Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(6):1466-1478. doi:10.7150/jca.29192 This issue Cite
Research Paper
Dual-Inhibition of mTOR and Bcl-2 Enhances the Anti-tumor Effect of Everolimus against Renal Cell Carcinoma In Vitro and In Vivo
Ayse Hande Nayman1, Halime Siginc1, Ebru Zemheri2, Faruk Yencilek3, Asif Yildirim4, Dilek Telci1 ![]()
1. Yeditepe University, Faculty of Engineering, Department of Genetics and Bioengineering, Kayisdagi Cad., 34755, Istanbul, Turkey
2. Department of Pathology, Umraniye Training and Research Hospital, Istanbul, Turkey
3. Yeditepe University, Faculty/School of Medicine, Yeditepe University Hospital, Istanbul, Turkey
4. Department of Urology/Faculty of Medicine, Medeniyet University, Istanbul, Turkey
Received 2018-8-13; Accepted 2019-1-3; Published 2019-2-23
Abstract
Renal cell carcinoma (RCC) is the predominant type of kidney cancer. Mammalian target of rapamycin (mTOR) inhibitor everolimus is currently used as a second-line therapy for sorafenib or sunitinib-refractory metastatic RCC patients. The clinical limitation confronted during everolimus therapy is the onset of drug resistance that decreases the efficacy of the drug. Elevated level of anti-apoptotic Bcl-2 protein is proposed to be an emerging feedback loop for the acquired drug-resistance in various cancer types. In this study, the Bcl-2 inhibitor ABT-737 was used in combination with everolimus to enhance its anti-tumor effectiveness in everolimus-resistant RCC cell lines. Everolimus and ABT-737 combination synergistically led to a decrease in the proliferation of primary site A-498 and metastatic site Caki-1 RCC cell lines, which was accompanied by a reduction in protein levels of cell cycle and mTOR pathway proteins. In both RCC cell lines, everolimus-ABT-737 combination not only induced apoptosis, caspase and PARP-1 cleavage but also a decrease in Bcl-2 protein levels in parallel with a concomitant increase in Bim and Noxa levels.
In order to confirm our in vitro findings, we have generated everolimus-resistant RenCa cell line (RenCares) to establish a RCC mouse xenograft model. Animals co-treated with everolimus and ABT-737 exhibited a complete suppression of tumor growth without any notable toxicity. This study thus proposes the everolimus-ABT-737 combination as a novel therapeutic strategy for the treatment of RCC to overcome the current clinical problem of everolimus resistance.
Keywords: Bcl-2, Everolimus-ABT 737 combination, mTOR, Renal Cell Carcinoma
Introduction
Renal cell carcinoma (RCC) is the prominent malignancy of kidney and accounts for 2-3% of all adult tumors (1). Histopathological analysis reveals that clear cell RCC (ccRCC) comprises the majority among the other RCC subtypes (2). It is a vascular neoplasm with a high metastatic potential. Indeed, the disease is mostly diagnosed at locally advanced stage, where almost 20-30% of patients suffer from metastasis (3). The identification of the molecular aspects underlining the tumor-associated vasculature of mRCC paved the way for the idea of the targeted therapy. In this regard, the molecular agents targeting the main signaling pathways playing a major role in RCC tumorigenesis are currently used in clinic. However, the acquired drug resistance is the main clinical limitation leading to a decrease in disease-free and cancer-specific survival outcomes (4). Therefore, novel therapeutic approaches are required to improve the anti-tumor effects of the molecular targeted agents used in RCC treatment.
Dysregulation of phosphoinositide-3-kinase (PI3K)/AKT/mTOR pathway is reported to be responsible for RCC tumorigenesis (5). The PI3K/ AKT/mTOR pathway is an intracellular signaling pathway regulating the crucial cellular processes including growth, proliferation and survival. The signaling cascade is activated by the binding of growth factors such as insulin-like growth factor (IGF), epidermal growth factor (EGF), platelet- derived growth factor (PDGF), and vascular endothelial growth factor (VEGF) to their receptor tyrosine kinases (RTK) (6). Upon ligand binding, RTKs are activated by auto-phosphorylation at their cytoplasmic domain resulting in PI3K activation (5). Once activated, PI3K catalyzes the formation of phosphatidylinositol-3,4,5- trisphosphate (PIP3) that recruits phosphoinositide-dependent kinase 1 (PDK1) and AKT to cell membrane, where AKT is phosphorylated and activated by PDK1 (7). Active AKT indirectly regulates mTOR kinase activity by phosphorylating and inhibiting TSC1/2 complex, an inhibitor of mTOR kinase (8). mTOR is the serine/ threonine kinase catalyzing the phosphorylation of downstream effectors such as the eukaryotic translation initiation factor 4E binding protein 1 (4E-BP1) and 70kDa ribosomal protein S6 kinase 1 (S6K1) to initiate the synthesis of proteins including Cyclin D1 and regulates HIF-1α activity (9, 10) and hence acts as the main molecular switch for the regulation of cell growth/proliferation and angiogenesis (11). Elevated HIF-1α protein level as well as constitutive activation of mTOR pathway was reported in solid tumors including breast and renal carcinoma (12, 13). Therefore targeting mTOR kinase activity provides a therapeutic advantage in cancer treatment, where elevated PI3K/AKT/mTOR signaling is prevalent (14).
mTOR inhibitor everolimus, a derivative of rapamycin, forms a complex with the cytoplasmic FKBP12 protein (15) and abrogates the mTOR- mediated initiation of translation by binding to the kinase FRB domain, which results in the attenuation of the tumor cell growth, cell proliferation and neovascularization (16). Following phase I and phase II trials (17, 18), the use of everolimus for the treatment of sorafenib/sunitinib-refractory mRCC patients as a second-line therapy was approved by FDA in 2009 (19). Although, consecutive phase III studies conducted with mRCC patients revealed a favorable risk-benefit ratio in comparison to tyrosine kinase inhibitors, complete response was not achieved within the patient groups (20, 21). High frequency of patient relapse due to the development of drug resistance is an important clinical limitation of everolimus therapy (22, 23).
There are several feedback loops proposed for the onset of everolimus resistance in RCC tumors, which coordinate the interplay between signaling cascades to circumvent the mTOR inhibition (24). The elevated level of anti-apoptotic Bcl-2 protein is suggested to be one of the feedback loops as its overexpression has been reported in hematological disorders and solid tumors as well as in RCC in association with the resistance to chemotherapy (25-28). Bcl-2 family proteins are the main regulators of the intrinsic apoptosis and clustered in three distinct groups according to their functional roles in the apoptotic process. The control of the apoptotic cell death was mediated by the tug of war between the opposing Bcl-2 family members through their interactions via BH3 domains (29). The induction of apoptosis is induced when pro-apoptotic members of the family could overcome the inhibitory effect of anti-apoptotic Bcl-2 proteins (30). In this regard, ABT-737, a small BH3 mimicking molecule, was developed to interrupt the interaction between anti-apoptotic Bcl-2 and their pro-apoptotic counterparts and thereby induce apoptosis (31, 32). The preclinical use of ABT-737 in solid tumors such as lymphoma, multiple myeloma, breast cancer, small cell lung cancer and various hematological cancers have been reported to be successful (33).
The aim of the present study was to enhance the anti-tumor effect of everolimus as well as to overcome the everolimus-resistance observed in RCC tumors by combining everolimus with ABT-737. Therefore, in vitro and in vivo experimental models were employed to investigate the efficacy of the combination therapy in RCC treatment.
Materials and Methods
Cell lines and inhibitors
The human RCC cell lines A-498, Caki-2, Caki-1, ACHN, HEK-293 and mouse murine RCC cell line RenCa were purchased from American Type Culture Collections (ATCC). All cell lines were cultured in appropriate media supplemented with 10% FBS (Gibco), 100 units/ml penicillin (Gibco), and 100 μg/ml streptomycin (Gibco) and maintained in a humidified incubator at 37ºC and 5% CO2.
Everolimus (S1120) and ABT-737 (S1002) were purchased from Selleck Chemicals. Stock concentrations were prepared in DMSO and stored according to the manufacturer's protocol.
Analysis of cell viability and cell death
A-498, Caki-1, HEK-293 and RenCa cells were serum starved for 4 hours prior to seeding into appropriate cell culture plates. After 24 hours, cells were treated with everolimus and/or ABT-737 for 24, 48 and 72 hours. At the indicated time points, cell proliferation reagent WST-1 (Roche 11644807001) and Annexin V-Fluos staining kit (Roche 11988549001) were used to analyze the cell viability and cell death according to the manufacturer's protocol, respectiveely. The number of apoptotic cells was determined by BD FACSCalibur (Becton Dickinson) flow cytometer.
Western blot analysis
Following the drug treatment, cells were scraped in RIPA buffer (Santa Cruz Biotechnology) and the whole cell lysates were sonicated. Samples were run at 6-15 % SDS-PAGE and transferred to 0.22µm nitrocellulose (Bio-Rad) or 0.45μm PVDF (Millipore) membranes. Membranes were probed for the indicated target proteins using primary and secondary antibodies as described before (34). 4E-BP1 (#9452), Bax (#5023), Bcl-2 (#2870), Bcl-xL (#2764), Bim (#2933), caspase 9 (#9502), CDK2 (# 2546), CDK4 (#12790), cleaved caspase 3 (#9664S), Cyclin D3 (#2936), Cyclin E1 (#4129S), Cyclin D1 (#2978S), Mcl-1 (#5453), mTOR (#2972), Noxa (#14766), p53 (#2524), Puma (#4976), p27Kip1 (#3686), p70S6K (#9202), pan-actin (#8456), PARP (#9542), p-4E-BP1 (#9455), p-mTOR (#5536), p-p70S6K (#9205), p-S6 (#4858 and #2215), and S6 (#2317) were purchased from Cell Signaling Technology, β-Actin (A5316) from Sigma Aldrich. The signal was detected by using Chemiluminescent detection kit (Advansta) and visualized by ChemiDoc XRS+ (Bio-Rad). Adobe Photoshop CC 2014 was used to process the images. One representative blot of at least three independent experiments was shown in figures.
Generation of drug resistant RenCa cells
RenCa cells were initially grown in complete RPMI-1640 (Sigma) medium containing 1 µM everolimus and sub-cultured in RPMI-1640 with increasing concentration of everolimus (10 µM). After 72 hours of drug treatment, dead cells were removed by washing and remaining attached cells were cultured in 1 µM everolimus containing growth medium until an exponential proliferation in the presence of everolimus was observed.
Mice xenograft model and pathological analysis
6-8 weeks old male BALB/c mice were bred and maintained in the animal facility of Yeditepe University (Turkey) in accordance with and approved by Animal Care and Welfare Committee of Yeditepe University (Turkey, approval number #355). 15x106 RenCares cells were injected subcutaneously into the dorsal side of mice. Following the fourth day of inoculations, mice were treated every other day by injection with vehicle control, everolimus (2 mg/kg), ABT-737 (75 mg/kg) or the combination of everolimus (2 mg/kg) and ABT-737 (75 mg/kg). After 21 days of treatment, mice were sacrificed and organs including brain, thymus, heart, lung, stomach, guts, liver, kidney, spleen, and testis were isolated and they were immediately stored in 10% formalin. Pathological analysis was performed according to hematoxylin and eosin (H&E) staining (35).
Statistical analysis
All data were obtained at least from three to six independent experiments and presented as the mean ± SD (error bars). The significant analysis of the treatment groups was performed by one-way ANOVA followed by Tukey post-hoc test using GraphPad Prism 6 (GraphPad Software) for in vitro experiments. Tumor weights of mice from different treatment groups were analyzed by two-tailed Student t-test. P value less than 0.05 was considered as statistically significant.
Results
Combination of everolimus and ABT-737 drastically decreased cell proliferation in RCC cell lines overexpressing Bcl-2
Western blot analysis to determine the basal Bcl-2 protein levels was performed in A-498, Caki-2, Caki-1, and ACHN cells. Elevated level of Bcl-2 was only found in the primary site A-498 and metastatic site Caki-1 cells, leading to their use as in vitro cell models for the study (Figure 1A).
Effect of everolimus and ABT-737 co-treatment on the proliferation of RCC cell lines. (A) Western blot analysis was performed to determine the basal levels of anti-apoptotic Bcl-2 proteins in A-498, Caki-2, Caki-1 and ACHN cells. β-Actin was used as loading control. A-498 and Caki-1 cells in 96-well plates were treated with increasing concentrations of everolimus (B and D) or ABT-737 (C and E) or DMSO (as control) for 24, 48 and 72 hours. The effect of monotherapies on cell proliferation was analyzed by WST-1 assay and the proliferation rate for control cells was considered as 100% at 24 hours. (F) A-498 cells were exposed to 1 µM everolimus and/or 5µM ABT-737, and (G) Caki-1 cells were subjected to 1 µM everolimus and/or 10 µM ABT-737 treatment for 24 to 72 hours. WST-1 assay was performed to analyze the effect of the combination therapy on A-498 and Caki-1 cells. Absorbance values obtained for control cells treated with DMSO at the end of 24 hours was set to 100%. Graphs show the mean ± S.D. of three independent experiments, each performed in triplicate. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001, **** P ≤ 0.0001.
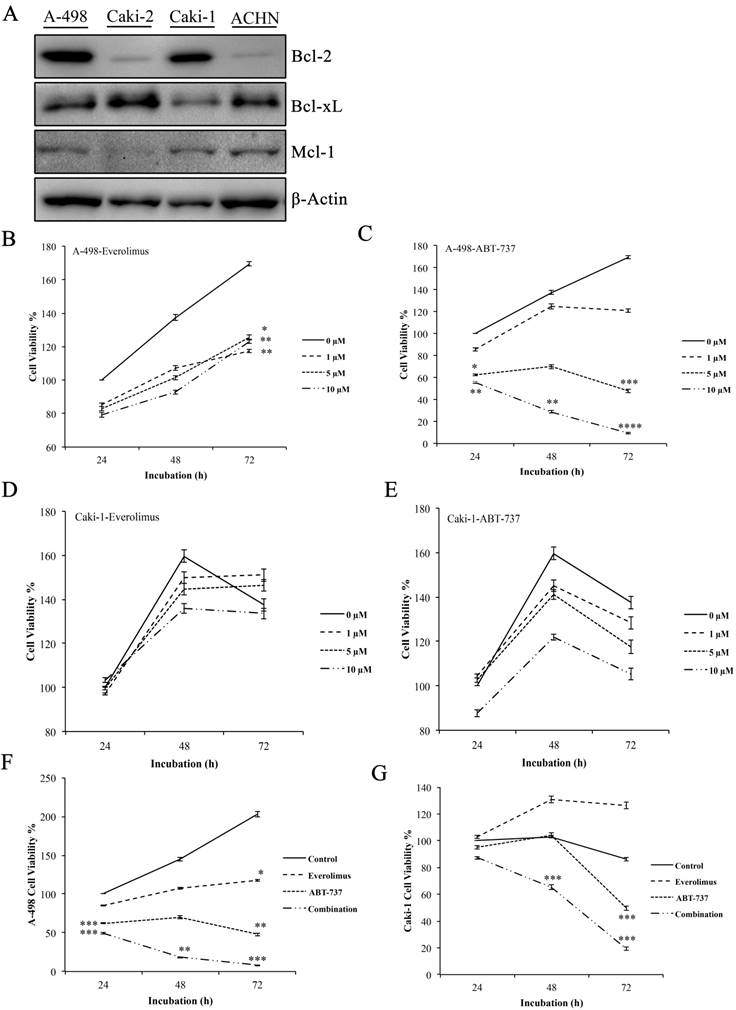
Next, we determined the cytotoxic concentrations of everolimus and ABT-737 for A-498 and Caki-1 cells. Increasing everolimus concentrations led to a decrease in the proliferation rate of both A-498 and Caki-1 cells (Figure 1B and 1D), suggesting that everolimus shows a cytostatic effect on these cell lines. While treatment with ABT-737 showed a dose- dependent cytotoxicity on A-498, an anti-proliferative activity was seen in Caki-1 cells albeit more potent than everolimus. A-498 cells exhibited 48% cell viability after 72 hours of 5µM ABT-737 treatment, when cell viability recorded for the vehicle-treated A-498 cells at 24 hours were considered as 100%. Decrease in the viability of A-498 cells was significantly pronounced upon 72 hours treatment with 10 µM ABT-737 (Figure 1C). When compared to A-498 cells, Caki-1 cells exhibited more resistant profile to ABT-737 regimen. WST-1 results demonstrated that Caki-1 cells treated with increasing doses of ABT-737 showed similar absorbance values recorded for the control cells at 24 hours, suggesting a cytostatic effect. When control cells reached to a 160% proliferation rate at 72 hours, Caki-1 cells exposed to 5 µM and 10 µM ABT-737 showed an average of 110% proliferation at 72 hours (Figure 1E).
The combination of everolimus and ABT-737 at fixed concentrations was synergistic for A-498 (combination index (CI), 0.32) and Caki-1 (CI, 0.685) cells (Figure S1). Co-treatment with everolimus and ABT-737 synergistically exhibited a cytotoxic effect on A-498 (Figure 1F) and Caki-1 (Figure 1G) cells at all time points. After treatment with the dual-drug combination for 72 hours, cell viability recorded for A-498 cells was 8% (Figure 1F), while the viability of Caki-1 cells was decreased to 20% (Figure 1G). Altogether this data demonstrated that the addition of ABT-737 to everolimus treatment exerted a more enhanced anti-cancer effect in Bcl-2 overexpressing RCC cell lines.
Combination of everolimus and ABT-737 attenuated the cell cycle and mTORC1 signaling
In order to determine whether the anti-proliferative effect of everolimus and ABT-737 co-treatment was mediated through changes in cell cycle dynamics, cell cycle proteins were analyzed in A-498 and Caki-1 cells subjected to either monotherapies or combination therapy for 24 hours (Figure 2A). Treatment of A-498 and Caki-1 cells with everolimus-ABT-737 combination diminished Cyclin D1 and Cyclin D3 protein levels (Figure 2A). Up to 5-fold reduction in CDK4, Cyclin E1 and CDK2 levels in both cell lines suggested that the combination of everolimus with ABT-737 could potentially induce the cell cycle arrest in the early G1-phase. Interestingly, a 7-fold decrease in G1/S-phase inhibitor p27Kip1 protein was evident in A-498 cells treated with DNA polymerase inhibitor aphidicolin or ABT-737 mono- and combination therapies. No change in p27Kip1 levels was observed for Caki-1 cells after the drug treatments.
Inhibitory effect of everolimus and ABT-737 combination on cell cycle and mTORC1 complex. (A) A-498 cells were treated either with 1 µM everolimus and/or 5 µM ABT-737 and Caki-1 cells were exposed to 1 µM everolimus and/or 10 µM ABT-737. After 24 hours of treatment, protein extracts of A-498 and Caki-1 cells were used to determine the protein levels of CDK4, Cyclin D1, Cyclin D3, CDK2, Cyclin E1, and p27Kip1 by Western blot analysis. β-Actin was used as loading control. (B) Effect of the combination treatment on mTOR pathway was investigated by the determination of protein levels of p-mTOR (Ser2448), mTOR, p-p70S6K (Thr389), p70S6K, p-S6 (Ser235/236), p-S6 (Ser240/244), S6, p-4E-BP1 (Ser65), and 4E-BP1 in A-498 cells treated with 1 µM everolimus and/or 5 µM ABT-737, and Caki-1 cells exposed to 1 µM everolimus and/or 10 µM ABT-737 for 24 hours. β-Actin was used as loading control.
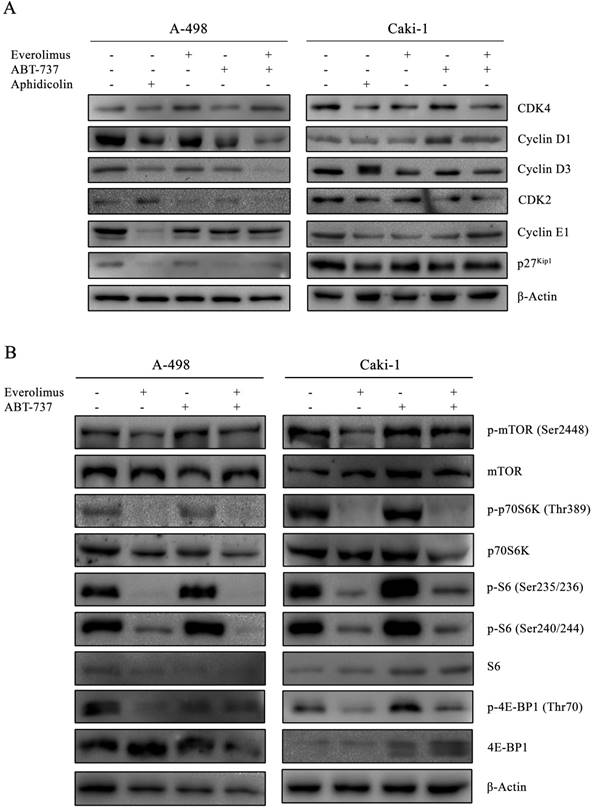
As the elevated activity of mTOR signal transduction is reported to be associated with tumorigenesis of advanced RCC, the effect of everolimus-ABT-737 combination on mTOR pathway was evaluated (36). A-498 and Caki-1 cells subjected either to single or combinatorial regimens for 24 hours showed no change in the basal mTOR levels, while only a slight increase was observed in the active phospho-Ser2448 mTOR levels (Figure 2B). In both cell lines, the combination therapy successfully inhibited the mTOR kinase activity as evidenced by the complete suppression of phosphorylation of mTOR kinase-downstream target proteins including phospho-Thr389 p70S6K1 kinase and phospho-Ser235/236 and -Ser240/244 S6 protein levels. Concomitantly, the dual-drug therapy also blocked phosphorylation of the translational repressors 4E-BP1 protein. Taken together, these findings suggest that the inhibition of mTOR pathway by the combination treatment might contribute to the decrease in survival rates for primary A-498 and metastatic Caki-1 RCC cell lines.
Combination of everolimus and ABT-737 induced apoptotic cell death in A-498 and Caki-1 cells
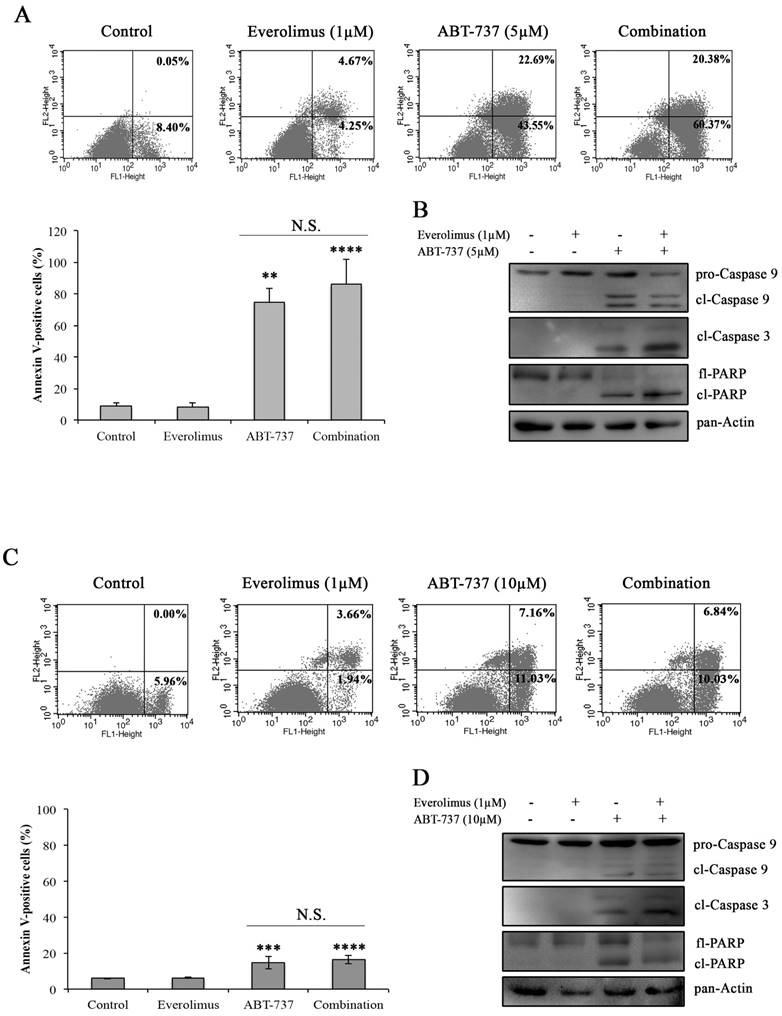
In order to determine whether everolimus and ABT-737 combination synergistically induces an apoptotic response in A-498 and Caki-1 cells, Annexin V-PI staining was carried out in A-498 and Caki-1 cells treated with everolimus and/or ABT-737 for 48 hours. In Figure 3A and 3C, no induction of apoptosis was observed in response to everolimus in both RCC cell lines after 48 hours treatment. However, a significant increase in the number of Annexin V-positive apoptotic A-498 cells was evident after ABT-737 monotherapy (Figure 3A). The increase in A-498 apoptotic cells was more pronounced when these cells were subjected to the combination therapy (Figure 3A). Similarly, Caki-1 cells were more prone to cell death once subjected to the combination therapy than ABT-737 regimen alone (Figure 3C). At molecular level, cleaved caspase 9 and caspase 3, the activators of apoptotic cell death, were analyzed in A-498 (Figure 3B) and Caki-1 cells (Figure 3D) upon ABT-737 monotherapy as well as everolimus-ABT-737 combination at 24 hours. Increased levels of cleaved caspase 3 and 9 and PARP protein was evident in response to ABT-737 suggesting that the Bcl-2 inhibitor alone or in combination with everolimus triggers the apoptotic process in A-498 and Caki-1 cells.
Induction of apoptosis by the combination regimen of everolimus and ABT-737. Flow cytometry analysis was performed with A-498 cells (A) and Caki-1 (C) exposed to everolimus and/or ABT-737 at indicated concentrations for 48 hours to analyze the effect of combinatorial drug treatment on cell death. The percentage of early apoptotic (bottom right quarter) and late apoptotic (top right) cells was presented in the histogram images and the graphical representation of percentages for Annexin V-positive cells (early and late apoptosis) was shown. Graphs represent percentage analysis of Annexin V-positive cells (n = 3). Each data point in bar graph represents the mean ± S.D. of three independent experiments, each performed in triplicate. N.S. (not significant); *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. The effect of drug treatments on apoptotic proteins including caspase 9 and 3, and PARP were analyzed in protein extracts of (B) A-498 or (D) Caki-1 cells exposed to everolimus and/or ABT-737 for 24 hours.
Effect of everolimus-ABT-737 combination on the expression levels of Bcl-2 family proteins
Next, we examined the effect of everolimus- ABT-737 combination on Bcl-2 family proteins. In both A-498 and Caki-1 cells, everolimus-ABT-737 treatment led up to a 80% decrease in the levels of Bcl-2 and Bcl-xL proteins without affecting Mcl-1 protein expression (Figure 4A), which was in correlation with apoptotic response.
Effect of everolimus-ABT-737 combination on the expression levels of Bcl-2 family proteins. Total cell lysates of A-498 cells treated with 1 µM everolimus and/or 5 µM ABT-737, and of Caki-1 cells exposed to 1 µM everolimus and/or 10 µM ABT-737 for 24 hours were used to determine the expression of (A) anti-apoptotic Bcl-2 family proteins (Bcl-2, Bcl-xL, and Mcl-1), and (B) p53, Bim, Puma, Noxa, and Bax proteins. As loading control, β-actin was used.
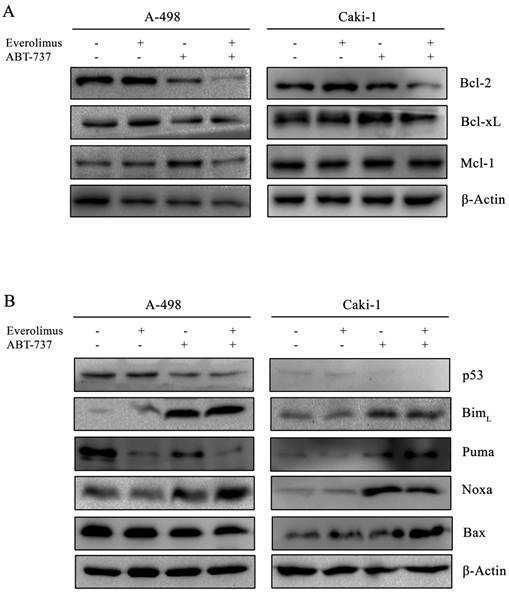
The combination therapy further reduced p53 protein levels about 60% in both cell lines. Albeit to the low p53 levels, a significant increase in the basal BimL (~ 10 fold) and Noxa (~ 20 fold) levels was recorded in A-498 and Caki-1 cells treated with everolimus-ABT-737 combination (Figure 4B). These data suggest that ABT-737 either alone or in combination with everolimus could induce the Bcl-2 family-mediated apoptosis by inducing BimL and Noxa expression in a p53-independent manner.
RenCares cells were generated to overcome everolimus sensitivity of RenCa cells
Next, we determined the cytotoxic effect of everolimus-ABT-737 combination on the mouse murine RCC cell line, RenCa, which were frequently used in in vivo xenograft RCC models (37). Increasing concentrations of everolimus led to a dose-dependent decrease up to 85% in the viability of RenCa cells compared to non-treated control cells (Figure 5A), underlining a possible everolimus sensitivity for this cell line. RenCa cells also displayed retardation in cell growth when treated with the high concentrations of ABT-737 (Figure 5B). To establish an everolimus- resistant RenCa cell line (RenCares), cells were grown in the presence of low everolimus concentrations. In comparison to parental RenCa cells, RenCares cells exhibited no decrease in cell proliferation rate when subjected to increased everolimus concentrations (Figure 5C). However, treatment of RenCares cell line with 20 µM ABT-737 led to a significant 81% decrease in the cell viability (Figure 5D) as a result of induced cell death (Figure 5G) at 72 hours.
Effect of everolimus and/or ABT-737 on RenCa and RenCares cells. (A) Everolimus response of parental RenCa cells was characterized by investigating the effect of increasing everolimus concentration on cell viability at 24, 48, and 72 hours. (B) RenCa cells were subjected to increasing concentrations of ABT-737 at indicated time points. (C) RenCares cells were subjected to increasing doses of everolimus for 24 to 72 hours to test acquired drug resistance by performing cell viability assay. (D) The response of RenCares cells to ABT-737 monotherapy was investigated by cell viability assay upon the treatment of cells with various ABT-737 concentrations for indicated time points. Cell viability of untreated parental and resistant control cells was considered as 100% for 24 hours and graphs show the mean ± S.D of three independent experiments, each performed in triplicate. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001. (E) The effect of the acquired everolimus resistance on mTOR pathway was determined in cell lysates of RenCa and RenCares cells by investigating the protein levels of p-mTOR (Ser2448), mTOR, p-p70S6K (Thr389), p70S6K, p-S6 (Ser235/236), p-S6 (Ser240/244), S6, p-4E-BP1 (Ser65), 4E-BP1. (F) Western blot analysis of Bcl-2, Bcl-xL, and Mcl-1 was performed with proteins isolated from RenCa and RenCares cells. β-Actin was used as loading control. (G) RenCares cells exposed to 20 µM ABT-737 were used for Annexin-V/PI staining to determine the effect of ABT-737 monotherapy for 72 hours. The graph shows the percentage of Annexin V-positive cells. Each data value represents the mean ± S.D. of three independent experiments, each performed in triplicate. *, P ≤ 0.05.
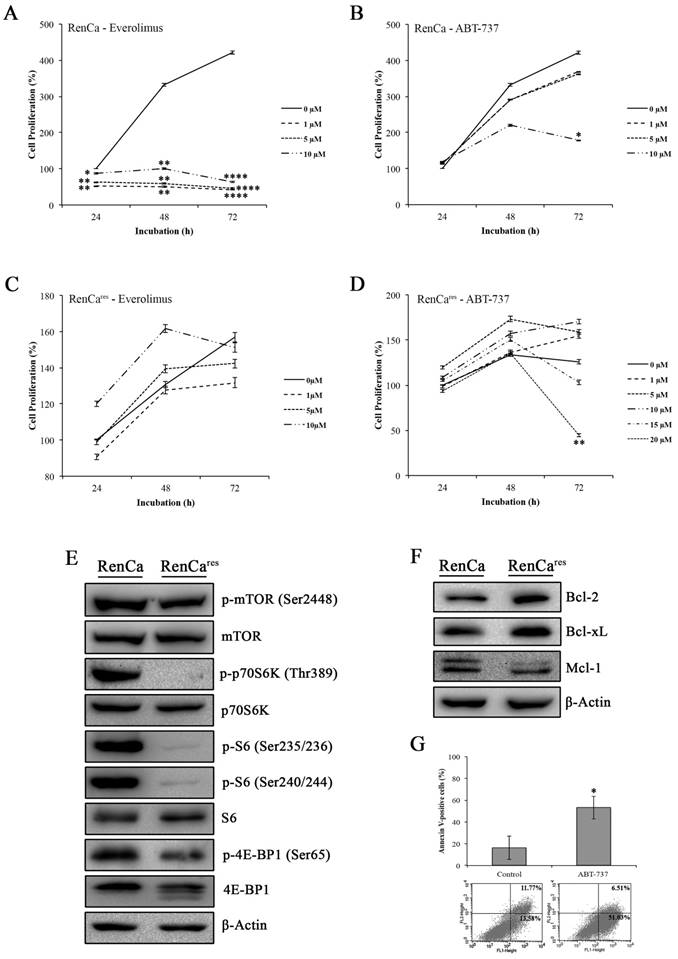
Investigation of mTOR pathway in RenCares cells revealed a decrease in the protein levels of mTOR kinase downstream targets when compared to the parental RenCa cells (Figure 5E). In support of our hypothesis, an average of 3 fold increase in Bcl-2 and Bcl-xL protein levels were observed in RenCares cells (Figure 5F), suggesting that Bcl-2 overexpression might be a key player in the development of everolimus resistance.
Combination of everolimus and ABT-737 could enhance the anti-tumor effect of everolimus
Using RenCares cells, an RCC xenograft BALB/c mouse model was established to elucidate the efficiency of combination therapy in vivo. The tumor formation was observed in the control group and groups subjected to the monotherapies (Figure 6A). The animals in the everolimus cohort developed smaller tumors than animals in the control and ABT-737 cohorts (Figure 6A). Additionally, the weight of tumors in the control group was significantly higher than the weight of tumors in everolimus cohort (Figure 6B). H&E staining of tumor tissues revealed nuclear pleomorphism and the histological architecture of RCC preserved in xenografts, indicating the successful generation of RCC xenograft mouse model (Table S1, Figure 6C). Mice subjected to combination therapy did not develop any tumors suggesting that the everolimus-ABT-737 combination markedly suppressed the tumor formation. In each animal cohort, no change in the body weight of mice was observed suggesting that injected doses of everolimus and ABT-737 were tolerable (Figure 6D). In addition, histopathological examination of brain, thymus, heart, lung, stomach, spleen, liver, kidney, intestine, and testis indicated no signs of toxicity or metastatic lesions in any of the animal groups.
Anti-tumor effect of everolimus and ABT-737 combination on RenCares xenograft mice model. BALB/c mice were randomized into four groups (n = 8) and administered every other day with the vehicle control, 2 mg/kg everolimus, 75 mg/kg ABT-737 or the combination of single drug doses. (A) Representative pictures of tumors isolated from each group after 21 days of treatment were shown. (B) Average weight of tumors from each cohort subjected to everolimus and/or ABT-737 treatment or control was shown. Scatter blot represents the mean ± S.D. The significant analysis of tumor weights of control groups compared to treatment groups was performed by two-tailed Student's t-test. *, P ≤ 0.05; ****, P ≤ 0.0001. (C) Images are the representatives for hematoxylin and eosin staining of tumor tissue sections. (D) The graph shows the change of mice body weight at the days of injection. Each data point in the graph represents the mean ± S.D. of body weights of eight mice in each group.
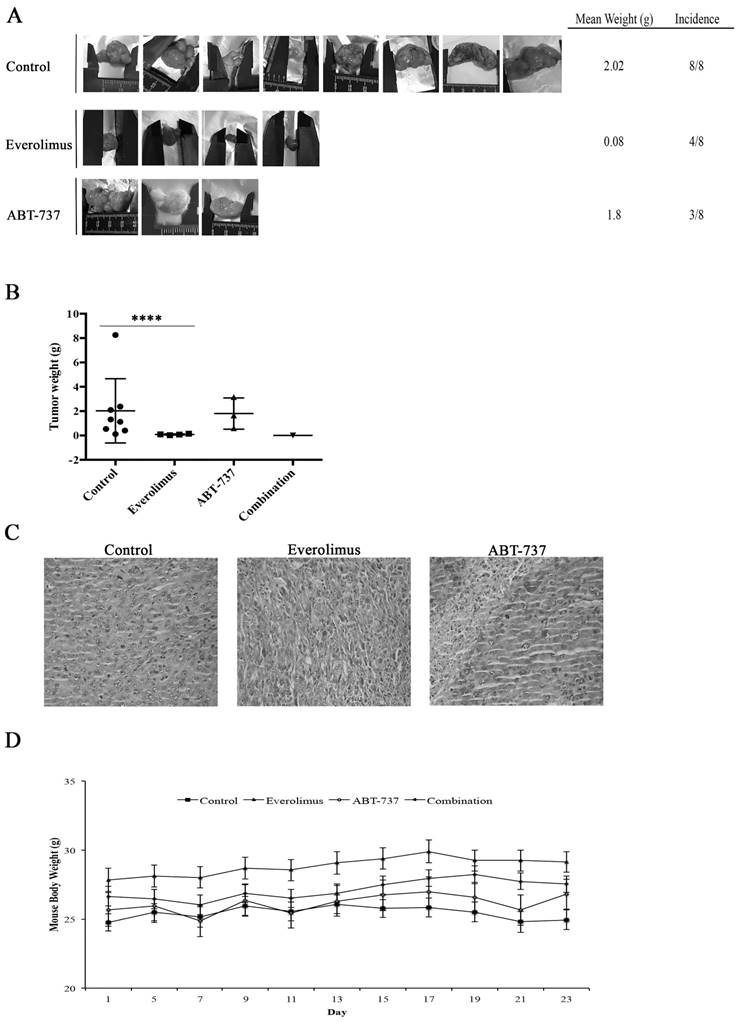
Collectively, our data suggest that Bcl-2 inhibitor ABT-737 could potentiate the anti-tumor effect of everolimus, when used in combination with everolimus.
Discussion
During the last decades, comprehensive research has been performed to find out the molecular mechanisms playing pivotal roles in RCC tumorigenesis. Everolimus, a molecular therapeutic, targets specifically one of these molecular mechanisms and was approved by FDA as a second-line therapy for the mRCC patients refractory to sorafenib or sunitinib therapy (19). The low survival rate and the acquired resistance to everolimus are the clinical limitations that represent an urgent need for the new therapy strategies (21, 23). In the current study, we showed that the combination of everolimus with the Bcl-2 inhibitor ABT-737 as a novel therapy enhanced the anti-tumor effect of everolimus in vitro ad in vivo models of RCC.
ABT-737 has been designed to suppress the anti-apoptotic effect of Bcl-2 protein whose elevated levels has been implicated in a number of solid tumors (32). In support of the literature, we chose primary site A-498 and metastatic site Caki-1 cells as in vitro RCC models due to their expression of high Bcl-2 basal protein levels. Our results suggested a possible everolimus resistance for these RCC cell lines, as no significant reduction in their cell viability was observed upon everolimus regimen, which was in consistence with the previous studies (38, 39). While ABT-737 monotherapy significantly decreased the proliferation of A-498 cells, only a cytostatic effect was evident for Caki-1 cells indicating that the overexpression of Bcl-2 might lead to ABT-737 resistance, which was also shown by Ni et al. in hepatocellular carcinoma cell lines (40). In comparison to monotherapies, everolimus-ABT-737 combination synergistically diminished the viability of A-498 and Caki-1 cells suggesting that ABT-737 enhances the anti-tumor effect of everolimus. Neither monotherapies (Figure S2A and S2B) nor the combination therapy (Figure S2C) resulted in a cytotoxic response on human embryonic kidney HEK-293 used as control healthy cell line.
One of the possibilities to explain the observed reduction in cell viability as a result of the combination therapy could be the inhibition of cell cycle process. Supporting literature showed that the decreased viability of non-small cell lung cancer (NSCLC) and esophageal squamous cell carcinoma cells was due to reduction in the levels of cell cycle proteins including CDK2, Cyclin D1, CDK4, and Cyclin E1 (41, 42). In agreement, we observed that the combination therapy decreased the basal levels of cell cycle proteins in both RCC cell lines. Everolimus- ABT-737 co-treatment led to the inactivation of mTOR pathway through loss of its phosphorylated downstream target proteins including S6K, S6, and 4E-BP1. Supporting our data the depletion of p-S6 resulted in G1 cell cycle arrest in RCC cells subjected to the combination of RAD001 and selumetinib, MEK1 inhibitor (43).
ABT-737 alone or in combination with everolimus triggered apoptosis in both A-498 and Caki-1 cells as evidenced by the increased number of apoptotic cells and detection of cleaved apoptosis activators, caspase 9 and 3, which is in agreement with the studies reporting the cell death-inducing effect of ABT-737 treatment for small cell lung cancer (SCLC) and thyroid carcinoma cell lines (44, 45). The significant decrease we observed in the basal Bcl-2 protein levels was in correlation with the upregulated BimL suggesting that ABT-737 led to the activation of intrinsic apoptosis that was regulated by the complex interplay between anti-apoptotic Bcl-2 and the BimL and Puma. Additionally, our data showed an indirect inhibitory effect of everolimus-ABT-737 combination on Mcl-1 protein, which is triggered through p53-independent protein expression of Noxa that neutralizes Mcl-1 (46).
The validation of our in vitro findings using RenCa xenograft mice model was not possible as previous literature reported that RenCa cell proliferation and viability was suppressed in response to everolimus (37). Therefore, we generated RenCares cells from the parental drug-sensitive RenCa cell line for our in vivo studies. Analysis of mTOR in RenCares cells showed that the phosphorylation of mTOR's downstream target proteins was completely abolished, although no decrease in RenCares cell viability was observed upon treatment with the increasing concentrations of everolimus. Supporting our hypothesis that the overexpression of anti-apoptotic Bcl-2 protein might correlate with the everolimus-resistance in A-498 and Caki-1 cells, RenCares cells displayed increased levels of Bcl-2 protein when compared to the parental drug sensitive RenCa cells, possibly giving a survival advantage to RenCares cells, even when mTOR pathway was abrogated. Indeed, a decrease in proliferation of RenCares cells was detected as a result of increased apoptosis upon treatment with Bcl-2 inhibitor ABT-737. We could successfully establish an RCC xenograft mouse model with RenCares cells as evidenced by the palpable tumor growth in all animals of the control cohort and monotherapy cohorts albeit with a lower incidence. In comparison to individual drug treatments, only everolimus-ABT- 737 combination could completely suppress the growth of RenCares xenograft tumors. No evidence of toxicity was found in any treatment groups as body weight was not altered and no pathological change was detected in the samples isolated. In agreement with our findings, recent literature reported enhanced therapeutic efficacy for the dual-drug combinations without any adverse toxicity (47, 48). Several reports in the literature also demonstrated the anti-tumor response of rapalogs and ABT-737 combination in lung cancer to overcome the acquired ABT-737 resistance observed in patients with SCLC (49, 50). The combination of ABT-737 with rapamycin could rescue the ABT-induced resistance in patient derived xenograft (PDX) SCLC model (49). In addition to SCLC, the therapeutic potential of rapamycin-ABT- 737 combination has been proposed to circumvent the clinical limitation of the resistance against radiotherapy observed in NSCLC patients (50).
In summary, we showed for the first time that targeting Bcl-2 protein by ABT-737 in a therapeutic combination with everolimus enhanced the anti- tumor effect of everolimus shown by the regression of RenCares tumor growth. According to these findings, our study might open a new avenue for the clinical evaluation of everolimus-ABT-737 combination to provide a therapeutic benefit for the treatment of advanced RCC patients.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was partly supported by The Scientific and Technological Research Council of Turkey (Grant number 114S332) and Yeditepe University Hospital. A. H. Nayman is a recipient of doctoral fellowships from the Yeditepe University. H. Siginc is a recipient of doctoral fellowship from The Scientific and Technological Research Council of Turkey. We thank Emrah Nikerel (Yeditepe University, Turkey) for advices concerning the statistical analysis. We would also like to acknowledge Burcin Asutay and Engin Sumer (Yeditepe University, Turkey) for their technical support.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69-90
2. Shuch B, Amin A, Armstrong AJ, Eble JN, Ficarra V, Lopez-Beltran A. et al. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. European urology. 2015;67(1):85-97
3. Cheville JC, Lohse CM, Zincke H, Weaver AL, Blute ML. Comparisons of outcome and prognostic features among histologic subtypes of renal cell carcinoma. Am J Surg Pathol. 2003;27(5):612-24
4. Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol. 2009;10(10):992-1000
5. Guo H, German P, Bai S, Barnes S, Guo W, Qi X. et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. J Genet Genomics. 2015;42(7):343-53
6. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274-93
7. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB. et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol. 1997;7(4):261-9
8. Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829-34
9. Takuwa N, Fukui Y, Takuwa Y. Cyclin D1 expression mediated by phosphatidylinositol 3-kinase through mTOR-p70(S6K)-independent signaling in growth factor-stimulated NIH 3T3 fibroblasts. Mol Cell Biol. 1999;19(2):1346-58
10. Land SC, Tee AR. Hypoxia-inducible factor 1alpha is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J Biol Chem. 2007;282(28):20534-43
11. Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17(6):596-603
12. Braunstein S, Karpisheva K, Pola C, Goldberg J, Hochman T, Yee H. et al. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol Cell. 2007;28(3):501-12
13. Krieg M, Haas R, Brauch H, Acker T, Flamme I, Plate KH. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene. 2000;19(48):5435-43
14. Azim H, Azim HA Jr, Escudier B. Targeting mTOR in cancer: renal cell is just a beginning. Target Oncol. 2010;5(4):269-80
15. Chen J, Zheng XF, Brown EJ, Schreiber SL. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc Natl Acad Sci U S A. 1995;92(11):4947-51
16. Oshiro N, Yoshino K, Hidayat S, Tokunaga C, Hara K, Eguchi S. et al. Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells. 2004;9(4):359-66
17. O'Donnell A, Faivre S, Burris HA 3rd, Rea D, Papadimitrakopoulou V, Shand N. et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol. 2008;26(10):1588-95
18. Amato RJ, Jac J, Giessinger S, Saxena S, Willis JP. A phase 2 study with a daily regimen of the oral mTOR inhibitor RAD001 (everolimus) in patients with metastatic clear cell renal cell cancer. Cancer. 2009;115(11):2438-46
19. Anandappa G, Hollingdale A, Eisen T. Everolimus - a new approach in the treatment of renal cell carcinoma. Cancer Manag Res. 2010;2:61-70
20. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S. et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: final results and analysis of prognostic factors. Cancer. 2010;116(18):4256-65
21. Grunwald V, Karakiewicz PI, Bavbek SE, Miller K, Machiels JP, Lee SH. et al. An international expanded-access programme of everolimus: addressing safety and efficacy in patients with metastatic renal cell carcinoma who progress after initial vascular endothelial growth factor receptor-tyrosine kinase inhibitor therapy. Eur J Cancer. 2012;48(3):324-32
22. Figlin RA, Kaufmann I, Brechbiel J. Targeting PI3K and mTORC2 in metastatic renal cell carcinoma: new strategies for overcoming resistance to VEGFR and mTORC1 inhibitors. Int J Cancer. 2013;133(4):788-96
23. Kroeger N, Choueiri TK, Lee JL, Bjarnason GA, Knox JJ, MacKenzie MJ. et al. Survival outcome and treatment response of patients with late relapse from renal cell carcinoma in the era of targeted therapy. European urology. 2014;65(6):1086-92
24. Santoni M, Pantano F, Amantini C, Nabissi M, Conti A, Burattini L. et al. Emerging strategies to overcome the resistance to current mTOR inhibitors in renal cell carcinoma. Biochim Biophys Acta. 2014;1845(2):221-31
25. Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S. et al. Expression of apoptosis-regulating proteins in chronic lymphocytic leukemia: correlations with In vitro and In vivo chemoresponses. Blood. 1998;91(9):3379-89
26. Robertson LE, Plunkett W, McConnell K, Keating MJ, McDonnell TJ. Bcl-2 expression in chronic lymphocytic leukemia and its correlation with the induction of apoptosis and clinical outcome. Leukemia. 1996;10(3):456-9
27. Kaiser U, Schilli M, Haag U, Neumann K, Kreipe H, Kogan E. et al. Expression of bcl-2-protein in small cell lung cancer. Lung Cancer. 1996;15(1):31-40
28. Maruyama R, Yamana K, Itoi T, Hara N, Bilim V, Nishiyama T. et al. Absence of Bcl-2 and Fas/CD95/APO-1 predicts the response to immunotherapy in metastatic renal cell carcinoma. Br J Cancer. 2006;95(9):1244-9
29. Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26(9):1324-37
30. Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2(9):647-56
31. Pellecchia M, Reed JC. Inhibition of anti-apoptotic Bcl-2 family proteins by natural polyphenols: new avenues for cancer chemoprevention and chemotherapy. Curr Pharm Des. 2004;10(12):1387-98
32. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA. et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677-81
33. Zheng L, Yang W, Zhang C, Ding WJ, Zhu H, Lin NM. et al. GDC-0941 sensitizes breast cancer to ABT-737 in vitro and in vivo through promoting the degradation of Mcl-1. Cancer Lett. 2011;309(1):27-36
34. Telci D, Wang Z, Li X, Verderio EA, Humphries MJ, Baccarini M. et al. Fibronectin-tissue transglutaminase matrix rescues RGD-impaired cell adhesion through syndecan-4 and beta1 integrin co-signaling. J Biol Chem. 2008;283(30):20937-47
35. Demirci S, Dogan A, Basak N, Telci D, Dede B, Orhan C. et al. A Schiff base derivative for effective treatment of diethylnitrosamine-induced liver cancer in vivo. Anticancer Drugs. 2015;26(5):555-64
36. Lieberthal W, Levine JS. The role of the mammalian target of rapamycin (mTOR) in renal disease. J Am Soc Nephrol. 2009;20(12):2493-502
37. Hirayama Y, Gi M, Yamano S, Tachibana H, Okuno T, Tamada S. et al. Anti-PD-L1 treatment enhances antitumor effect of everolimus in a mouse model of renal cell carcinoma. Cancer Sci. 2016;107(12):1736-44
38. Juengel E, Nowaz S, Makarevi J, Natsheh I, Werner I, Nelson K. et al. HDAC-inhibition counteracts everolimus resistance in renal cell carcinoma in vitro by diminishing cdk2 and cyclin A. Mol Cancer. 2014;13:152
39. Hassan B, Akcakanat A, Sangai T, Evans KW, Adkins F, Eterovic AK. et al. Catalytic mTOR inhibitors can overcome intrinsic and acquired resistance to allosteric mTOR inhibitors. Oncotarget. 2014;5(18):8544-57
40. Ni Z, Wang B, Dai X, Ding W, Yang T, Li X. et al. HCC cells with high levels of Bcl-2 are resistant to ABT-737 via activation of the ROS-JNK-autophagy pathway. Free Radic Biol Med. 2014;70:194-203
41. Chen B, Tan Z, Gao J, Wu W, Liu L, Jin W. et al. Hyperphosphorylation of ribosomal protein S6 predicts unfavorable clinical survival in non-small cell lung cancer. J Exp Clin Cancer Res. 2015;34:126
42. Kim SH, Jang YH, Chau GC, Pyo S, Um SH. Prognostic significance and function of phosphorylated ribosomal protein S6 in esophageal squamous cell carcinoma. Mod Pathol. 2013;26(3):327-35
43. Zou Y, Wang J, Leng X, Huang J, Xue W, Zhang J. et al. The selective MEK1 inhibitor Selumetinib enhances the antitumor activity of everolimus against renal cell carcinoma in vitro and in vivo. Oncotarget. 2017;8(13):20825-33
44. Hann CL, Daniel VC, Sugar EA, Dobromilskaya I, Murphy SC, Cope L. et al. Therapeutic efficacy of ABT-737, a selective inhibitor of BCL-2, in small cell lung cancer. Cancer Res. 2008;68(7):2321-8
45. Broecker-Preuss M, Becher-Boveleth N, Muller S, Mann K. The BH3 mimetic drug ABT-737 induces apoptosis and acts synergistically with chemotherapeutic drugs in thyroid carcinoma cells. Cancer Cell Int. 2016;16:27
46. Zall H, Weber A, Besch R, Zantl N, Hacker G. Chemotherapeutic drugs sensitize human renal cell carcinoma cells to ABT-737 by a mechanism involving the Noxa-dependent inactivation of Mcl-1 or A1. Mol Cancer. 2010;9:164
47. Zhu Y, Zhang X, Liu Y, Zhang S, Liu J, Ma Y. et al. Antitumor effect of the mTOR inhibitor everolimus in combination with trastuzumab on human breast cancer stem cells in vitro and in vivo. Tumour Biol. 2012;33(5):1349-62
48. Paoluzzi L, Gonen M, Bhagat G, Furman RR, Gardner JR, Scotto L. et al. The BH3-only mimetic ABT-737 synergizes the antineoplastic activity of proteasome inhibitors in lymphoid malignancies. Blood. 2008;112(7):2906-16
49. Gardner EE, Connis N, Poirier JT, Cope L, Dobromilskaya I, Gallia GL. et al. Rapamycin rescues ABT-737 efficacy in small cell lung cancer. Cancer Res. 2014;74(10):2846-56
50. Kim KW, Moretti L, Mitchell LR, Jung DK, Lu B. Combined Bcl-2/mammalian target of rapamycin inhibition leads to enhanced radiosensitization via induction of apoptosis and autophagy in non-small cell lung tumor xenograft model. Clin Cancer Res. 2009;15(19):6096-105
Author contact
![]() Corresponding author: Dilek Telci, Yeditepe University, Faculty of Engineering, Department of Genetics and Bioengineering, Kayisdagi Cad., 34755, Istanbul, Turkey. Phone: +902165781592; Fax: +902165780829; E-mail: dilek.telciedu.tr
Corresponding author: Dilek Telci, Yeditepe University, Faculty of Engineering, Department of Genetics and Bioengineering, Kayisdagi Cad., 34755, Istanbul, Turkey. Phone: +902165781592; Fax: +902165780829; E-mail: dilek.telciedu.tr