Impact Factor ISSN: 1837-9664
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Volume 17; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Special Issues
Introduction
Notch signaling pathway
Activation and regulation of...
Notch signaling and HCC...
Notch signaling and HCC...
Notch signaling and HCC...
Notch signaling and HCC invasion...
Notch signaling and HCC prognosis
Notch signaling as a therapeutic...
Therapy with a single agent
Treatment with a combination of...
Conclusion and future prospective
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(6):1570-1579. doi:10.7150/jca.26847 This issue Cite
Review
The Carcinogenic Role of the Notch Signaling Pathway in the Development of Hepatocellular Carcinoma
Qinfeng Huang1, Junhong Li2, Jinghui Zheng3, Ailing Wei2 ![]()
1. Graduate School, Hunan University of Chinese Medicine, Changsha 410208, Hunan, China;
2. The First Affiliated Hospital, Guangxi University of Chinese Medicine, Nanning 530023, Guangxi, China;
3. Discipline Construction Office, Guangxi University of Chinese Medicine, Nanning 530200, Guangxi, China.
Received 2018-4-23; Accepted 2019-1-12; Published 2019-2-26
Abstract
The Notch signaling pathway, known to be a highly conserved signaling pathway in embryonic development and adult tissue homeostasis, participates in cell fate decisions that include cellular differentiation, cell survival and cell death. However, other studies have shown that aberrant in Notch signaling is pro-tumorigenic, particularly in hepatocellular carcinoma (HCC). HCC is one of the most common malignant tumors in the world and has a high mortality rate. Growing evidence supports that Notch signaling plays a critical role in the development of HCC by regulating the tumor microenvironment, tumorigenesis, progression, angiogenesis, invasion and metastasis. Accordingly, overexpression of Notch is closely associated with poor prognosis in HCC. In this review, we focus on the pro-tumorigenic role of Notch signaling in HCC, summarize the current knowledge of Notch signaling and its role in HCC development, and outline the therapeutic potential of targeting Notch signaling in HCC.
Keywords: hepatocellular carcinoma, Notch signaling pathway, carcinogenesis, therapeutics
Introduction
Primary liver cancer is one of the most common aggressive malignancies, which presents a serious threat to human health [1]. Hepatocellular carcinoma (HCC) is the major histological subtype among primary liver cancers, representing over 90% of total cases of primary liver malignancies [2]. China is home to more than 50% of the word's HCC patients [3, 4]. Hepatitis B virus, Hepatitis C virus, and nonalcoholic fatty liver disease are major contributing factors to this high incidence of HCC [5]. Treatment options for HCC include surgery, chemotherapy, radiation and liver transplantation. However, high incidence of recurrence and poor prognosis still prevail, particularly for patients with advanced HCC. Therefore, it is imperative that we better understand the molecular pathogenesis of HCC in order to develop more effective treatment options for HCC patients.
Notch signaling plays a significant role in both normal liver development and liver tumorigenesis [6]. The available data that the role of Notch signaling in HCC were controversial. Some reports suggest that Notch signaling functions as a tumor suppressor in HCC. Notch1 can induce apoptosis HCC cells by altering the balance between B-cell lymphoma-2 and p53 [7]. Consistent with the finding, upregulated p53 expression induced by Notch1 enhanced HCC cells to sensitive to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis [8]. Furthermore, activation of Notch signaling reduced cell proliferation and tumor growth of HCC by inactivating the retinoblastoma pathway [9]. However, more evidence supports the carcinogenic role of Notch in the development of HCC. Notch1, Notch3, and Notch4 are commonly overexpressed in HCC, with Notch3 and Notch4 detected in 78% and 68% of HCC tissues, respectively [10]. Activated Notch signaling has also been shown to promote liver tumor formation in mouse models [11]. Upregulation of Notch1 signaling can also increase the oncogenic potential of human HCC cells [12]. Due to the involvement of Notch signaling in HCC proliferation, invasion, and metastasis, numerous studies have demonstrated the potential of Notch as an HCC therapeutic target [13, 14]. This article reviews the advances in understanding Notch signaling and its pro-tumorigenic role in HCC, as well as the current progress in targeted therapy.
Notch signaling pathway
Notch signaling is composed of receptors, ligands, and DNA-binding proteins. At present, 4 transmembrane Notch receptors (Notch1, 2, 3, and 4), and 2 families of Notch ligands (Jagged-1, 2 and Delta-like (DLL-1, 3, 4)) have been found in mammals. These homologous Notch receptors function as transmembrane heterodimers. Each Notch receptor consists of 3 parts, namely, the intracellular domain of Notch (ICN), transmembrane fragment, and the extracellular domain of Notch (ECN). The intracellular domain has an RBP-J kappa-associated module (RAM) domain and 7 ankyrin repeats. The activity of specific transcription factors is regulated by the RAM domain and ankyrin repeats of ICN. The intracellular domain also contains several nuclear localization sequences, a transactivation region, and a proline-glutamate-serine-threonine-rich region. The extracellular domain of Notch proteins includes 29-36 tandem epidermal growth factor (EGF)-like repeats, which are followed by the negative regulatory region with 3 cysteine-rich repeat regions and a heterodimerization domain [15]. Modification of the extracellular Notch domain by binding of calcium ions, fucosylation, or glycosylation can increase the regulatory capacity of Notch.
The ECN of the canonical and noncanonical ligands also include a few EGF repeats, which are more plentiful in the jagged-like ligands [16, 17]. The canonical Notch ligands are Delta/Serrate/Lag-2 (DSL) proteins, which share a conserved N-terminal region in the ECN necessary for receptor ligation and activation of Notch signaling. The DSL domain together with the amino terminal domain represents the EGF-binding domain region, which is essential for ligand interactions with specific Notch EGF-like repeats [15]. The Notch ligands Delta-like 1(Dll1) and Jagged1/2 also contain EGF-like repeats, termed the Delta and OSM-11(DOS) motif, which functions to enhance ligand affinity for the Notch receptors. Despite lacking the DSL domain, Delta-like non-canonical Notch ligand 1 and 2 (DLK1and DLK2) also belong to the EGF-like family of membrane proteins and act as functional inhibitors of Notch signaling. In addition to DLK1 and DLK2, other soluble proteins can compete with the canonical Notch ligands Jagged1 and Dll4 for Notch receptor binding [18].
Activation and regulation of Notch signaling pathway
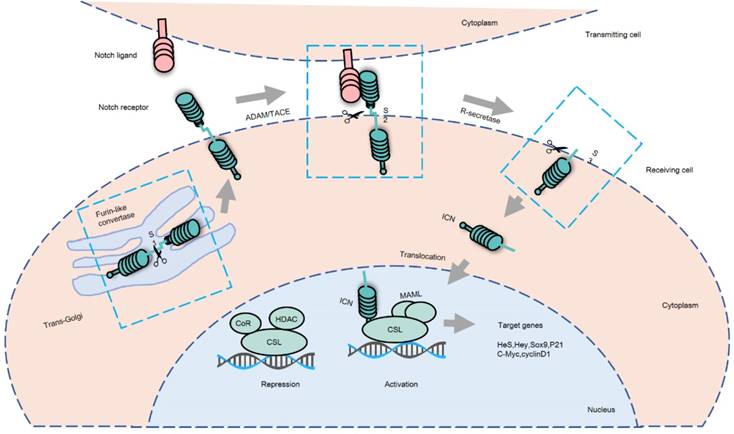
The activation of Notch signaling can take place via either of two pathways; the non-canonical Notch signaling pathway and a more frequently used Notch-dependent processes called the canonical Notch signaling pathway (Figure 1). Activation of Notch signaling requires cell-to-cell contact through interaction of the Notch receptor and its ligand [19], which are expressed by the “receiver” cell and the “transmitter” cell, respectively. Sustained ligand-receptor interactions result in a switch where signal transmitting cells start expressing the receptors, and the signal receiving cells start expressing the ligands [20]. The ICN is released from the cell membrane after it is cleaved by the γ--secretase complex containing presenilin, nicastrin, APH1 and PEN2. Subsequently, the ICN in a complex with DNA-binding proteins of the CSL (CBF1/RBPJ-kappa/Su (H)/Lag1) family is transported to the nucleus of receiving cells where it serves as a transcriptional co-activator. This activates the transcription of Notch target genes, including members of the Hairy and Enhancer of Split 1(HES1), and HES1-related (HESR1) families of basic helix-loop- helix transcription factors [21]. These transcription factors control a number of cellular processes, including proliferation, differentiation, and apoptosis, which are deregulated in cancer development and progression [22, 23].
Canonical Notch signaling pathway. Notch signaling is initiated by cell-to-cell contact allowing for interaction of Notch receptors and ligands. There are 3 major cleavage processes: S1 cleavage occurs in the trans-Golgi when a Furin-like convertase cleaves Notch receptors to produce heterodimeric receptors. Then, Notch receptors are transported to the cell surface of the receiving cell. When the extracellular domain of Notch receptor combines with its ligand, the Notch receptor is cleaved by a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10), ADAM17 or TNF-α-converting enzyme (TACE) metalloprotease in S2 cleavage site, releasing an extracellular fragment. Afterwards, the remnant receptor is cleaved again by the γ-secretase complex in its transmembrane domain at S3 cleavage site, releasing the active intracellular domain of Notch (ICN). ICN translocates to the nucleus of the receiving cell and binds to the DNA-binding transcription factor CSL (CBF1/RBPJ-kappa/Su (H)/Lag1). Thereafter, ICN competitively replaces the co-repressor (CoR) followed by the recruitment of the co-activator mastermind-like 1(MAML) to form a complex, initiating the transcription of CSL-dependent Notch target genes.
Notch signaling and HCC microenvironment
Published studies have alluded to the importance of the interaction between tumor cells and the tumor microenvironment and its role in the etiopathogenesis of HCC. The tumor microenvironment plays an integral role in modulating hepatic fibrosis, liver carcinogenesis, epithelial-mesenchymal transition, neoplasm invasion, and neoplasm metastasis [24, 25]. HCC is closely associated with liver fibrosis, with fibrotic or cirrhotic livers contributing to approximately 90% of HCC cases. This suggests that hepatic fibrosis is a crucial factor in the liver premalignant environment. Experiments in mice have shown that hepatic stellate cells (HSCs) contribute to over 90% of the hepatic myofibroblasts in fibrosis induced by biliary injury, hepatotoxins, or non-alcoholic fatty liver disease, demonstrating that activation of HSCs can mediate fibrogenesis [26]. The major cell types found in HCC tumor microenvironment include immune cells, macrophages, fibroblasts, HSCs, endothelial cells, and cancer stem cells (CSCs). Macrophages are believed to be derived from monocytes and can further differentiate into tumor associated macrophages (TAMs). TAMs contribute to the development of tumors by releasing pro-tumorigenic cytokines [27]. Notch signaling ligand Dll4 promotes pro-inflammatory activation of macrophages in vitro. Silencing of Notch1-4 by RNA interference attenuated Dll4-triggered expression of inducible nitric oxide synthase in human macrophages [28]. Activated TAMs accelerate angiogenesis and hepatocarcinogenesis through multiple mechanisms. Additionally, density of TAMs is associated with large tumor size, advanced TNM stage, intrahepatic metastasis, disease recurrence, and poor overall survival in HCC patients [29]. Cancer- associated fibroblasts (CAFs) can modulate the biological activities of HCC [30]. The presence of CAFs is associated with HCC cell growth, metastasis, and intravasation. In turn, HCC cells stimulate CAF proliferation, suggesting that CAFs play a critical role in mediating the interaction between cancer cells and the tumor microenvironment [31]. CAFs can secrete high levels of IL-6. This cytokine facilitates stem cell-like characteristics in HCC cells by activating Notch signaling pathway through STAT3 phosphorylation. Notch1 silencing may abolish the pro-tumorigenic effect of CAFs on HCC progression [32]. More importantly, fibroblasts and macrophages can increase the tumorigenesis and malignancy of HCC due in part to Notch signaling-involved M1/M2 polarization of macrophages and the activation of HSC in liver fibrosis [33, 34]. HSCs are responsible for collagen synthesis in the liver [35]; HSCs are activated in response to the repeated liver damage and then trans-differentiated into myofibroblast-like cells. This transformation is considered as a key process in the development of hepatic fibrosis [36]. Notch3 can regulate HSC activation in hepatic fibrosis, suggesting that selective interruption of Notch3 signaling may be a potential strategy to treat liver fibrosis [37]. During liver fibrosis, activated HSCs trigger accumulation of the extracellular matrix and infiltrate the stroma of hepatic tumors situated around the fibrous septa, tumor sinusoids, and capsules. Therefore, HSCs may play a fundamental role in the progression of HCC [38]. Additionally, HCC cells have been shown to promote the activation of HSCs in a rat model [39]. The mRNA levels of Notch1 and Notch target gene HES1 was significantly reduced in the activation of HSCs [40]. Furthermore, the interaction between activated HSCs and monocytes has been shown to promote the development and progression of HCC by enhancing HCC cell proliferation, migration and tumor sphere formation [41].
The cancer stem cell (CSC) niche, which is concept unique aspect of the HCC microenvironment, actively participates in the growth of primary HCC tumors and HCC metastasis [42]. CSCs, with their capacity for self-renewal and differentiation have been identified in HCC [43]. Several cell markers of hepatic CSCs have been identified, including CD90, CD44, CD133, EpCAM, CD13, and CD24. Notch signaling has been shown to be associated with the recurrence of CSCs after radiation therapy, due to its involvement in CSC self-renewal [44]. Notch signaling activity has been detected in isolated liver CSCs, which contribute to the heterogeneity of HCC [45]. Additionally, Notch signaling activated by Jagged1 can enhance the cancer stem cell-like features of CD90- HCC cells, which demonstrate rapid G1/S transition phase after activation of Notch signaling [46]. In liver development, liver progenitor cells as well as hepatoblasts, differentiate into two cell lineages: hepatocytes and cholangiocytes, which may lead to tumor formation by acquiring dedifferentiation-induced cancer stem cell-like features [47]. Notch2 and Notch4 are implicated in the proliferation of hepatoblasts [48]. Notch2 and Jagged1 facilitates liver progenitor cell differentiation into biliary epithelial cell. However, biliary differentiation is antagonized by hepatocyte growth factor, which promotes hepatocyte differentiation [49]. Notch 2-expressing cells include bile duct epithelial cell precursors, indicating that Notch signaling participates in the formation of the bile duct in mice [49]. Jagged1 is highly expressed in fibroblast in the portal region, suggesting that Notch signaling is activated and contributes to cholangiocyte differentiation [50]. In normal liver tissue, Notch1, 2, and -3 were coexpressed on bile duct epithelium; however, with the exception of Notch3 in livers with primary sclerosing cholangitis, expression was absent from proliferating ductules in all disease states examined. This study suggested that Notch signaling may be important to normal bile duct formation [51]. Additionally, cell proliferation of bile duct cells is induced by activation of Notch signaling, leading to the formation of bile canaliculi [52]. It has been reported that genetic aberrations in Jagged1 can enhance HCC malignancy [53]. Therefore, the development and progression of HCC is heavily impacted by the tumor microenvironment, suggesting that this interaction needs to be further explored.
Notch signaling and HCC tumorigenesis
Aberrant Notch signaling is an important factor in HCC tumorigenesis and progression. Disrupted Notch genes are frequently found in HCC tissues in comparison to normal liver tissues. Four Notch receptors have been reported to be expressed in liver cancer cells, with varying sub-cellular distributions. Specifically, Notch1 and Notch4 were both expressed in the cytoplasm and nucleus, while Notch2 and Notch3 were expressed only in the cytoplasm. In comparison with non-tumor adjacent tissues, HCC tissues expressed a higher level of Notch1 in the cytoplasm and Notch4 in nucleus, and a low level of Notch2 in the cytoplasm. There was no significant difference between the expression levels of Notch3 and Notch4 [54]. The heterogeneous expression pattern of Notch receptors has also been observed in another study [55]. More than 80% of human HCC specimens demonstrate high Notch expression in comparison to the adjacent normal tissues [12]. Notch1 signaling has been shown to facilitate HCC formation in a genetically engineered mouse models [11]. A positive correlation has also been observed between the expression of Notch1 and the concentration of Alpha Fetal Protein (AFP); whereas there is a negative correlation between Notch1 expression and the degree of differentiation in HepG2 and QGY7701 hepatoma cells [56]. Jagged1 expression and genome amplification was also associated with AFP value in clinical samples [53].
Studies have shown that Notch1 gene expression is similar to the expression of Jagged1 in HCC, suggesting that the progression of HCC may be sustained by downregulation of Notch1/Jagged1 signaling [57]. Yes-associated protein (YAP) is a transcriptional regulator, and overexpression of YAP promoted HCC development and progression by depending on Jagged1 to trigger activation of Notch-signaling in vivo [58]. Hepatitis B virus X protein (HBx) can induce human hepatic L02 cell malignant transformation and activated Notch1 and Wnt/β-catenin signaling pathways. The L02 cell proliferation and activation of Wnt/β-catenin pathway were decreased by inhibition of the Notch1 signaling. The inhibition effect of knockdown Notch1 on proliferation and survival in L02/HBx cells was overcame by inhibition of the Wnt/β-catenin pathway. Therefore, Notch1 promotes HBx protein-induced hepatocarcinogenesis via Wnt/β-catenin pathway [59]. Furthermore, HBx has also been found to contribute to HCC development through upregulating Notch target genes HES1 and JAGGED1 [60, 61]. Notch2 is a key regulator of self-renewal and tumorigenicity in human HCC cells, with genetic depletion of Notch2 impairing the tumor formation in vivo [62]. In a study of human HCC samples, abnormally high levels of Notch3 and Hes1 were detected; higher expression levels of Notch3 in the tumor tissues may result in upregulation of HES1 gene expression [63]. The Notch target gene Sox9 is expressed in multi-potent cells in the liver. Overexpression of Sox9 was frequently detected in patients with HCCs, and the Notch signaling pathway may activate via extending a pre-existing progenitor- like cell or giving the progenitor-like capabilities to differentiated cells during hepatocarcinogenesis [11, 64]. Therefore, the aberrant expression of Notch signaling closely correlate with liver oncogenesis.
Notch signaling and HCC angiogenesis
HCC is one of the most hypervascular solid neoplasms, in which angiogenesis plays a critical role in neoplasm progression. Angiogenesis also contributes to the high recurrence and disappointing survival rates associated with HCC [65]. HCC's vascularity is unique and vastly different from the peripheral parenchyma of liver. Notch signaling acts as a key role in the process of vascular development, directly affecting vascular stability, differentiation of vascular smooth muscle cells, and arteriovenous formation [66]. While 4 Notch receptors are expressed in the adult liver, only 2 key ligands of Notch signaling have been implicated in HCC angiogenesis, namely, Dll4 and Jagged1 [67]. Notch1 is primarily responsible for angiogenesis and maturity of the vasculature, and Notch3 supports the maturation of vascular smooth muscle cells. However, the role of Notch4 in the formation of blood vessels has not been established. Dll4 is expressed in arterial endothelium during embryogenesis in mammals. High levels of Dll4 has also been observed in the endothelium of tumor blood vessels in comparison to neighboring normal tissue vessels [68]. Notch1 expression is very high in primary HCC cases and distant metastasis, and vasculogenic mimicry structures have only been observed in HCC patients with metastasis. This study suggested that Notch1 may act on endothelial progenitor cells to initiate cancer angiogenesis [69]. Hypersprouting and disruption of the vasculature has been associated with reduction in the expression of Dll4 [70]. Dll4 allele deletion inhibited hepatic vascular alterations [71]. Jagged1, which has been found to be implicated in early cardiovascular development via regulation of endothelial cells and vascular smooth muscle cells, can stimulate Notch-dependent angiogenesis in tumor cells [72, 73].
Notch1 can maintain hepatic sinusoid epithelial cell homeostasis, inhibit abnormal differentiation of endothelial cells, and prevent tumor carcinogenesis [74]. Dll4 expression is also regulated by vascular endothelial growth factor (VEGF), and its expression in cancers is associated with VEGF expression levels [75]. The level of angiogenesis regulators, VEGF-A and VEGF-R2 were significantly decreased in HCC tumor with inhibited Dll4 compared with the control group. Moreover, the inhibition of Dll4 expression in the tumor cells reduced the formation of new blood vessels in the tumor and cell proliferation [76]. Intratumoral microvessel density in HCC xenograft mouse tumors was profoundly reduced by Livistonae chinensis seeds, and this anti-angiogenic effect mediated by significantly decreased expression levels of Notch, Dll4, Jagged1, VEGF-A, and VEGF-R2 [77]. Additionally, the key mediators of Notch signaling, including Notch1, Dll4 and Jagged1, can serve as biomarkers of the inhibitory effect of Rubus alceifolius Poir on HCC angiogenesis in vivo [78].
Dll4 and Jagged1 seem to play opposing roles in angiogenesis. Dll4 is thought to act on endothelial cells and restrict new vessel sprout formation in tumors. Tumor growth was suppressed by the inhibition of Dll4 in tumor endothelial cells through inducing non-productive angiogenesis [79]. In contrast, Jagged1 expressed by tumor cells may engage Notch signaling in neighboring cells. Jagged1 can bind activated Notch1 to promote its expression by antagonizing Dll4 activity and enhancing VEGF activity. In turn, this effect stimulates tumor angiogenesis and promotes tumor growth, invasion, and metastasis [73]. Antagonism of Jagged1 regulates the activation of Notch signaling mediated by Dll4 in stalk-cell, to increase the number and activity of terminal cells and promote angiogenesis. Thus, further studies are required to validate the effects of Dll4 and Jagged1 on angiogenesis in HCC, and to assess their potential as therapeutic targets in HCC.
Notch signaling and HCC invasion and metastasis
Notch signaling is not only critical to physiologic angiogenesis, but also correlated with tumor angiogenesis and metastasis. Despite the development of modern medicine and technologies adopted in HCC treatment, overall survival remains low due to high rates of intrahepatic and extrahepatic metastasis [80]. Tumor invasion and metastasis consist of a series of discrete biological processes involving the complex interactions in tumor cells themselves, and between the host tissue and tumor cells. However, tumor cells must overcome a series of sequential rate-limiting steps in order to metastasize. The change in biochemical characteristics in tumor cells, including intravasation, arrest and extravasation, dormancy, angiogenesis, and metastatic colonization results in the transduction of abnormal signal, maintaining and promoting cancer invasion [81]. The expression of Notch1 and Jagged1 is increased in HCC, and HCC tumor node metastasis (TNM) staging is significantly correlated with increased Notch1 mRNA levels. In comparison to patients TNM Stage I-II disease and those without tumor venous invasion, HCC patients with TNM Stage III-IV and tumor venous invasion, had higher expression levels of Notch1. Knockout of Notch1 by shRNA abolished Snail1 expression and effectively inhibited cancer cell motility in vitro and pulmonary metastasis. Finally, this study confirmed that Notch signaling affected the invasion and metastasis of HCC by the Notch1-Snail1-E-cadherin association [13]. High Jagged1 copy number variation (CNV) was associated with advanced TMN classification and positive vascular invasion in liver cancer patients who had undergone surgical resection [53]. In another study, downregulation of Notch1 in MHCC97H and HepG2 cells inhibited the invasion of HCC by upregulating the expression of phosphatase and tensin homolog (PTEN) and phospho-PTEN and downregulating the expression of focal adhesion kinase (FAK) and phospho-FAK [82]. In H2O2 HCC cells and Stage II/III HCC tissue samples, upregulation of Notch1 induced a higher expression of Snail and enhanced the malignancy and invasiveness of HCC by cooperating with reactive oxygen species-induced activation of the phosphoinositide 3-kinase/ Akt pathway [83]. Downregulation of Notch signaling caused by N-[N-(3,5-difluorophenyl acetyl)-L-alanyl]-sphenylglycine butyl ester (DAPT), a synthetic γ-secretase inhibitor, can also inhibit HCC cell invasion through the extracellular signal-regulated kinases 1 and 2 signaling pathway, which contributes to the down-regulation of matrix metalloproteinase-2 (MMP2) and -9 (MMP9) and VEGF. MMP2/9 and VEGF are key factors in invasion and metastasis of tumor in blood vessel formation [84]. Additionally, suppression of Notch1 has been demonstrated to prevent HCC metastasis both in vitro and in vivo [85, 86]. Activated Notch2 has been detected in metastatic HCC, suggesting that Notch2 plays a functional role in HCC progression and metastasis [87]. Notch4 has also been found to enhance the aggressiveness and metastasis of HCC [6].
Furthermore, hypoxia, which is a common feature of HCC, can accelerate metastasis of tumor cells. Numerous factors are stimulated to participate in tumor invasion and metastasis in a hypoxic microenvironment. Hypoxia-inducible factor-1α and HBx were the factors that caused the elevated expression of Notch1-4 at both the protein and mRNA levels. Additionally, the overexpression of Notch3 was closely associated with the vascular invasiveness of HCC [88]. Studies have suggested that anti-angiogenic therapy that blocks primary tumor growth with inhibitors of VEGF and platelet derived growth factor, may elicit resistance in these tumor cells and may paradoxically promote increased invasion and metastasis in mouse models [89, 90]. By inhibiting neovascularization in tumors, it is likely that anti-angiogenic therapy may result in clonal selection of tumor cells through upregulation of genes involved in survival and invasion. The possibility that Notch proteins may be functionally relevant in this setting is increased by the interaction between Notch signaling and hypoxic regulation of gene expression. However, more work is needed to further explore this relationship.
Notch signaling and HCC prognosis
HCC is a common aggressive neoplasm, with increasing mortality and poor prognosis. Notch signaling activated in human HCC samples accelerates liver cancer formation in mice, suggesting that a Notch signaling signature may be a biomarker of response to Notch inhibition in vitro [11]. Higher expression levels of Notch3 has been shown to be consistent with shorter recurrence time and poorer overall survival in HBV-related HCC postsurgical patients [91]. The roles of Notch1 and Notch3 in malignant transformation have been explored. Patients whose tumors had higher levels of Notch1 and Notch3 experienced poorer overall survival outcomes. Additionally, studies have suggested that Notch1 may be more crucial to HCC progression and metastasis than Notch3, due to Notch1 interaction with signaling pathways and correlation to HCC metastasis [85]. Therefore, Notch1 may be a more relevant therapeutic target for HCC than Notch3. The activation of Notch2 can be enhanced by the cooperation of long noncoding RNA (lncAKHE) and YEATS4. In addition, higher expression of either Notch2 or lncAKHE in HCC patients has been associated with greater clinical severity and poorer survival rates [92, 93]. Overexpression of Notch1, Notch3, and Notch4 proteins have been observed in a total of 288 primary HCC patients who had undergone curative resection. Both Notch1 and Notch4 overexpression detected by immunohistochemistry showed an unfavorable correlation to disease-specific survival. Therefore, Notch1 and Notch4 have been identified as biomarkers of poor prognosis for HCC patients [6]. Higher AFP levels was frequently detected in advanced and end-stage liver cancer clinical cases, and was connected with high Jagged1 CNV. Shorter overall survival was shown in higher Jagged1 CNV cases. However, even if AFP is not elevated and Jagged1 CNV is high, the clinical course is worse, earlier recurrences of liver cancer occur [53]. HCC patients with overexpression of HES1 experienced longer median overall survival time in comparison to patients with low levels of HES1. However, the time to recurrence showed an inverse relationship with the expression of HES1 [14]. The marker of biliary epithelial cells Sox9 is regulated by Notch signaling. Overexpression of Sox9 was correlated with lower 5-year disease-free survival and lower 5-year overall survival. Therefore, high levels of Sox9 was an independent factor for poor prognosis of HCC patients [94]. According to extensive studies, the prognosis of HCC patients may be predicted by Notch signaling factors, such as Notch1, Notch3, Notch4, Jagged1, Hes-1, and Sox9. Increasing numbers of studies have shown that Notch signaling is associated with a disappointing prognosis in HCC. Therefore, Notch signaling may be a novel prognostic indicator for HCC patients.
Notch signaling as a therapeutic target for HCC
Currently, chemotherapeutic agents are used for treating HCC, but are only modestly effective because of significant non-selective cytotoxic side effects. Sorafenib is the only approved molecular-targeted agent for HCC, and this drug is widely believed to be a small multi-targeted tyrosine kinase inhibitor. However, the outcome of sorafenib-treated HCC patients remains disappointing. This highlights the need for novel therapies to be developed for HCC therapy. As previously mentioned, aberrant upregulation of the Notch signaling pathway is observed in HCC tissues. Consequently, downregulation of Notch signaling can inhibit HCC cell growth in vitro and tumor growth in vivo by Notch specific γ- secretase inhibitors [11, 95]. These studies indicate that targeting Notch signaling may be a potential novel option for HCC treatment, including the use of a single agent or a combination of anticancer agents.
Therapy with a single agent
The activation of Notch signaling relies on γ-secretase activity. Thus, γ-secretase has become a promising target for Notch inhibition. γ -secretase inhibitors have been studied in various cancers, including HCC. Suppression of activated Notch signaling by γ -secretase inhibitors decreased cell growth and blocked tumor progression in SMMC7721 cells in vivo [95]. RBP-J-interacting and tubulin-associated (RITA) protein can downregulate Notch-mediated transcription and may also be a potent antitumor agent for HCC therapy. It has been shown that RITA may inhibit tumor growth and induce apoptosis in HCC cells in vitro [96]. In addition, Numb segregates asymmetrically and acts as a cell fate determinant, and can block the progress of malignant liver cancer by inhibiting the activation of Notch signaling [97]. Therefore, it is possible that it may be a novel therapeutic target for HCC by promoting Numb-mediated endocytosis of Notch receptors. As have been reported, Notch receptors can be cleaved by ADMA17. ZLDI-8 (previously named as inhibitor of ADAM-17 compound No. 8 or IAC-8) was found to enhance the anti-tumor effect of traditional chemotherapeutic agents and sorafenib in HCC tumors in vivo or in vitro. Treatment of HCC cells with ZLDI-8 inhibited the activation of ADAM-17 and decreased the expression of 2 downstream proteins in Notch signaling pathway, namely, Survivin and cIAP1/2. Therefore, ZLDI-8 may be a promising therapeutic agent for HCC patients, particularly for patients with multi-drug resistance [98]. Lidamycin (LDM), an anticancer antibiotic, has demonstrated distinct antitumor effects in liver cancer. LDM reduced the mRNA levels of Hes1, Hey1 and Notch1 genes. Additionally, the protein levels of Notch1, Notch intracellular domain, Hey1, and Hes1 were also decreased in LDM-treated in Huh7 cells. However, further clinical evaluation of LDM is required [99].
Treatment with a combination of anticancer agents
The combination of different agents or treatment approaches is attractive for improving clinical efficacy. Co-delivery of platinum drugs and siRNA with Notch1-targeting micelleplex enhanced tumor control in the SMMC7721 xenograft model. This phenotype was due to the increased sensitivity of HCC cells to platinum drugs induced by siRNA-mediated inhibition of Notch1 [100]. The anticonvulsant drug valproic acid, acting as a histone deacetylase inhibitor or a Notch signaling inhibitor, induced downregulation of Notch signaling by inhibiting Notch1 expression and its target gene HES1 in HCC. Additionally, the combination of valproic acid and the peptide-drug conjugate camptothecin-somatostatin resulted in more potent anti-proliferative effects on HCC cells in vitro than either single agent [101, 102]. Doxorubicin has reportedly been used to treat HCC with reduced Notch signaling. In vitro, the depletion of Notch3 by Notch3 shRNA can increase the expression of p53 and p27, decrease the expression of p21, enhance the uptake of doxorubicin, and improve cell apoptosis induced by doxorubicin. Therefore, the combination of Notch3 silencing and doxorubicin treatment can produce an enhanced antitumor effect in HCC cells by a p53-dependent mechanism [60]. In addition, silybin, an antioxidant derived from the milk thistle plant, has been reported to exert antitumor effects. Notch1 siRNA (in vitro) or Notch1 inhibitor DAPT has been confirmed to enhance the antitumor effect of silybin in HCC HepG2 cells [60]. Despite these promising preliminary studies demonstrating the antitumor activity of Notch-targeting combination strategies, further studies are needed to enhance the development of rational combination strategies to target HCC.
Conclusion and future prospective
Based on the studies discussed in this review, Notch signaling has a pro-tumorigenic role in HCC progression (Table 1). However, the role of Notch signaling in HCC is still controversial. Some reports suggest the opposite viewpoint that Notch signaling serves as a tumor suppressor in liver neoplasms [7, 8]. The conflict may be due to the highly context-dependent nature of the Notch pathway [103]. In addition, HCCs exhibit various subtypes, including bi-phenotypic HCCs with combined hepatocellular and cholangiocarcinoma tumors, clear cell HCC, cirrhotomimetic HCC, fibrolamellar carcinoma, lymphocyte-rich HCC, scirrhous HCC, myxoid HCC, steatohepatitic HCC, sarcomatoid HCC, and granulocyte colony-stimulating factor HCC with major neutrophilic infiltrates [104]. The relationship between Notch signaling abnormalities and HCC subtypes has not been well established. Therefore, more studies are needed to further investigate the precise role of Notch signaling in HCC subtypes.
Abnormal genes of Notch signaling in cancer progression
| Cancer progression | Abnormal genes | References |
|---|---|---|
| Tumorigenesis | Notch1, Notch2, Notch3, Notch4, Jagged1, Hes1, Sox9 | [11,53-64] |
| Angiogenesis | Dll4 and Jagged1 | [67,71, 76-78] |
| Invasion and metastasis | Notch1, Notch2, Notch3, Notch4, Jagged1 | [6, 13,53,82-88] |
| Prognosis | Notch1, Notch2, Notch3, Notch4, Jagged1, Hes1, Sox9 | [6,11,14,53,85,91-94] |
In summary, HCC is an aggressive tumor with poor prognosis and high mortality. Currently, the knowledge base demonstrates the activity and function of Notch receptors and ligands in the development and progression of HCC. Notch signaling has been demonstrated to regulate the HCC tumor microenvironment, tumorigenesis, progresssion, angiogenesis, invasion and metastasis, and prognosis. Notch signaling represents a potential therapeutic target in HCC. Points of therapeutic intervention may include 1) the expression of Notch ligands or receptors, 2) ligand-receptor binding, 3) γ-secretase-mediated cleavage of Notch, and 4) regulation of Notch activation. Importantly, the effect of cross-talk between Notch signaling and other signaling pathways (e.g., sonic hedgehog, TGF-β, PI3K/AKT, Wnt, mTOR, and Ras/Raf/MEK/ ERK) related to HCC cannot be ignored. Therefore, Notch signaling is a potential diagnostic and prognostic biomarker, and a promising target for HCC therapy. Additional studies are urgently needed to further investigate Notch signaling in HCC in order to foster the development of effective therapeutic approaches for HCC treatment.
Abbreviations
HCC: hepatocellular carcinoma; ICN: intracellular domain of Notch; ECN: extracellular domain of Notch; RAM: RBP-Jkappa-associated module; EGF: epidermal growth factor; DSL: Delta/Serrate/Lag-2; DOS: Delta and OSM-11-like; DLK1: Delta-like 1; DLK2: Delta-like 2; CSL: CBF1/RBPJ-kappa/Su (H)/Lag1; HES1: Hairy and Enhancer of Split 1; HSCs: hepatic stellate cells; CSCs: cancer stem cells; TAMs: tumor associated macrophages; CAFs: cancer associated fibroblasts; YAP: Yes-associated protein; HBx: Hepatitis B virus X protein; VEGF: vascular endothelial growth factor; TNM: tumor node metastasis; CNV: copy number variation; PTEN: phosphatase and tensin homolog; FAK: focal adhesion kinase; DAPT: N-[N-(3,5-difluorophenyl acetyl)-L-alanyl]-Sphenylglycine butyl ester; MMP2: matrix metalloproteinase-2; MMP9: matrix metalloproteinase-9; lncAKHE: long noncoding RNA; RITA: RBP-J-interacting and tubulin-associated; LDM: Lidamycin.
Acknowledgements
This paper was supported by the National Natural Science Foundation of China (Grant No.81760851), Foundation of Guangxi University of Chinese Medicine (Grant No. P2012116), and Guangxi Science Research and Technology Development Project (Grant No. guikegong: 1598013-5).
Competing Interests
The authors have declared that no competing interest exists.
References
1. El-Serag HB, Kanwal F. Epidemiology of hepatocellular carcinoma in the United States: where are we? Where do we go? Hepatology. 2014;60:1767-75
2. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225-49
3. Mittal S, Elserag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47(Suppl):S2-6
4. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
5. Dhanasekaran R, Limaye A, Cabrera R. Hepatocellular carcinoma: current trends in worldwide epidemiology, risk factors, diagnosis, and therapeutics. Hepat Med. 2012;4:19-37
6. Ahn S, Hyeon J, Park CK. Notch1 and Notch4 are markers for poor prognosis of hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2013;12:286-94
7. Qi R, An H, Yu Y, Zhang M, Liu S, Xu H. et al. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003;63:8323-9
8. Wang C, Qi R, Li N, Wang Z, An H, Zhang Q. et al. Notch1 signaling sensitizes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human hepatocellular carcinoma cells by inhibiting Akt/Hdm2-mediated p53 degradation and up-regulating p53-dependent DR5 expression. J Biol Chem. 2009;284:16183-90
9. Viatour P, Ehmer U, Saddic LA, Dorrell C, Andersen JB, Lin C. et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J Exp Med. 2011;208:1963-76
10. Gramantieri L, Giovannini C, Lanzi A, Chieco P, Ravaioli M, Venturi A. et al. Aberrant Notch3 and Notch4 expression in human hepatocellular carcinoma. Liver Int. 2007;27:997-1007
11. Villanueva A, Alsinet C, Yanger K, Hoshida Y, Zong Y, Toffanin S. et al. Notch signaling is activated in human hepatocellular carcinoma and induces tumor formation in mice. Gastroenterology. 2012;143:1660-9
12. Ning L, Wentworth L, Chen H, Weber SM. Down-regulation of Notch1 signaling inhibits tumor growth in human hepatocellular carcinoma. Am J Transl Res. 2009;1:358-66
13. Wang XQ, Zhang W, Lui EL, Zhu Y, Lu P, Yu X. et al. Notch1-Snail1-E-cadherin pathway in metastatic hepatocellular carcinoma. Int J Cancer. 2012;131:E163-72
14. Zou JH, Xue TC, Sun C, Li Y, Liu BB, Sun RX. et al. Prognostic significance of hes-1, a downstream target of notch signaling in hepatocellular carcinoma. Asian Pac J Cancer Prev. 2015;16:3811-6
15. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216-33
16. D'Souza B, Melotykapella L, Weinmaster G. Canonical and non-canonical Notch ligands. Curr Top Dev Biol. 2010;92:73-129
17. Falix FA, Aronson DC, Lamers WH, Gaemers IC. Possible roles of DLK1 in the Notch pathway during development and disease. Biochim Biophys Acta. 2012;1822:988-95
18. Sánchezsolana B, Nueda ML, Ruvira MD, Ruizhidalgo MJ, Monsalve EM, Rivero S. et al. The EGF-like proteins DLK1 and DLK2 function as inhibitory non-canonical ligands of NOTCH1 receptor that modulate each other's activities. Biochim Biophys Acta. 2011;1813:1153-64
19. Gridley T. Notch signaling and inherited disease syndromes. Hum Mol Genet. 2003:12 Spec No 1: R9-13
20. Fortini ME. Notch Signaling: The Core Pathway and Its Posttranslational Regulation. Dev Cell. 2009;16:633-47
21. Nowell C, Radtke F. Cutaneous Notch signaling in health and disease. Cold Spring Harb Perspect Med. 2013;3:a017772
22. Licciulli S, Avila JL, Hanlon L, Troutman S, Cesaroni M, Kota S. et al. Notch1 is required for Kras-induced lung adenocarcinoma and controls tumor cell survival via p53. Cancer Res. 2013;73:5974-84
23. Sonoshita M, Itatani Y, Kakizaki F, Sakimura K, Terashima T, Katsuyama Y. et al. Promotion of colorectal cancer invasion and metastasis through activation of NOTCH-DAB1-ABL-RHOGEF protein TRIO. Cancer Discov. 2015;5:198-211
24. Yang JD, Nakamura I, Roberts LR. The tumor microenvironment in hepatocellular carcinoma: current status and therapeutic targets. Semin Cancer Biol. 2011;21:35-43
25. Gotzmann J, Mikula M, Eger A, Schulte-Hermann R, Foisner R, Beug H. et al. Molecular aspects of epithelial cell plasticity: implications for local tumor invasion and metastasis. Mutat Res. 2004;566:9-20
26. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH. et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. 2013;4:2823
27. Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605-12
28. Fung E, Tang SM, Canner JP, Morishige K, Arboleda-Velasquez JF, Cardoso AA. et al. Delta-like 4 induces notch signaling in macrophages: implications for inflammation. Circulation. 2007;115:2948-56
29. Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ. et al. High Expression of Macrophage Colony-Stimulating Factor in Peritumoral Liver Tissue Is Associated With Poor Survival After Curative Resection of Hepatocellular Carcinoma. J Clin Oncol. 2008;26:2707-16
30. Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010;17:135-47
31. Mazzocca A, Fransvea E, Dituri F, Lupo L, Antonaci S, Giannelli G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology. 2010;51:523-34
32. Xiong S, Wang R, Chen Q, Luo J, Wang J, Zhao Z. et al. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am J Cancer Res. 2018;8:302-16
33. Bansal R, Baarlen JV, Storm G, Prakash J. The interplay of the Notch signaling in hepatic stellate cells and macrophages determines the fate of liver fibrogenesis. Sci Rep. 2015;5:18272
34. Lau EY, Lo J, Cheng BY, Ma MK, Lee JM, Ng JK. et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016;15:1175-89
35. Malik R, Selden C, Hodgson H. The role of non-parenchymal cells in liver growth. Semin Cell Dev Biol. 2002;13:425-31
36. Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199-210
37. Chen YX, Weng ZH, Zhang SL. Notch3 regulates the activation of hepatic stellate cells. World J Gastroenterol. 2012;18:1397-403
38. Hellerbrand C. Hepatic stellate cells—the pericytes in the liver. Pflugers Arch. 2013;465:775-8
39. Xia Y, Chen R, Song Z, Ye S, Sun R, Xue Q. et al. Gene expression profiles during activation of cultured rat hepatic stellate cells by tumoral hepatocytes and fetal bovine serum. J Cancer Res Clin Oncol. 2010;136:309-21
40. Zhang K, Zhang YQ, Ai WB, Hu QT, Zhang QJ, Wan LY. et al. Hes1,an important gene for activation of hepatic stellate cells,is regulated by Notch1 and TGF-β/BMP signaling. World J Gastroenterol. 2015;21:878-87
41. Ji J, Eggert T, Budhu A, Forgues M, Takai A, Dang H. et al. Hepatic stellate cell and monocyte interaction contributes to poor prognosis in hepatocellular carcinoma. Hepatology. 2015;62:481-95
42. Borovski T, De SEMF, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res. 2011;71:634-9
43. Yamashita T, Wang XW. Cancer stem cells in the development of liver cancer. J Clin Invest. 2013;123:1911-8
44. Clement V, Sanchez P, De TN, Radovanovic I, Ruiz iAA. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165-72
45. Mishra L, Banker T, Murray J, Byers S, Thenappan A, He AR. et al. Liver stem cells and hepatocellular carcinoma. Hepatology. 2009;49:318-29
46. Jing L, Peng W, Wang R, Wang J, Man L, Si X. et al. The Notch pathway promotes the cancer stem cell characteristics of CD90+cells in hepatocellular carcinoma. Oncotarget. 2016;7:9525-37
47. Chen Y, Wong PP, Sjeklocha L, Steer CJ, Sahin MB. Mature Hepatocytes Exhibit Unexpected Plasticity by Direct Dedifferentiation into Liver Progenitor Cells in Culture. Hepatology. 2012;55:563-74
48. Ortica S, Tarantino N, Aulner N, Israël A, Gupta-Rossi N. The 4 Notch receptors play distinct and antagonistic roles in the proliferation and hepatocytic differentiation of liver progenitors. FASEB J. 2014;28:603-14
49. Mccright B, Lozier J, Gridley T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development. 2002;129:1075-82
50. Tanimizu N, Miyajima A. Notch signaling controls hepatoblast differentiation by altering the expression of liver-enriched transcription factors. J Cell Sci. 2004;117:3165-74
51. Nijjar SS, Crosby HA, Wallace L, Hubscher SG, Strain AJ. Notch receptor expression in adult human liver: a possible role in bile duct formation and hepatic neovascularization. Hepatology. 2001;34:1184-92
52. Xie G, Karaca G, Swiderskasyn M, Michelotti GA, Krüger L, Chen Y. et al. Cross-talk between Notch and Hedgehog regulates hepatic stellate cell fate in mice. Hepatology. 2013;58:1801-13
53. Kawaguchi K, Honda M, Yamashita T, Okada H, Shirasaki T, Nishikawa M. et al. Jagged1 DNA Copy Number Variation Is Associated with Poor Outcome in Liver Cancer. Am J Pathol. 2016;186:2055-67
54. Gao J, Song Z, Chen Y, Xia L, Wang J, Fan R. et al. Deregulated expression of Notch receptors in human hepatocellular carcinoma. Dig Liver Dis. 2008;40:114-21
55. Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP. et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343-52
56. Lu CJ, He YF, Yuan WZ, Xiang LJ, Zhang J, Liang YR. et al. Dihydromyricetin-mediated inhibition of the Notch1 pathway induces apoptosis in QGY7701 and HepG2 hepatoma cells. World J Gastroenterol. 2017;23:6242-51
57. Wang M, Xue L, Cao Q, Lin Y, Ding Y, Yang P. et al. Expression of Notch1, Jagged1 and beta-catenin and their clinicopathological significance in hepatocellular carcinoma. Neoplasma. 2009;56:533-41
58. Tschaharganeh DF, Chen X, Latzko P, Malz M, Gaida MM, Felix K. et al. Yes-associated protein up-regulates Jagged-1 and activates the Notch pathway in human hepatocellular carcinoma. Gastroenterology. 2013;144:1530-42
59. Sun Q, Wang R, Luo J, Wang P, Xiong S, Liu M. et al. Notch1 promotes hepatitis B virus X protein-induced hepatocarcinogenesis via Wnt/β-catenin pathway. Int J Oncol. 2014;45:1638-48
60. Giovannini C, Gramantieri L, Chieco P, Minguzzi M, Lago F, Pianetti S. et al. Selective ablation of Notch3 in HCC enhances doxorubicin's death promoting effect by a p53 dependent mechanism. J Hepatol. 2009;50:969-79
61. Wang F, Zhou H, Yang Y, Xia X, Sun Q, Luo J. et al. Hepatitis B virus X protein promotes the growth of hepatocellular carcinoma by modulation of the Notch signaling pathway. Oncol Rep. 2012;27:1170-6
62. Wu WR, Zhang R, Shi XD, Yi C, Xu LB, Liu C. Notch2 is a crucial regulator of self-renewal and tumorigenicity in human hepatocellular carcinoma cells. Oncol Rep. 2016;36:181-8
63. Zhang Q, Lu C, Fang T, Wang Y, Hu W, Qiao J. et al. Notch3 functions as a regulator of cell self-renewal by interacting with the β-catenin pathway in hepatocellular carcinoma. Oncotarget. 2015;6:3669-79
64. Furuyama K, Kawaguchi Y, Akiyama H, Horiguchi M, Kodama S, Kuhara T. et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet. 2011;43:34-41
65. Muto J, Shirabe K, Sugimachi K, Maehara Y. Review of angiogenesis in hepatocellular carcinoma. Hepatol Res. 2015;45:1-9
66. Dou GR, Wang L, Wang YS, Han H. Notch Signaling in Ocular Vasculature Development and Diseases. Mol Med. 2012;18:47-55
67. Nijjar SS, Wallace L, Crosby HA, Hubscher SG, Strain AJ. Altered Notch ligand expression in human liver disease: further evidence for a role of the Notch signaling pathway in hepatic neovascularization and biliary ductular defects. Am J Pathol. 2002;160:1695-703
68. Mailhos C, Modlich U, Lewis J, Harris A, Bicknell R, Ish-Horowicz D. Delta4, an endothelial specific Notch ligand expressed at sites of physiological and tumor angiogenesis. Differentiation. 2001;69:135-44
69. Zhu MS, Xu LB, Zeng H, Shi XD, Wu WR, Liu C. Association of Notch1 with vasculogenic mimicry in human hepatocellular carcinoma cell lines. Int J Clin Exp Pathol. 2014;7:5782-91
70. Gale NW, Dominguez MG, Noguera I, Pan L, Hughes V, Valenzuela DM. et al. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci U S A. 2004;101:15949-54
71. Djokovic D, Trindade A, Gigante J, Badenes M, Silva L, Liu R. et al. Combination of Dll4/Notch and Ephrin-B2/EphB4 targeted therapy is highly effective in disrupting tumor angiogenesis. BMC Cancer. 2010;10:641
72. Skrtic A, Sokolic L, Borovecki A, Rosa J, Fenzl V. Immunohistochemical localization of CD31, NOTCH1 and JAGGED1 proteins in experimentally induced polycystic ovaries of immature rats. Acta Histochem. 2011;113:262-9
73. Benedito R, Roca C, Sörensen I, Adams S, Gossler A, Fruttiger M. et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137:1124-35
74. Dill MT, Rothweiler S, Djonov V, Hlushchuk R, Tornillo L, Terracciano L. et al. Disruption of Notch1 Induces Vascular Remodeling, Intussusceptive Angiogenesis, and Angiosarcomas in Livers of Mice. Gastroenterology. 2012;142:967-77
75. Patel NS, Li JL, Generali D, Poulsom R, Cranston DW, Harris AL. Up-regulation of delta-like 4 ligand in human tumor vasculature and the role of basal expression in endothelial cell function. Cancer Res. 2005;65:8690-7
76. Kunanopparat A, Issara-Amphorn J, Leelahavanichkul A, Sanpavat A, Patumraj S, Tangkijvanich P. et al. Delta-like ligand 4 in hepatocellular carcinoma intrinsically promotes tumour growth and suppresses hepatitis B virus replication. World J Gastroenterol. 2018;24:3861-70
77. Lin W, Zhao J, Cao Z, Zhuang Q, Zheng L, Zeng J. et al. Livistona chinensis seeds inhibit hepatocellular carcinoma angiogenesis in vivo via suppression of the Notch pathway. Oncol Rep. 2014;31:1723-8
78. Zhao J, Lin W, Cao Z, Zhuang Q, Zheng L, Peng J. et al. Total alkaloids of Rubus alceifolius Poir inhibit tumor angiogenesis through suppression of the Notch signaling pathway in a mouse model of hepatocellular carcinoma. Mol Med Rep. 2015;11:357-61
79. Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y. et al. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature. 2006;444:1083-7
80. Aravalli R N, Steer C J, Cressman E N. Molecular Mechanisms of Hepatocellular Carcinoma. Hepatology. 2008;48:2047-63
81. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895-904
82. Hu YJ, Li HY, Qiu KJ, Li DC, Zhou JH, Hu YH. et al. Downregulation of Notch1 inhibits the invasion of human hepatocellular carcinoma HepG2 and MHCC97H cells through the regulation of PTEN and FAK. Int J Mol Med. 2014;34:1081-6
83. Kim HS, Jung G. Notch1 increases Snail expression under high reactive oxygen species conditions in hepatocellular carcinoma cells. Free Radic Res. 2014;48:806-13
84. Zhou L, Wang DS, Li QJ, Sun W, Zhang Y, Dou KF. Downregulation of the Notch signaling pathway inhibits hepatocellular carcinoma cell invasion by inactivation of matrix metalloproteinase-2 and -9 and vascular endothelial growth factor. Oncol Rep. 2012;28:874-82
85. Zhou L, Zhang N, Song W, You N, Li Q, Sun W. et al. The significance of Notch1 compared with Notch3 in high metastasis and poor overall survival in hepatocellular carcinoma. Plos One. 2013;8:e57382
86. Zhou L, Wang DS, Li QJ, Sun W, Zhang Y, Dou KF. The down-regulation of Notch1 inhibits the invasion and migration of hepatocellular carcinoma cells by inactivating the cyclooxygenase-2/Snail/E-cadherin pathway in vitro. Dig Dis Sci. 2013;58:1016-25
87. Hayashi Y, Osanai M, Lee GH. NOTCH2 signaling confers immature morphology and aggressiveness in human hepatocellular carcinoma cells. Oncol Rep. 2015;34:1650-8
88. Yang SL, Ren QG, Zhang T, Pan X, Wen L, Hu JL. et al. Hepatitis B virus X protein and hypoxia-inducible factor-1α stimulate Notch gene expression in liver cancer cells. Oncol Rep. 2016;37:348-56
89. Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F. et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220-31
90. Ebos JM, Lee CR, Cruzmunoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232-9
91. Yu T, Han C, Zhu G, Liao X, Qin W, Yang C. et al. Prognostic value of Notch receptors in postsurgical patients with hepatitis B virus-related hepatocellular carcinoma. Cancer Med. 2017;6:1587-600
92. Huang G, Jiang H, Lin Y, Wu Y, Cai W, Shi B. et al. lncAKHE enhances cell growth and migration in hepatocellular carcinoma via activation of NOTCH2 signaling. Cell Death Dis. 2018;9:487
93. Zhu P, Wang Y, Du Y, He L, Huang G, Zhang G. et al. C8orf4 negatively regulates self-renewal of liver cancer stem cells via suppression of NOTCH2 signalling. Nat Commun. 2015;6:7122
94. Guo X, Lu X, Sun T, Peng R, Lin Z, Zhu H. et al. Expression features of SOX9 associate with tumor progression and poor prognosis of hepatocellular carcinoma. Diagn Pathol. 2012;7:44
95. Gao J, Dong Y, Zhang B, Xiong Y, Xu W, Cheng Y. et al. Notch1 activation contributes to tumor cell growth and proliferation in human hepatocellular carcinoma HepG2 and SMMC7721 cells. Int J Oncol. 2012;41:1773-81
96. Wang H, Chen G, Wang H, Liu C. RITA inhibits growth of human hepatocellular carcinoma through induction of apoptosis. Oncol Res. 2013;20:437-45
97. Giebel B, Wodarz, Andreas. Notch signaling: numb makes the difference. Curr Biol. 2012;22:R133-5
98. Zhang Y, Li D, Jiang Q, Cao S, Sun H, Chai Y. et al. Novel ADAM-17 inhibitor ZLDI-8 enhances the in vitro and in vivo chemotherapeutic effects of Sorafenib on hepatocellular carcinoma cells. Cell Death Dis. 2018;9:743
99. Chen Y, Sun W, He R, Zhang F, Wang H, Li P. et al. Lidamycin decreases CD133 expression in hepatocellular carcinoma via the Notch signaling pathway. Oncol Lett. 2017;14:7889-95
100. Shen S, Sun CY, Du XJ, Li HJ, Liu Y, Xia JX. et al. Co-delivery of platinum drug and siNotch1 with micelleplex for enhanced hepatocellular carcinoma therapy. Biomaterials. 2015;70:71-83
101. Machado MC, Bellodi-Privato M, Kubrusly MS, Molan NA Jr TT, de Oliveira ER. et al. Valproic acid inhibits human hepatocellular cancer cells growth in vitro and in vivo. J Exp Ther Oncol. 2011;9:85-92
102. Sun G, Mackey LV, Coy DH, Yu CY, Sun L. The Histone Deacetylase Inhibitor Vaproic Acid Induces Cell Growth Arrest in Hepatocellular Carcinoma Cells via Suppressing Notch Signaling. J Cancer. 2015;6:996-1004
103. Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer. 2011;11:338-51
104. Torbenson MS. Morphologic Subtypes of Hepatocellular Carcinoma. Gastroenterol Clin North Am. 2017;46:365-91
Author contact
![]() Corresponding author: Dr. Ailing Wei, Department of liver disease, the First Affiliated Hospital, Guangxi University of Chinese Medicine, 89-9 Dongge Road, Nanning 530023, Guangxi, China. Tel and Fax: 86-0771-3126658; E-mail: 100365edu.cn.
Corresponding author: Dr. Ailing Wei, Department of liver disease, the First Affiliated Hospital, Guangxi University of Chinese Medicine, 89-9 Dongge Road, Nanning 530023, Guangxi, China. Tel and Fax: 86-0771-3126658; E-mail: 100365edu.cn.