Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(5):1077-1085. doi:10.7150/jca.29440 This issue Cite
Research Paper
Annexin A2 Silencing Inhibits Proliferation and Epithelial-to-mesenchymal Transition through p53-Dependent Pathway in NSCLCs
Minhua Wu1*, Yanqin Sun2*, Feipeng Xu3*, Yanqing Liang1, Hao Liu4, Yanmei Yi1 ![]()
1. Department of Histology and Embryology, Guangdong Medical University, Zhanjiang 524023, Guangdong, China
2. Department of Pathology, Guangdong Medical University, Dongguan 523808, Guangdong, China
3. Department of Gastrointestinal Surgery, Affiliated hospital of Guangdong Medical University, Zhanjiang 524000, Guangdong, China
4. Cancer Hospital and Cancer Research Institute, Guangzhou Medical University, Guangzhou 510095, Guangdong, China
*These authors contributed equally to this work.
Received 2018-8-23; Accepted 2018-12-15; Published 2019-1-29
Abstract
Annexin A2 has been involved in cancer cell adhesion, invasion and metastasis. However, the exact function and mechanism of Annexin A2 in tumor progression of NSCLCs have not been elucidated. In this study, we showed that Annexin A2 was evidently overexpressed in human NSCLCs cell lines and NSCLCs tissues. Clinicopathologic analysis showed that Annexin A2 expression was significantly correlated with clinical stage, and lymph node metastasis. Kaplan-Meier analysis revealed that patients with high Annexin A2 expression had poorer overall survival rates than those with low Annexin A2 expression. Moreover, we found that knockdown of Annexin A2 significantly suppressed cell proliferation and invasion of NSCLCs cells. Mechanistically, our studies showed that knockdown of Annexin A2 increased the expression of p53, which in turn, induced cell cycle G2 arrest and inhibited epithelial-to-mesenchymal transition (EMT). Taken together, these data suggest that Annexin A2 plays an important role in NSCLCs progression, which could serve as a potential prognosis marker and a novel therapeutic target for NSCLCs.
Keywords: Annexin A2, NSCLCs, cell proliferation, epithelial-to-mesenchymal transition, p53
Introduction
Lung cancer is the most common cancer worldwide with high incidence and mortality [1]. It could be divided into two categories, small cell lung cancers (SCLCs) and non-small cell lung cancers (NSCLCs). NSCLCs including adenocarcinoma, squamous cell carcinoma and large cell carcinoma, constitute 80% of all lung cancers [2]. Although great achievements have been made in NSCLCs treatment recent years, 5-year survival rate after surgery is less than 18%. Unfortunately, the majority of NSCLCs patients have been diagnosed with a late stage of cancer and lose the best chance for operative treatment [3]. Therefore, it is necessary to find more effective diagnostic and therapeutic targets for NSCLCs.
Annexin A2 (also called ANXA2) is mainly expressed in endothelial cells. It exists not only as a soluble monomer in cytoplasm, but also as a complex associated with cellular membranes [4, 5]. It always combines with S100A10 to form a tetramer which promotes plasmin generation [6, 7]. As a result, Annexin A2 normally contributes to clearance of fibrin formation on the blood vessel surface in the case of minor vascular injury [8]. Intriguingly, high expression of Annexin A2 has been found in various types of cancer including ovarian cancer [9], hepatocellular carcinoma [10], laryngeal cancer [11] and so on, and is correlated with poor tumor prognosis. Extensive researches demonstrate that Annexin A2 plays important roles in tumor proliferation, invasion and metastasis [12-14]. However, the exact function and mechanism of Annexin A2 in tumor progression of NSCLCs need to be further determined.
In the present study, we found that Annexin A2 was overexpressed in human NSCLCs cell lines and NSCLCs tissues. Knockdown of Annexin A2 significantly suppressed cell proliferation and invasion of NSCLCs cells, and the biological effect of Annexin A2 on proliferation and invasion was mediated by the inhibition of p53, which in turn, promoted cell cycle progression and EMT.
Materials and Methods
Cell lines and cell culture
Lung cancer cell line A549 (CCL-185), H460 (HTB-177), H1299 (CRL-5803), H1975 (CRL-5908) and normal lung epithelial cell line Beas-2B (CRL-9609) were all obtained from ATCC. Cells were cultured in RPMI 1640 medium (Gibco, Thermo Fisher Scientific, USA) supplemented with 10% fetal bovin serum (Gibco, Thermo Fisher Scientific, USA), 100U/ml penicillin, and 10μg/ml streptomycin at 37°C in a 5% CO2 atmosphere.
Antibodies
Antibodies for Annexin A2, β-actin, p53, Vimentin, E-cadherin, N-cadherin, Zeb1, Snail and Slug were all from Cell Signaling Technology (USA). Antibodies for p21, p27, Cyclin B1, CDK1 and CDK2 were all from Santa Cruz Biotechnology (USA).
Western blot assay
Western blot assay was mainly performed as the method in reference [15]. Cells were harvested and proteins were extracted using RIPA buffer (50 mM Tris-Cl, pH 8.0, 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 1% NP-40) supplemented with protease inhibitor cocktail. Cell lysates were centrifuged at 12000 rpm for 30 min at 4 °C, supernatants were saved, and protein concentrations were determined by BCA protein assay (Thermo Fisher Scientific, USA). 50μg of protein samples were loaded per well and separated on SDS-polyacrylamide gels, and then electrophoretically transferred onto PVDF membranes. Following blocking with 5% non-fat milk at room temperature for 2h, membranes were incubated with primary antibodies overnight at 4 °C and further incubated with secondary horseradish peroxidase-conjugated antibodies. Finally, protein bands were detected with the enhanced chemiluminescence western blot detection kit (Engreen Biosystem, China).
Immunohistochemistry
71 tumor tissues and 20 adjacent normal tissues were obtained from NSCLCs patients who underwent complete resection in the Affiliated Hospital of Guangdong Medical University between 2011 and 2013. Follow-up information was obtained from review of the patients' medical record. This study was approved by the Ethics Committee of Guangdong Medical University.
Paraffin-embedded sections were dewaxed in xylene, rehydrated through graded alcohol and placed in an endogenous peroxide block for 15 min. Antigen retrieval was done using a microwave in 0.01M (pH 6.0) citrate buffer. Nonspecific staining was blocked by goat serum for 20 min. Annexin A2 rabbit polyclonal antibody (1:100 dilutions) was applied. Subsequent operations were conducted using IHC kit (UNIV, China).
The intensity of Annexin A2 staining was scored as 0 (no signal), 1 (weak), 2 (moderate), and 3 (marked). Percentage scores were assigned as 1, 1-25%; 2, 26-50%; 3, 51-75%; and 4, 76-100%. The scores of each tumor sample were multiplied to give a final score of 0-12, and the tumors were finally determined as negative (-), score 0; lower expression (+), score ≤4; moderate expression (++), score 5-8; and high expression (+++), score ≥9. An optimal cutoff value was identified: a staining index of five or greater was used to define tumors of high expression, and four or lower for low expression.
RNA interference and plasmid transfection
Annexin A2 shRNA-lentivirus vectors and a non-targeting control shRNA were obtained from GeneCopeia (Guang zhou, China). HEK293FT cells were co-transfected with lentivirus and packaging vectors using Lipofectamine 2000. After 48 hours, media containing lentiviruses were harvested and filtered through 0.45 μm syringe filters. Lentiviruses were transduced in 50% confluent A549 or H460 cells. Annexin A2 stable knockdown A549 or H460 cells were selected by puromycin (1 μg/mL).
The small-interfering RNA (siRNA) targeting p53 (SignalSilence® p53 siRNA I #6231) were from Cell Signaling Technology (USA). A549 cells were plated into 6-well plate at a density of 1.5×105 cells per well. After 24 hours of incubation, negative control siRNA, siRNAs targeting p53 were transiently transfected to A549 cells with Lipofectamine 2000 Transfection Reagent (Invitrogen, USA) and incubated for 48 h.
BrdU incorporation assay
A549 or H460 cells were transfected with Annexin A2 shRNA or control shRNA for 24h, then treated with 0.03 mg/ml BrdU (Thermo Fisher Scientific, USA) for 12 h, at 37 °C, fixed with 4% paraformaldehyde, washed in 0.1 M PBS (phosphate-buffered saline, pH 7.4) with 1% Triton X-100, and incubated with 1 M Hcl (hydrochloric acid) and 2 M Hcl. A borate buffer (0.1 M) was added and cells were blocked with 5% normal goat serum in 0.1 M PBS in the presence of 1% Triton X-100, 1.0 M glycine. The cells were sequentially incubated with anti-BrdU and secondary antibodies.
Cell proliferation assay
A549 or H460 cells were seeded into 6-well plates and then transfected with Annexin A2 shRNA or control shRNA, cell numbers were counted after 0, 24, 48, 72 and 96 h of incubation using a Coulter Counter (Beckman Coulter, Fullerton, CA, USA) in triplicate.
Colony formation assays
A549 or H460 cells were transfected with Annexin A2 shRNA or control shRNA for 24 h. About 500 cells were seeded per well in six-well-plates. After 14 days, the cells were fixed in methanol and stained with 0.2% crystal violet. Number of colonies was counted using Quantity One software (Bio-Rad, Hercules, CA, USA).
Cell Cycle Analysis
A549 or H460 cells were transfected with Annexin A2 shRNA or control shRNA for 3 days. The cell cycle was detected using nuclear propidium iodide (Sigma-Aldrich, USA) staining, then analyzed using flow cytometry (BD Biosciences, USA) with excitation set at 488 nm and emission detected at the FL-2 channel (565-610 nm). Distribution of cells in different phases of the cell cycle was calculated using FlowJo software (TreeStar, Ashland, OR, USA).
Cell invasion assay
The polycarbonate filters (Thermo Fisher Scientific, USA) were pre-coated with Matrigel Matrix (BD Biosciences, USA). A549 or H460 cells (1×105) transfected with control shRNA or Annexin A2 shRNA for 24 h were suspended in 150μl serum free RPMI 1640 and then added into the upper chamber, while 600μl of complete media was added to the lower chamber. After 24h of incubation, the cells migrated through the matrigel and adhered onto the lower chamber were fixed in 4% paraformaldehyde for 20min, stained with Mayer's hematoxylin (Sigma-Aldrich, USA) and counted under microscope (five fields per chamber). Each invasion assay was carried out in triplicate and repeated in three independent experiments.
Wound healing scratch assay
A549 or H460 cells transfected with control shRNA or Annexin A2 shRNA for 24 h were grown as monolayers in triplicates in 12-well plates (2×105/well). Cells were then serum-starved overnight and an artificial scratch wound was created. Cell debris was removed by washing with PBS. Cell migration at indicated time was photographed.
Statistical analysis
Statistical analyses were performed using GraphPad Prism6 software (GraphPad Software Inc., CA). Unpaired t tests were used to determine statistical significance. Values shown are the Means ± SD. Kaplan-Meier method was used to analyze the survival curves for patients. Statistical significance was defined as p < 0.05.
Results
Annexin A2 is overexpressed and associated with poor prognosis in human NSCLCs
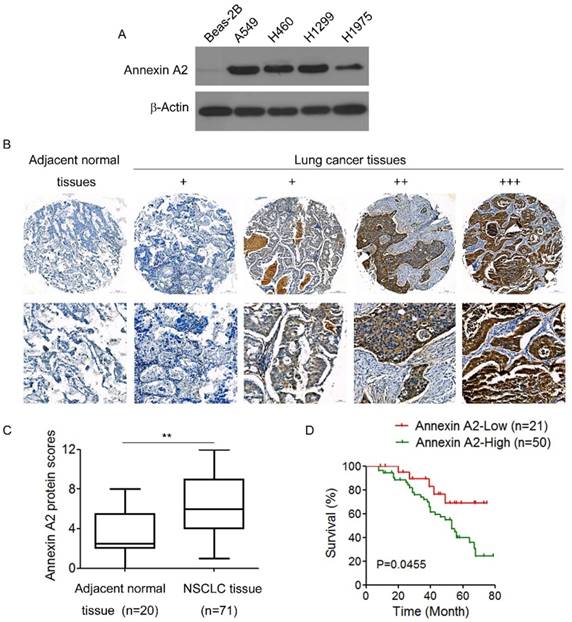
We first examined Annexin A2 expression in a panel of 4 human NSCLCs lines and 1 normal human lung epithelial cell line BEAS-2B. Western blot results showed that there was almost no Annexin A2 expression in normal lung epithelial cells Beas-2B, but abundant expression of Annexin A2 in NSCLCs cells (Figure 1A). We further detected the expression of Annexin A2 in NSCLCs tissues by IHC in 72 NSCLCs specimens and 20 adjacent normal tissues, the results showed that Annexin A2 was high expressed in lung cancer tissues compared with adjacent normal tissues (Figure 1B-C). Next, we analyzed the relationship between Annexin A2 expression levels and clinic pathological characteristics. As shown in Table 1, no statistically significant correlations were observed between the expression of Annexin A2 and age, or gender. However, statistically significant correlations were found between high levels of Annexin A2 expression and clinical stage, as well as lymph node metastasis (p<0.01). Kaplan-Meier survival analysis demonstrated that NSCLCs patients with high Annexin A2 expression had poorer overall survival than those with low Annexin A2 expression (p=0.0455) (Figure 1D). Altogether, our present data suggest that Annexin A2 is overexpressed in NSCLCs and high level of Annexin A2 expression is a predictor of progression and poor prognosis of NSCLCs.
Annexin A2 is overexpressed and associated with poor prognosis in human NSCLCs. (A) Annexin A2 expression in Beas-2B, A549, H460, H1299 and H1975 cells was analyzed by Western blot. β-actin was employed as an inner control. (B) Representative immunohistochemical staining examples of Annexin A2 protein expression in adjacent normal tissues and NSCLCs tissues (100×, 400×). The NSCLCs tissue sections were quantitatively scored according to the percentage of positive cells and staining intensity as described in Materials and Methods. The percentage and intensity scores were multiplied to obtain a total score (range, 0-12), and the tumors were finally determined as negative (-), score 0; lower expression (+), score ≤4; moderate expression (++), score 5-8; and high expression (+++), score ≥9. (C) Annexin A2 protein scores in NSCLCs tissues and adjacent normal tissues. **p<0.01. (D) Kaplan-Meier OS curves of 71 NSCLCs patients relative to different expression levels of Annexin A2, p=0.0455.
Correlation between the clinical pathologic features of NSCLCs patients and expression of Annexin A2
| Characteristics | Number of patients (n=71) | Annexin A2 expression | P-valuea | |
|---|---|---|---|---|
| Low (n=21) | High (n=50) | |||
| Age | ||||
| <50 | 38 | 11 | 27 | 0.436 |
| >50 | 33 | 10 | 23 | |
| Gender | ||||
| Male | 52 | 16 | 36 | 0.579 |
| Female | 19 | 5 | 14 | |
| Clinical stage | ||||
| I | 16 | 8 | 8 | <0.01b |
| II | 35 | 10 | 25 | |
| III+IV | 20 | 3 | 17 | |
| Lymph node metastasis | ||||
| N0 | 32 | 14 | 18 | <0.01c |
| N1-3 | 39 | 7 | 32 | |
aX2 test.
bComparing clinical stages I versus II, III-IV.
cComparing Lymph node metastasis N0 versus N1-3.
Knockdown of Annexin A2 inhibits NSCLCs cell proliferation
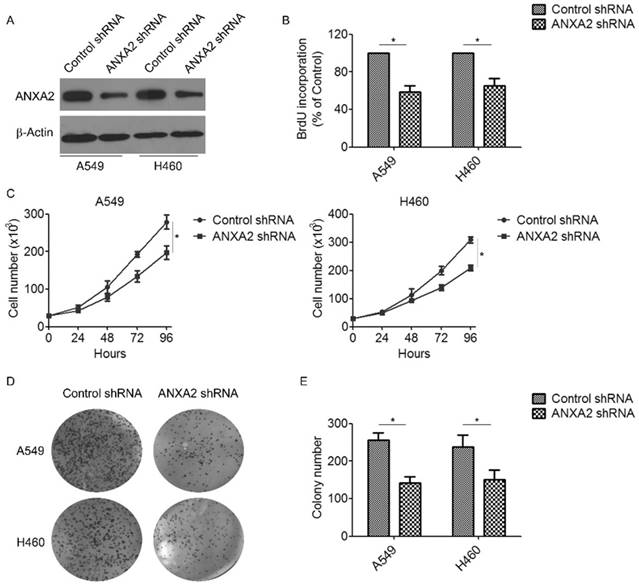
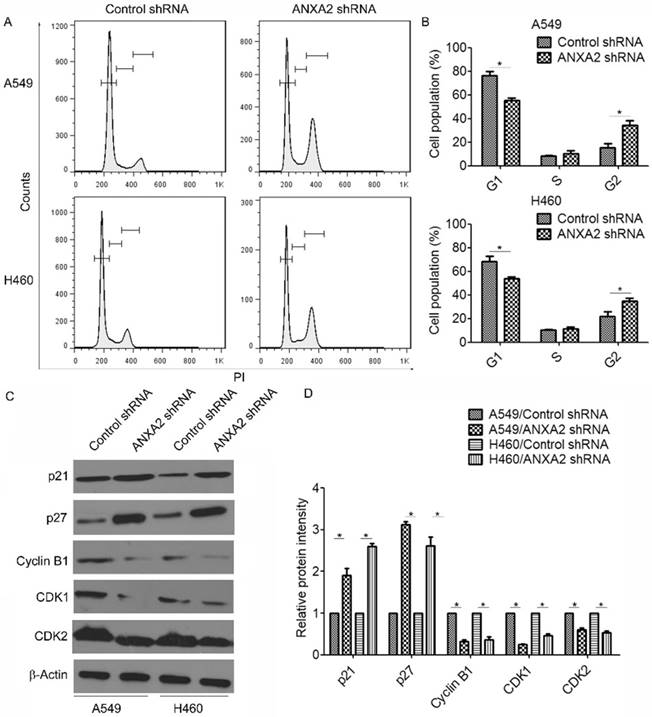
To investigate the biological effect of Annexin A2 deregulation on NSCLCs cells, lentivirus-based shRNA was used to silence Annexin A2 in NSCLCs cells A549 and H460 (Figure 2A). Using BrdU incorporation assays and direct cell counting, we found that Annexin A2 silencing significantly inhibited cell proliferation in A549 and H460 cells (Figure 2B-C). Moreover, colony formation assay also revealed that Annexin A2 knockdown remarkably decreased the colony number of the A549 and H460 cells (Figure 2D-E). To further explore the mechanism by which Annexin A2 promoted the cell proliferation, we investigated the cell cycle by PI staining and flow cytometric analysis. Results showed that Annexin A2 knockdown significantly decreased the cell number in G1 phase and increased the cell number in G2 phase in A549 and H460 cells (Figure 3A-B). Moreover, western blot results indicated that Annexin A2 deficiency upregulated the expression of CKI p21 and p27, and downregulated the expression of CDK1, CDK2 and Cyclin B1 (Figure 3C-D). Together, these results suggest that knockdown of Annexin A2 inhibits cell proliferation by inducing cell cycle G2 arrest in NSCLCs cells.
Knockdown of Annexin A2 inhibits proliferation of NSCLCs cells. (A) A549 or H460 cells were transfected with Annexin A2 shRNA or control shRNA, Annexin A2 expression was analyzed by Western blot. β-actin was employed as an inner control. (B) A549 or H460 cells were transfected with Annexin A2 shRNA or control shRNA, cell proliferation was detected by BrdU incorporation assay. *p<0.05. (C) A549 or H460 cells were transfected with Annexin A2 shRNA or control shRNA, cell numbers were counted at indicated time by Coulter Counter. *p<0.05. (D-E) A549 or H460 cells were transfected with Annexin A2 shRNA or control shRNA. After 14 days, the cells were fixed in methanol and stained with crystal violet. Colonies were photographed and counted. *p<0.05.
Knockdown of Annexin A2 induces G2 arrest in NSCLCs cells. (A) A549 or H460 cells were transfected with Annexin A2 shRNA or control shRNA, the cell cycle stages were determined using PI staining followed by flow cytometry 3 days post-transfection. One representative data set is shown. (B) Distribution of cells in different phases of the cell cycle is shown from three individual experiments. *p<0.05. (C) A549 or H460 cells were transfected with Annexin A2 shRNA or control shRNA, expression of p21, p27, Cyclin B1, CDK1 and CDK2 was analyzed by Western blot. β-actin was employed as an inner control. (D) Bands of Western blot were analyzed by ImageJ software. Results were obtained from the ratio of target band to β-actin. *p<0.05.
Knockdown of Annexin A2 suppresses invasion and migration of NSCLCs cells
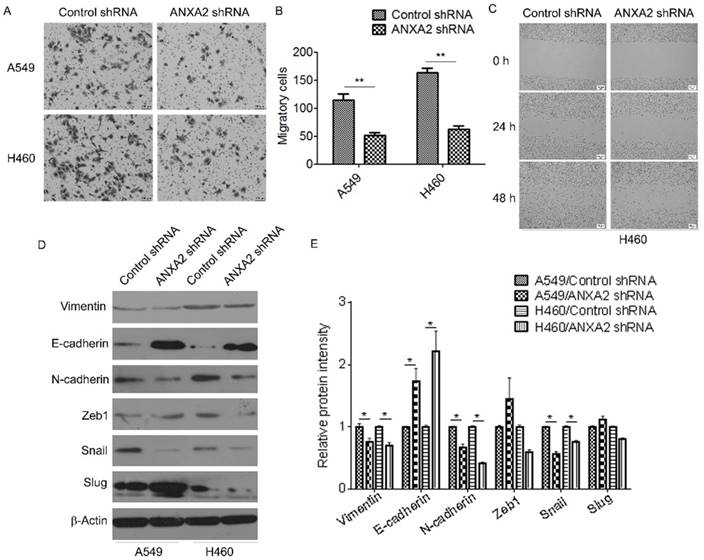
We further evaluated the effect of Annexin A2 silencing on invasion and migration of NSCLCs cells. Transwell assay showed that knockdown of Annexin A2 drastically reduced the invasive ability of A549 and H460 cells compared with that of the control (Figure 4A-B). Wound healing scratch assay also demonstrated that knockdown of Annexin A2 significantly decreased the migratory speed of H460 cells (Figure 4C). Furthermore, we evaluated the effects of Annexin A2 on the expression of several EMT-related markers. Western blot results showed that Annexin A2 silencing downregulated the expression of Vimentin and N-cadherin, but upregulated the expression of E-cadherin. Correspondingly, expression of EMT-related transcription factor Snail was significantly decreased in Annexin A2-knockdown cells, while expression of Zeb1 and Slug had no marked change (Fig. 4D-E). These results suggest that Annexin A2 silencing suppresses invasion and migration of NSCLCs cells via inhibition of EMT.
Knockdown of Annexin A2 suppresses invasion and migration of NSCLCs cells. (A-B) A549 or H460 cells were transfected with Annexin A2 shRNA or control shRNA, cell migration ability was measured by Transwell assay. **p<0.01. (C) H460 cells were transfected with Annexin A2 shRNA or control shRNA, cell migration ability was measured by wound healing scratch assay. (D) A549 or H460 cells were transfected with Annexin A2 shRNA or control shRNA, expression of Vimentin, E-cadherin, N-cadherin, Zeb1, Snail and Slug was analyzed by Western blot. β-actin was employed as an inner control. (E) Bands of Western blot were analyzed by ImageJ software. Results were obtained from the ratio of target band to β-actin. *p<0.05.
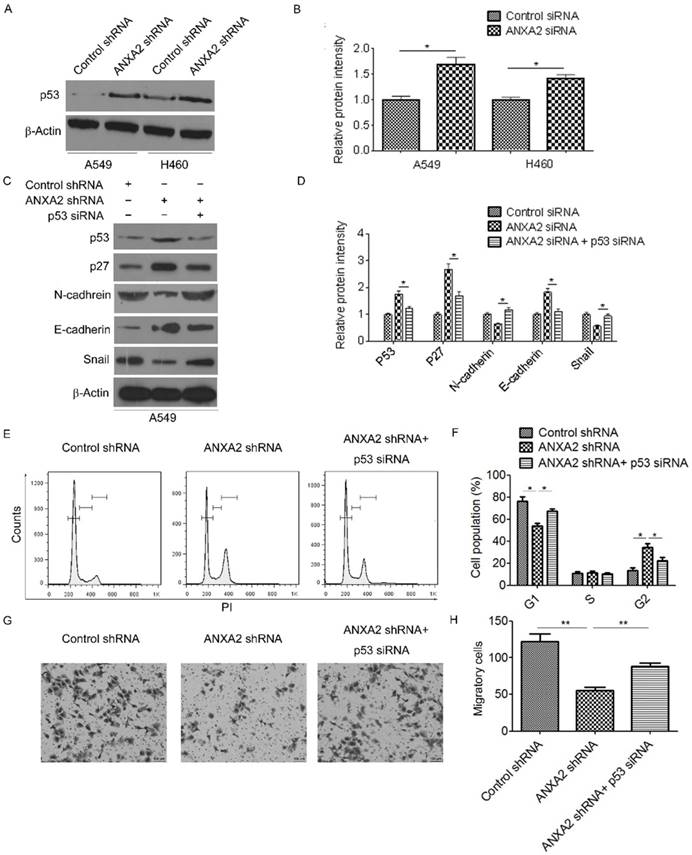
Knockdown of Annexin A2 induces cell cycle G2 arrest and suppresses EMT via inhibition of p53
Previous studies indicate that Annexin A2 inhibits p53 expression [16]. Indeed, we found that Annexin A2 silencing remarkably increased p53 protein expression (Figure 5A-B). To evaluate whether knockdown of Annexin A2 induced cell cycle G2 arrest and EMT suppression were due to inhibition of p53, Annexin A2-knockdown cells were transfected with p53-specific small interfering RNA (p53 siRNA). The results showed that knockdown of p53 significantly decreased the expression of p27 in Annexin A2-knockdown cells (Figure 5C-D). More importantly, Annexin A2 shRNA-induced cell cycle G2 arrest was abolished by knockdown of p53 (Figure 5E-F). Moreover, co-transfection with p53 siRNA and Annexin A2 shRNA upregulated the expression of N-cadherin, but downregulated the expression of E-cadherin compared with transfection with Annexin A2 shRNA alone in A549 cells (Figure 5C-D). Meanwhile, knockdown of p53 significantly increased the amount of invasive cells in Annexin A2-knockdown cells (Figure 5G-H). These results indicate that knockdown of Annexin A2 induces cell cycle G2 arrest and suppresses EMT via inhibition of p53 in NSCLCs.
Knockdown of Annexin A2 induces cell cycle G2 arrest and suppresses EMT via inhibition of p53. (A) A549 or H460 cells were transfected with Annexin A2 shRNA or control shRNA, p53 expression was analyzed by Western blot. β-actin was employed as an inner control. (B) Bands of Western blot were analyzed by ImageJ software. Results were obtained from the ratio of target band to β-actin. *p<0.05. (C) A549 cells were transfected with control shRNA, Annexin A2 shRNA or Annexin A2 shRNA and p53 siRNA, expression of p53, p27, E-cadherin, N-cadherin and Snail was analyzed by Western blot. β-actin was employed as an inner control. (D) Bands of Western blot were analyzed by ImageJ software. Results were obtained from the ratio of target band to β-actin. *p<0.05. (E) A549 cells were transfected with control shRNA, Annexin A2 shRNA or Annexin A2 shRNA and p53 siRNA, the cell cycle stages were determined using PI staining followed by flow cytometry 3 days post-transfection. One representative data set is shown. (F) Distribution of cells in different phases of the cell cycle is shown from three individual experiments. *p<0.05. (G-H) A549 cells were transfected with control shRNA, Annexin A2 shRNA or Annexin A2 shRNA and p53 siRNA, cell migration ability was measured by Transwell assay. **p<0.01.
Discussion
In this study, our data provided the evidences that Annexin A2 was overexpressed in NSCLCs and deregulation of Annexin A2 played an oncogenic role in promoting cancer progression. We found that high expression of Annexin A2 was correlated with advanced clinical stage and lymph node metastasis, as well as adverse clinical outcomes. Furthermore, our results demonstrated that knockdown of Annexin A2 induced cell cycle G2 arrest and inhibited EMT via p53-dependent manner.
Annexin A2 overexpression in patients with various solid tumors, including lung cancer [16], colorectal cancer [17], pancreatic cancer [18, 19], breast cancer [20] and hepatocellular cancer [21], shows promising therapeutic implications and determines poor clinical outcome. Our present study confirmed that Annexin A2 was high expressed in NSCLCs. Notably, we found that high Annexin A2 expression was strongly correlated with the clinical stages and lymph node metastasis in NSCLCs, which suggested that Annexin A2 was closely associated with NSCLCs progression.
Several studies show that aberrant Annexin A2 expression has oncogenic effects, including promoting cancer cell proliferation[16], inhibiting cell apoptosis [22] and increasing drug resistance [15, 23]. In this study, our results showed that Annexin A2 knockdown significantly reduced cell proliferation properties and colony formation in NSCLCs cells. More importantly, we found that Annexin A2 knockdown significantly increased the expression of p21 and p27, but decreased the expression of CDK1, CDK2 and Cyclin B1, which in turn induced cell cycle arrest at G2-phase. These results were consistent with previous studies, showing that Annexin A2 knockdown induced cell cycle arrest in A549 cells [16].
Annexin A2 has also been shown to play an important role in cancer cell invasion and metastasis, thus playing a crucial role in cancer development [24, 25]. We found that downregulation of Annexin A2 significantly inhibited NSCLCs cell invasion. The effect of Annexin A2 on promoting the invasiveness of NSCLCs cells was verified by the IHC results, which showed a significant correlation between Annexin A2 expression and lymph node metastasis in NSCLCs. EMT, characterized by the loss of epithelial characteristics and the acquisition of mesenchymal phenotypes, plays an important role in cancer invasion and metastasis [26]. Indeed, we found that knockdown of Annexin A2 significantly suppressed EMT of NSCLCs cells, with the evidence of increasing the expression of epithelial marker E-cadherin, and decreasing the expression of mesenchymal marker (N-cadherin), thereby decreasing cell motility and invasion.
Previous studies show that Annexin A2 knockdown could inactivate JNK which negatively regulates p53 through c-Jun stabilization, thereby causing p53 upregulation [27-29]. p53 is a well-known tumor suppressor in regulating cell cycle progression [30]. Moreover, wild-type p53 could reduce the expression of snail [31-34], while p53 mutation enhances snail expression by inhibiting its degradation [35]. Thus, the study was further investigated to determine whether p53 played a role in Annexin A2 shRNA-mediated cell cycle arrest and EMT inhibition in NSCLCs cells. Strikingly, our results indicated that silencing of Annexin A2 induced cell cycle G2 arrest and inhibited EMT through a p53-dependent pathway, with the evidence that knockdown of p53 in Annexin A2-silencing NSCLCs cells reversed the effects of Annexin A2 shRNA on cell cycle arrest and EMT inhibition. Of course, recent studies have provided important insights into several potential candidates, such as NF-kB and β-catenin, involving in Annexin A2-mediated cancer progression [36-38], whether these candidates play a role in Annexin A2-induced cell proliferation and invasion should be further investigated.
Conclusions
In summary, the results of this study show that Annexin A2 overexpression is an important molecular event that plays an oncogenic role in NSCLCs. Inhibition of Annexin A2 suppresses NSCLCs cell proliferation and invasion through activating p53 pathway, which in turn induces cell cycle arrest and inhibits EMT. Therefore, Annexin A2 could serve as a potential prognosis marker and a novel therapeutic target for NSCLCs.
Abbreviations
NSCLCs: non-small cell lung cancers; EMT: epithelial-to-mesenchymal transition; CKI: cyclin-dependent kinase inhibitor; CDK: cyclin-dependent kinase; IHC: immunohistochemistry.
Acknowledgements
This work was supported by grant from the National Natural Science Foundation of China (81201763); Natural Science Foundation of Guangdong Province, China (2018A030310116); Science Research Foundation of Guangdong Medical University, China (B2017027, GDMUZ201808).
Ethical approval
This study was approved by the institutional review board of Guangdong Medical University. All procedures performed in this study were in accordance with the 1964 Declaration of Helsinki and its later amendments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Brambilla E, Travis WD, Colby TV. et al. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18:1059-68
3. Wang T, Nelson RA, Bogardus A. et al. Five-year lung cancer survival: which advanced stage nonsmall cell lung cancer patients attain long-term survival? Cancer. 2010;116:1518-25
4. Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449-61
5. Gerke V, Moss SE. Annexins: from structure to function. Physiol Rev. 2002;82:331-71
6. Rescher U, Gerke V. S100A10/p11: family, friends and functions. Pflugers Arch. 2008;455:575-82
7. O'Connell PA, Surette AP, Liwski RS. et al. S100A10 regulates plasminogen-dependent macrophage invasion. Blood. 2010;116:1136-46
8. Cesarman-Maus G, Hajjar KA. Molecular mechanisms of fibrinolysis. Br J Haematol. 2005;129:307-21
9. Deng Y, Chen C, Hua M. et al. Annexin A2 plays a critical role in epithelial ovarian cancer. Arch Gynecol Obstet. 2015;292:175-82
10. Zhang H, Yao M, Wu W. et al. Up-regulation of annexin A2 expression predicates advanced clinicopathological features and poor prognosis in hepatocellular carcinoma. Tumour Biol. 2015;36:9373-83
11. Luo S, Xie C, Wu P. et al. Annexin A2 is an independent prognostic biomarker for evaluating the malignant progression of laryngeal cancer. Exp Ther Med. 2017;14:6113-8
12. Wang CY, Lin CF. Annexin A2: its molecular regulation and cellular expression in cancer development. Dis Markers. 2014;2014:308976
13. Zhang F, Zhang L, Zhang B. et al. Anxa2 plays a critical role in enhanced invasiveness of the multidrug resistant human breast cancer cells. J Proteome Res. 2009;8:5041-7
14. Han F, Shrestha S, Huang H. et al. Expression of annexin II in gastric carcinoma and its role in gastric cancer metastasis. World J Gastroenterol. 2017;23:7009-15
15. Feng X, Liu H, Zhang Z. et al. Annexin A2 contributes to cisplatin resistance by activation of JNK-p53 pathway in non-small cell lung cancer cells. J Exp Clin Cancer Res. 2017;36:123
16. Wang CY, Chen CL, Tseng YL. et al. Annexin A2 silencing induces G2 arrest of non-small cell lung cancer cells through p53-dependent and -independent mechanisms. J Biol Chem. 2012;287:32512-24
17. Duncan R, Carpenter B, Main LC. et al. Characterisation and protein expression profiling of annexins in colorectal cancer. Br J Cancer. 2008;98:426-33
18. Jung H, Kim JS, Kim WK. et al. Intracellular annexin A2 regulates NF-kappaB signaling by binding to the p50 subunit: implications for gemcitabine resistance in pancreatic cancer. Cell Death Dis. 2015;6:e1606
19. Vishwanatha JK, Chiang Y, Kumble KD. et al. Enhanced expression of annexin II in human pancreatic carcinoma cells and primary pancreatic cancers. Carcinogenesis. 1993;14:2575-9
20. Chuthapisith S, Bean BE, Cowley G. et al. Annexins in human breast cancer: Possible predictors of pathological response to neoadjuvant chemotherapy. Eur J Cancer. 2009;45:1274-81
21. Mohammad HS, Kurokohchi K, Yoneyama H. et al. Annexin A2 expression and phosphorylation are up-regulated in hepatocellular carcinoma. Int J Oncol. 2008;33:1157-63
22. Huang Y, Jin Y, Yan CH. et al. Involvement of Annexin A2 in p53 induced apoptosis in lung cancer. Mol Cell Biochem. 2008;309:117-23
23. Yi Y, Zeng S, Wang Z. et al. Cancer-associated fibroblasts promote epithelial-mesenchymal transition and EGFR-TKI resistance of non-small cell lung cancers via HGF/IGF-1/ANXA2 signaling. Biochim Biophys Acta. 2018;1864:793-803
24. Rocha MR, Barcellos-de-Souza P, Sousa-Squiavinato ACM. et al. Annexin A2 overexpression associates with colorectal cancer invasiveness and TGF-ss induced epithelial mesenchymal transition via Src/ANXA2/STAT3. Sci Rep. 2018;8:11285
25. Zheng L, Foley K, Huang L. et al. Tyrosine 23 phosphorylation-dependent cell-surface localization of annexin A2 is required for invasion and metastases of pancreatic cancer. PLoS One. 2011;6:e19390
26. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97-110
27. Das M, Jiang F, Sluss HK. et al. Suppression of p53-dependent senescence by the JNK signal transduction pathway. Proc Natl Acad Sci U S A. 2007;104:15759-64
28. Kruse JP, Gu W. Modes of p53 regulation. Cell. 2009;137:609-22
29. Hollstein M, Hainaut P. Massively regulated genes: the example of TP53. J Pathol. 2010;220:164-73
30. Vazquez A, Bond EE, Levine AJ. et al. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979-87
31. Chang CJ, Chao CH, Xia W. et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317-23
32. Hermeking H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010;17:193-9
33. Kim T, Veronese A, Pichiorri F. et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208:875-83
34. Siemens H, Jackstadt R, Hunten S. et al. miR-34 and SNAIL form a double-negative feedback loop to regulate epithelial-mesenchymal transitions. Cell Cycle. 2011;10:4256-71
35. Wang SP, Wang WL, Chang YL. et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat Cell Biol. 2009;11:694-704
36. Christensen MV, Hogdall CK, Jochumsen KM. et al. Annexin A2 and cancer: A systematic review. Int J Oncol. 2018;52:5-18
37. Liu Y, Li H, Ban Z. et al. Annexin A2 inhibition suppresses ovarian cancer progression via regulating beta-catenin/EMT. Oncol Rep. 2017;37:3643-50
38. Wang Y, Chen K, Cai Y. et al. Annexin A2 could enhance multidrug resistance by regulating NF-kappaB signaling pathway in pediatric neuroblastoma. J Exp Clin Cancer Res. 2017;36:111
Author contact
![]() Corresponding author: Dr. Yanmei Yi, Department of Histology and Embryology, Guangdong Medical University, No.2 Wenmingdong Road, Xiashan District, Zhanjiang 524023, Guangdong, China. Email: yiymeiedu.cn.
Corresponding author: Dr. Yanmei Yi, Department of Histology and Embryology, Guangdong Medical University, No.2 Wenmingdong Road, Xiashan District, Zhanjiang 524023, Guangdong, China. Email: yiymeiedu.cn.