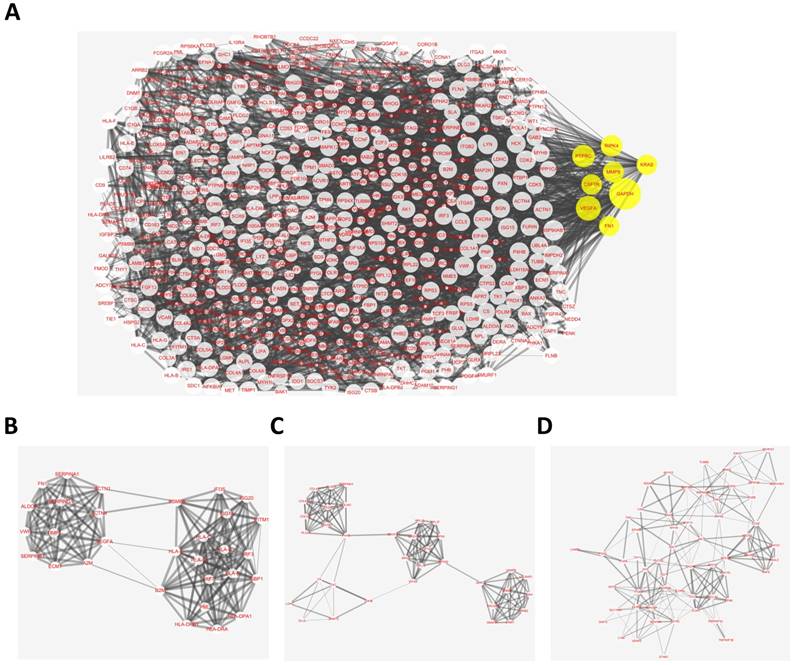
According the degree of importance, 3 significant modules (Fig. 3B-D) from PPI network were further analyzed by App Mcode. The KEGG pathway enrichment analysis of the top 3 module genes using DAVID showed enrichment in the cell cycle, cell proliferation, pathways in cancer, all of which have a close relationship with tumor biology (supplemental table).
The mRNA expression levels of 8 hub genes in TGCT
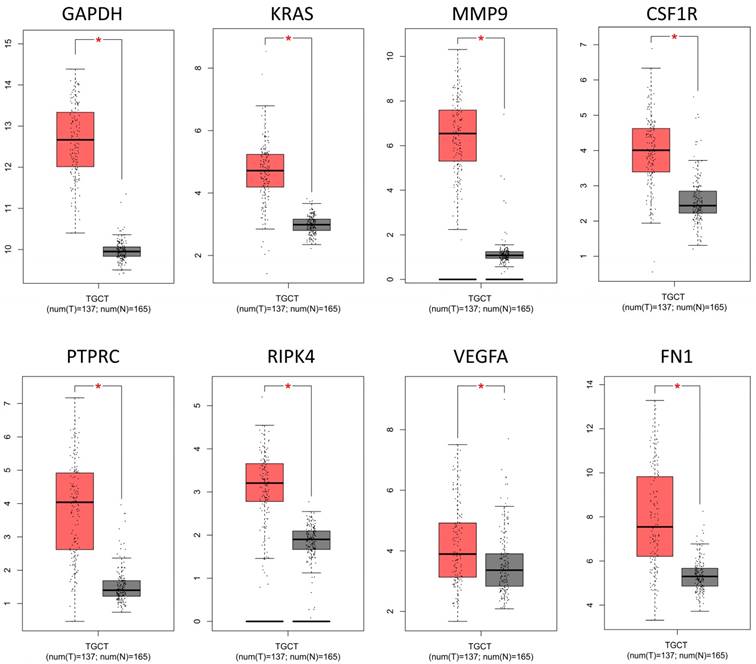
To confirm the expression levels of identified 8 hub genes, we collected the related published data from TCGA datasets and analyzed by GEPIA platform. As expected, the results revealed that all hub genes had higher levels in tumor tissues than that in normal tissues (Fig. 4), suggesting these 8 genes might be candidate genes affected by aberrant methylation during TGCT development.
The mRNA expression levels of 8 hub genes in TGCT. The published online data of gene mRNA expression level were analyzed by GEPIA platform. These 8 hub genes were all up-regulated in the TGCT tissues comparing with the normal tissues, including GAPDH, VEGFA, PTPRC, RIPK4, MMP9, CSF1R, KRAS, FN1. *P<0.05.
Methylation analysis of 8 hub genes in TGCT
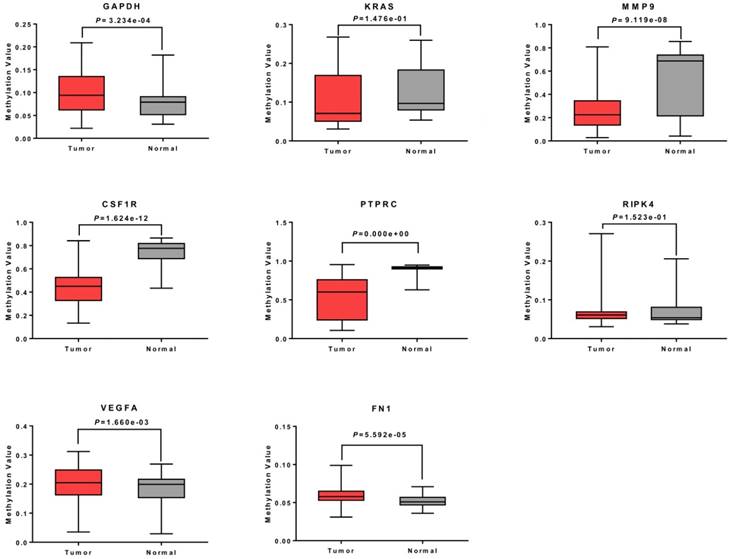
To further verify the methylation levels of GAPDH, VEGFA, PTPRC, RIPK4, MMP9, CSF1R, KRAS, and FN1, the data of DNA methylation status in TGCT was obtained from DiseaseMeth database. We found that DNA hypomethylation in the promoters of MMP9, CSF1R, and PTPRC, which might be one of the main causes of aberrant high expression in TGCT. Unexpectedly, for KRAS and RIPK4, there was no significant difference in DNA methylation value between TGCT and normal tissues. Inversely, GAPDH, VEGFA and FN1 even showed aberrant promoters hypermethylation in TGCT. The results were summarized in Fig. 5.
Methylation analysis of 8 hub genes in TGCT. The promoters of MMP9, CSF1R and PTPRC were hypomethylated. No obvious change was observed in methylation levels of KRAS and RIPK4, whereas GAPDH, VEGFA and FN1 showed gene hypermethylation in TGCT.
The prognostic significance of hub genes in TGCT
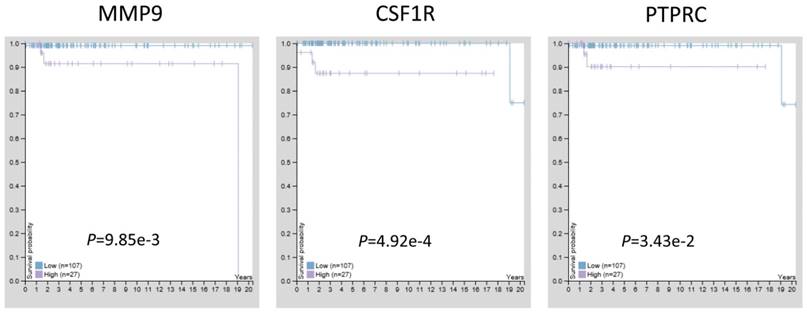
Previous studies have confirmed that MMP9, CSF1R, and PTPRC were up-regulated in TGCT with hypomethylation, suggesting these three genes may act as tumor promoters and involve in TGCT procession. To estimate the prognostic significance of abnormal expressed MMP9, CSF1R, and PTPRC, the survival time and gene expression levels were acquired from the Human Protein Atlas database (https://www.proteinatlas.org/). Kaplan-Meier method was used to evaluate the survival time data of each identified gene. The analysis results showed that higher levels of MMP9, CSF1R, and PTPRC were related to shorter survival time (Fig. 6), these three key genes may provide potential makers for TGCT diagnosis and prognosis evaluation.
The prognostic significance of hub genes in TGCT. Higher levels of MMP9, CSF1R and PTPRC were associated with poorer overall survival time, respectively.
Discussion
Testicular germ cell tumor (TGCT) is one of the most common malignancies among young men in large parts of the world [19, 20]. According to the statistics, white Caucasian populations in industrialized countries usually have the highest incidence of TGCT [21]. However, the reality is that the genetic etiology and pathogenicity related genes for TGCT are a lot of unknowns. Therefore, revealing the underlying molecular mechanism of TGCT is a key approach to greatly benefit the diagnosis and treatment. Fortunately, the emerging microarray and next-generation sequencing technology help us gain deeper insight into the thousands or millions of human genome sequences concurrently [22-30]. As a consequence, these related technologies have been adopted extensively to screen the potential diagnostic indicator and treatment target points for multiple diseases, such as cancer, and metabolic disease [31-37].
In present study, we analyzed two gene mRNA expression profiles (GSE3218 and GSE18155) and one DNA methylation dataset (GSE72444) of human TGCT tissues from the available GEO database and identified 604 hypomethylation-high expression genes and 147 hypermethylation-low expression genes. These identified abnormal DNA methylated-differentially expressed genes were imported into online biological classification tool DAVID for further functional analysis. The hypomethylation-upregulated genes were enriched in biological processes of cell migration and proliferation. GO cell component analysis showed that the upregulated genes were significantly enriched in the extracellular exosome, focal adhesion, endoplasmic reticulum, cell surface [38-42]. Besides, for molecular function, the hypomethylation-upregulated genes were significantly enriched in calcium ion binding, GTP binding, extracellular matrix structural constituent, GTPase activator activity [43]. It is reasonable because high capacity of cell migration and proliferation are two apparent hallmarks of tumor cells including TGCT [21], which promote tumor development. GO cell component analysis suggested that these genes are mainly located in membranaceous structures of cell and organelle. Endoplasmic reticulum stress has been known to create a profound effect on cancer cell proliferation and survival, which also increase injury of spermatogonia and decrease of spermatocytes [44]. Notably, KEGG pathway enrichment analysis suggested significant enrichment in pathways including ECM-receptor interaction, microRNA in cancer [45-48] and PI3K-Akt signaling pathway. The previous studies reported that PI3K-Akt signaling activation is associated with tumor cell growth, proliferation, migration and angiogenesis of TGCT [49-52]. It is well known that increased cell proliferation and migration are the leading causes for malignant tumor initiation and progression [53-55], thus our biological analysis results were consistent with previous researches.
PPI network of hypomethylation-high expression genes illustrated the protein-protein interactome of the top 8 hub genes, named GAPDH, VEGFA, PTPRC, RIPK4, MMP9, CSF1R, KRAS, and FN1, which may provide a new sight for the therapeutic strategy in TGCT by constructing the PPI network. Module analysis of the PPI network revealed that the development of TGCT was associated with cell cycle, cell proliferation and pathways in cancer, which was consistent with our pathway analysis. Seven of the eight hub genes are reported to directly participate in the identified pathways, except for RIPK4. Additionally, our previous study found that upregulation of RIPK4 increases the expression of VEGFA and contributes to the progression of bladder urothelial carcinoma [56].
To further confirm the top 8 hub genes expression levels in TGCT tissues, the related published data from TCGA datasets were collected and analyzed by GEPIA platform. As expected, all hub genes had higher levels in tumor tissues than that in normal tissues. However, whether DNA hypomethylation contribute to the abnormal high expression status was still unclear. Then, we analyzed the methylation data of TGCT tissues acquired from DiseaseMeth database. Only MMP9, CSF1R, and PTPRC were confirmed with hypomethylation. No obvious change was observed in methylation level of KRAS, RIPK4, whereas VEGFA, GAPDH and FN1 showed gene hypermethylation in TGCT. These differences reflect that some other reasons may affect the expression level of these five genes. Except DNA methylation, histone modification is also an important way of regulating of gene expression and associated with transcriptional activity [57, 58]. Enrichment for histone H3 Lys 27 acetylation (H3K27Ac) has been found in the DNA regions of VEGFA, GAPDH and FN1 as confirmed by UCSC Genome Browser (https://genome.ucsc.edu/), which may cause a significant increase of mRNA levels of VEGFA, GAPDH and FN1 when their promoters are hypermethylated. In our future work, we will focus on this issue to deeper insight into the gene regulatory mechanism.
MMP9, CSF1R, and PTPRC, are hypomethylation-high expressed genes, suggesting they may involve in TGCT procession. The Kaplan-Meier analysis results showed that higher levels of MMP9, CSF1R, and PTPRC were related to shorter survival time of TGCT patients. MMP9 is a class of enzymes involved in the degradation of the extracellular matrix, which was up-regulated in tumor tissue samples of multi cancer [59-61], such as TGCT [62], breast cancer [63], lung cancer [64]. Moreover, MMP9 overexpression was significantly associated with poor survival of nasopharyngeal carcinoma [65] and glioblastoma [66]. CSF1R is a cell-surface protein, while PTPRC is a member of the protein tyrosine phosphatase (PTP) family [67, 68]. High expression levels of CSF1R were associated with higher lung cancer-specific mortality [69]. CSF1R and PTPRC are known to regulate a variety of cellular processes including cell growth, differentiation, and oncogenic transformation [70]. The present study is the first to identify the prognostic significance of these three genes, which may serve as valuable prognostic indicators of TGCT.
In conclusion, with the microarray gene expression profiling and gene methylation datasets, this study provides a network-based approach to identify abnormal DNA methylated-differentially expressed genes, which may play important roles in initiation and progression of TGCT. Hub genes including PTPRC, MMP9 and CSF1R might serve as potential biomarkers for diagnosis, treatment and prognosis evaluation of TGCT in the future.
Acknowledgements
This work was supported by the National Key Research and Development Program of China (2016YFC1000600), the National Natural Science Foundation of China (No. 31472054), the Fundamental Research Funds for the Central Universities of Central South University (1053320171023) and the Special Fund of Clinical Medicine of Chinese Medical Association (17020280697). Fundamental Research Funds for the Central Universities of Central South University (2017zzts220).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Manku G, Hueso A, Brimo F, Chan P, Gonzalez-Peramato P, Jabado N. et al. Changes in the expression profiles of claudins during gonocyte differentiation and in seminomas. Andrology. 2016;4:95-110
2. Das MK, Furu K, Evensen HF, Haugen OP, Haugen TB. Knockdown of SPRY4 and SPRY4-IT1 inhibits cell growth and phosphorylation of Akt in human testicular germ cell tumours. Sci Rep. 2018;8:2462
3. International Prognostic Factors Study G, Lorch A, Beyer J, Bascoul-Mollevi C, Kramar A, Einhorn LH. et al. Prognostic factors in patients with metastatic germ cell tumors who experienced treatment failure with cisplatin-based first-line chemotherapy. J Clin Oncol. 2010;28:4906-11
4. Turnbull C, Rahman N. Genome-wide association studies provide new insights into the genetic basis of testicular germ-cell tumour. Int J Androl. 2011;34:e86-96 discussion e-7
5. Hanna NH, Einhorn LH. Testicular cancer-discoveries and updates. N Engl J Med. 2014;371:2005-16
6. Kanetsky PA, Mitra N, Vardhanabhuti S, Li M, Vaughn DJ, Letrero R. et al. Common variation in KITLG and at 5q31.3 predisposes to testicular germ cell cancer. Nat Genet. 2009;41:811-5
7. Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6-21
8. Loginov VI, Pronina IV, Burdennyy AM, Filippova EA, Kazubskaya TP, Kushlinsky DN. et al. Novel miRNA genes deregulated by aberrant methylation in ovarian carcinoma are involved in metastasis. Gene. 2018;662:28-36
9. Shao L, Chen Z, Peng D, Soutto M, Zhu S, Bates A. et al. Methylation of the HOXA10 Promoter Directs miR-196b-5p-Dependent Cell Proliferation and Invasion of Gastric Cancer Cells. Mol Cancer Res. 2018;16:696-706
10. Smiraglia DJ, Szymanska J, Kraggerud SM, Lothe RA, Peltomaki P, Plass C. Distinct epigenetic phenotypes in seminomatous and nonseminomatous testicular germ cell tumors. Oncogene. 2002;21:3909-16
11. Netto GJ, Nakai Y, Nakayama M, Jadallah S, Toubaji A, Nonomura N. et al. Global DNA hypomethylation in intratubular germ cell neoplasia and seminoma, but not in nonseminomatous male germ cell tumors. Mod Pathol. 2008;21:1337-44
12. Brait M, Maldonado L, Begum S, Loyo M, Wehle D, Tavora FF. et al. DNA methylation profiles delineate epigenetic heterogeneity in seminoma and non-seminoma. Br J Cancer. 2012;106:414-23
13. Kushwaha R, Jagadish N, Kustagi M, Tomishima MJ, Mendiratta G, Bansal M. et al. Interrogation of a context-specific transcription factor network identifies novel regulators of pluripotency. Stem Cells. 2015;33:367-77
14. Palmer RD, Murray MJ, Saini HK, van Dongen S, Abreu-Goodger C, Muralidhar B. et al. Malignant germ cell tumors display common microRNA profiles resulting in global changes in expression of messenger RNA targets. Cancer Res. 2010;70:2911-23
15. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A. et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808-15
16. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98-W102
17. Xiong Y, Wei Y, Gu Y, Zhang S, Lyu J, Zhang B. et al. DiseaseMeth version 2.0: a major expansion and update of the human disease methylation database. Nucleic Acids Res. 2017;45:D888-D95
18. Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A. et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419
19. Chia VM, Quraishi SM, Devesa SS, Purdue MP, Cook MB, McGlynn KA. International trends in the incidence of testicular cancer, 1973-2002. Cancer Epidemiol Biomarkers Prev. 2010;19:1151-9
20. Wei F, Wu Y, Tang L, Xiong F, Guo C, Li X. et al. Trend analysis of cancer incidence and mortality in China. Sci China Life Sci. 2017;60:1271-5
21. Trabert B, Chen J, Devesa SS, Bray F, McGlynn KA. International patterns and trends in testicular cancer incidence, overall and by histologic subtype, 1973-2007. Andrology. 2015;3:4-12
22. Bo H, Gong Z, Zhang W, Li X, Zeng Y, Liao Q. et al. Upregulated long non-coding RNA AFAP1-AS1 expression is associated with progression and poor prognosis of nasopharyngeal carcinoma. Oncotarget. 2015;6:20404-18
23. Zeng Z, Bo H, Gong Z, Lian Y, Li X, Li X. et al. AFAP1-AS1, a long noncoding RNA upregulated in lung cancer and promotes invasion and metastasis. Tumour Biol. 2016;37:729-37
24. Lian Y, Xiong F, Yang L, Bo H, Gong Z, Wang Y. et al. Long noncoding RNA AFAP1-AS1 acts as a competing endogenous RNA of miR-423-5p to facilitate nasopharyngeal carcinoma metastasis through regulating the Rho/Rac pathway. J Exp Clin Cancer Res. 2018;37:253
25. Gong Z, Yang Q, Zeng Z, Zhang W, Li X, Zu X. et al. An integrative transcriptomic analysis reveals p53 regulated miRNA, mRNA, and lncRNA networks in nasopharyngeal carcinoma. Tumour Biol. 2016;37:3683-95
26. Xu K, Xiong W, Zhou M, Wang H, Yang J, Li X. et al. Integrating ChIP-sequencing and digital gene expression profiling to identify BRD7 downstream genes and construct their regulating network. Mol Cell Biochem. 2016;411:57-71
27. Wang Y, Xue D, Li Y, Pan X, Zhang X, Kuang B. et al. The Long Noncoding RNA MALAT-1 is A Novel Biomarker in Various Cancers: A Meta-analysis Based on the GEO Database and Literature. J Cancer. 2016;7:991-1001
28. Yang L, Tang Y, He Y, Wang Y, Lian Y, Xiong F. et al. High Expression of LINC01420 indicates an unfavorable prognosis and modulates cell migration and invasion in nasopharyngeal carcinoma. J Cancer. 2017;8:97-103
29. Yu J, Liu Y, Guo C, Zhang S, Gong Z, Tang Y. et al. Upregulated long non-coding RNA LINC00152 expression is associated with progression and poor prognosis of tongue squamous cell carcinoma. J Cancer. 2017;8:523-30
30. Fan C, Wang J, Tang Y, Wang Y, Xiong F, Zhang S. et al. Long non-coding RNA LOC284454 promotes migration and invasion of nasopharyngeal carcinoma via modulating the Rho/Rac signaling pathway. Carcinogenesis. 2018
31. He Y, Jing Y, Wei F, Tang Y, Yang L, Luo J. et al. Long non-coding RNA PVT1 predicts poor prognosis and induces radioresistance by regulating DNA repair and cell apoptosis in nasopharyngeal carcinoma. Cell Death Dis. 2018;9:235
32. Tu C, Zeng Z, Qi P, Li X, Guo C, Xiong F. et al. Identification of genomic alterations in nasopharyngeal carcinoma and nasopharyngeal carcinoma-derived Epstein-Barr virus by whole genome sequencing. Carcinogenesis. 2018;39:1517-28
33. He R, Liu P, Xie X, Zhou Y, Liao Q, Xiong W. et al. circGFRA1 and GFRA1 act as ceRNAs in triple negative breast cancer by regulating miR-34a. J Exp Clin Cancer Res. 2017;36:145
34. Wang Y, Mo Y, Yang X, Zhou R, Wu Z, He Y. et al. Long non-coding RNA AFAP1-AS1 is a novel biomarker in various cancers: a systematic review and meta-analysis based on the literature and GEO datasets. Oncotarget. 2017;8:102346-60
35. Tu C, Zeng Z, Qi P, Li X, Yu Z, Guo C. et al. Genome-Wide Analysis of 18 Epstein-Barr Viruses Isolated from Primary Nasopharyngeal Carcinoma Biopsy Specimens. J Virol. 2017:91
36. Tang Y, He Y, Shi L, Yang L, Wang J, Lian Y. et al. Co-expression of AFAP1-AS1 and PD-1 predicts poor prognosis in nasopharyngeal carcinoma. Oncotarget. 2017;8:39001-11
37. Yu J, Liu Y, Gong Z, Zhang S, Guo C, Li X. et al. Overexpression long non-coding RNA LINC00673 is associated with poor prognosis and promotes invasion and metastasis in tongue squamous cell carcinoma. Oncotarget. 2017;8:16621-32
38. Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y. et al. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017;8:761-73
39. Wei F, Tang L, He Y, Wu Y, Shi L, Xiong F. et al. BPIFB1 (LPLUNC1) inhibits radioresistance in nasopharyngeal carcinoma by inhibiting VTN expression. Cell Death Dis. 2018;9:432
40. Wei F, Wu Y, Tang L, He Y, Shi L, Xiong F. et al. BPIFB1 (LPLUNC1) inhibits migration and invasion of nasopharyngeal carcinoma by interacting with VTN and VIM. Br J Cancer. 2018;118:233-47
41. Wang JP, Tang YY, Fan CM, Guo C, Zhou YH, Li Z. et al. The role of exosomal non-coding RNAs in cancer metastasis. Oncotarget. 2018;9:12487-502
42. Wang YA, Li XL, Mo YZ, Fan CM, Tang L, Xiong F. et al. Effects of tumor metabolic microenvironment on regulatory T cells. Mol Cancer. 2018;17:168
43. Tang Y, He Y, Zhang P, Wang J, Fan C, Yang L. et al. LncRNAs regulate the cytoskeleton and related Rho/ROCK signaling in cancer metastasis. Mol Cancer. 2018;17:77
44. Zhang S, Jiang C, Liu H, Guan Z, Zeng Q, Zhang C. et al. Fluoride-elicited developmental testicular toxicity in rats: roles of endoplasmic reticulum stress and inflammatory response. Toxicol Appl Pharmacol. 2013;271:206-15
45. Fan C, Tang Y, Wang J, Xiong F, Guo C, Wang Y. et al. The emerging role of Epstein-Barr virus encoded microRNAs in nasopharyngeal carcinoma. J Cancer. 2018;9:2852-64
46. Zhong Y, Du Y, Yang X, Mo Y, Fan C, Xiong F. et al. Circular RNAs function as ceRNAs to regulate and control human cancer progression. Mol Cancer. 2018;17:79
47. Song Y, Li X, Zeng Z, Li Q, Gong Z, Liao Q. et al. Epstein-Barr virus encoded miR-BART11 promotes inflammation-induced carcinogenesis by targeting FOXP1. Oncotarget. 2016;7:36783-99
48. Zeng Z, Huang H, Huang L, Sun M, Yan Q, Song Y. et al. Regulation network and expression profiles of Epstein-Barr virus-encoded microRNAs and their potential target host genes in nasopharyngeal carcinomas. Sci China Life Sci. 2014;57:315-26
49. Yang NQ, Luo XJ, Zhang J, Wang GM, Guo JM. Crosstalk between Meg3 and miR-1297 regulates growth of testicular germ cell tumor through PTEN/PI3K/AKT pathway. Am J Transl Res. 2016;8:1091-9
50. Yang Y, Liao Q, Wei F, Li X, Zhang W, Fan S. et al. LPLUNC1 inhibits nasopharyngeal carcinoma cell growth via down-regulation of the MAP kinase and cyclin D1/E2F pathways. PLoS One. 2013;8:e62869
51. Liang F, Li Q, Li X, Li Z, Gong Z, Deng H. et al. TSC22D2 interacts with PKM2 and inhibits cell growth in colorectal cancer. Int J Oncol. 2016;49:1046-56
52. Li Q, Chen P, Zeng Z, Liang F, Song Y, Xiong F. et al. Yeast two-hybrid screening identified WDR77 as a novel interacting partner of TSC22D2. Tumour Biol. 2016;37:12503-12
53. Gong Z, Zhang S, Zeng Z, Wu H, Yang Q, Xiong F. et al. LOC401317, a p53-regulated long non-coding RNA, inhibits cell proliferation and induces apoptosis in the nasopharyngeal carcinoma cell line HNE2. PLoS One. 2014;9:e110674
54. Deng X, Xiong F, Li X, Xiang B, Li Z, Wu X. et al. Application of atomic force microscopy in cancer research. J Nanobiotechnology. 2018;16:102
55. Tang L, Wei F, Wu Y, He Y, Shi L, Xiong F. et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J Exp Clin Cancer Res. 2018;37:87
56. Liu JY, Zeng QH, Cao PG, Xie D, Chen X, Yang F. et al. RIPK4 promotes bladder urothelial carcinoma cell aggressiveness by upregulating VEGF-A through the NF-kappaB pathway. Br J Cancer. 2018;118:1617-27
57. Tang Y, Wang J, Lian Y, Fan C, Zhang P, Wu Y. et al. Linking long non-coding RNAs and SWI/SNF complexes to chromatin remodeling in cancer. Mol Cancer. 2017;16:42
58. Zhou R, Wu Y, Wang W, Su W, Liu Y, Wang Y. et al. Circular RNAs (circRNAs) in cancer. Cancer Lett. 2018;425:134-42
59. He B, Li W, Wu Y, Wei F, Gong Z, Bo H. et al. Epstein-Barr virus-encoded miR-BART6-3p inhibits cancer cell metastasis and invasion by targeting long non-coding RNA LOC553103. Cell Death Dis. 2016;7:e2353
60. Yan Q, Zeng Z, Gong Z, Zhang W, Li X, He B. et al. EBV-miR-BART10-3p facilitates epithelial-mesenchymal transition and promotes metastasis of nasopharyngeal carcinoma by targeting BTRC. Oncotarget. 2015;6:41766-82
61. Yang L, Tang Y, Xiong F, He Y, Wei F, Zhang S. et al. LncRNAs regulate cancer metastasis via binding to functional proteins. Oncotarget. 2018;9:1426-43
62. Gilbert DC, Chandler I, Summersgill B, McIntyre A, Missiaglia E, Goddard NC. et al. Genomic gain and over expression of CCL2 correlate with vascular invasion in stage I non-seminomatous testicular germ-cell tumours. Int J Androl. 2011;34:e114-21 discussion e21
63. Weiler J, Mohr M, Zanker KS, Dittmar T. Matrix metalloproteinase-9 (MMP9) is involved in the TNF-alpha-induced fusion of human M13SV1-Cre breast epithelial cells and human MDA-MB-435-pFDR1 cancer cells. Cell Commun Signal. 2018;16:14
64. He W, Zhang H, Wang Y, Zhou Y, Luo Y, Cui Y. et al. CTHRC1 induces non-small cell lung cancer (NSCLC) invasion through upregulating MMP-7/MMP-9. BMC Cancer. 2018;18:400
65. Zhou DN, Deng YF, Li RH, Yin P, Ye CS. Concurrent alterations of RAGE, RECK, and MMP9 protein expression are relevant to Epstein-Barr virus infection, metastasis, and survival in nasopharyngeal carcinoma. Int J Clin Exp Pathol. 2014;7:3245-54
66. Li Q, Chen B, Cai J, Sun Y, Wang G, Li Y. et al. Comparative Analysis of Matrix Metalloproteinase Family Members Reveals That MMP9 Predicts Survival and Response to Temozolomide in Patients with Primary Glioblastoma. PLoS One. 2016;11:e0151815
67. Zhang Y, Xia M, Jin K, Wang S, Wei H, Fan C. et al. Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol Cancer. 2018;17:45
68. Fan C, Tang Y, Wang J, Xiong F, Guo C, Wang Y. et al. Role of long non-coding RNAs in glucose metabolism in cancer. Mol Cancer. 2017;16:130
69. Inamura K, Shigematsu Y, Ninomiya H, Nakashima Y, Kobayashi M, Saito H. et al. CSF1R-Expressing Tumor-Associated Macrophages, Smoking and Survival in Lung Adenocarcinoma: Analyses Using Quantitative Phosphor-Integrated Dot Staining. Cancers (Basel). 2018:10
70. Brahmi M, Vinceneux A, Cassier PA. Current Systemic Treatment Options for Tenosynovial Giant Cell Tumor/Pigmented Villonodular Synovitis: Targeting the CSF1/CSF1R Axis. Curr Treat Options Oncol. 2016;17:10
Author contact
![]() Corresponding authors: Jingjing Li, Department of Plastic Surgery of Third Xiangya Hospital, Central South University, Changsha, China; Liqing Fan, Institute of Reproductive and Stem Cell Engineering, School of Basic Medical Science, Central South University, Changsha, China; Reproductive & Genetic Hospital of CITIC-Xiangya, Changsha, China. E-mail: jingjingliedu.cn and liqingfanedu.cn
Corresponding authors: Jingjing Li, Department of Plastic Surgery of Third Xiangya Hospital, Central South University, Changsha, China; Liqing Fan, Institute of Reproductive and Stem Cell Engineering, School of Basic Medical Science, Central South University, Changsha, China; Reproductive & Genetic Hospital of CITIC-Xiangya, Changsha, China. E-mail: jingjingliedu.cn and liqingfanedu.cn