Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(8):1846-1854. doi:10.7150/jca.28809 This issue Cite
Research Paper
CDCA5, Transcribed by E2F1, Promotes Oncogenesis by Enhancing Cell Proliferation and Inhibiting Apoptosis via the AKT Pathway in Hepatocellular Carcinoma
Hao Chen*, Jun Chen*, Long Zhao, Wenfeng Song, Zefeng Xuan, Jian Chen, Zequn Li, Guangyuan Song, Liangjie Hong, Penghong Song ![]() , Shusen Zheng
, Shusen Zheng ![]()
Division of Hepatobiliary and Pancreatic Surgery, Department of Surgery, First Affiliated Hospital, School of Medicine, Zhejiang University; NHCPRC Key Laboratory of Combined Multi-organ Transplantation; Key Laboratory of the diagnosis and treatment of organ Transplantation, CAMS; Key Laboratory of Organ Transplantation, Zhejiang Province, Hangzhou 310003, China; Collaborative innovation center for Diagnosis treatment of infectious diseases
*These authors contributed equally to this work.
Received 2018-7-27; Accepted 2019-1-28; Published 2019-4-21
Abstract
Cell division cycle associated 5 (CDCA5) is an important element for the interaction between cohesin and chromatin in interphase. It is abnormally expressed in many types of cancer and works as an indicator of poor prognosis, but little is known about its activity in hepatocellular carcinoma (HCC). In the present study, we found that the expression of CDCA5 was upregulated in HCC tissues compared to paracancerous tissues and had a negative correlation with patient survival. Cell proliferation and tumorigenesis were inhibited and cell apoptosis was induced with the knockdown of CDCA5, suggesting an oncogenic role of CDCA5 in liver cancer. Luciferase reporter assay and chromatin immunoprecipitation showed that CDCA5 was transcribed by E2F1. Furthermore, we confirmed that CDCA5 interrupted cell behavior via the AKT pathway. These findings demonstrated that CDCA5 plays an important role in HCC progression.
Keywords: hepatocellular carcinoma, CDCA5, proliferation, apoptosis, E2F1, AKT
Introduction
HCC is one of the most lethal cancers worldwide [1]. Despite improvements in therapy over the past decades, morbidity and mortality remain high [2]. It is urgent to explore the molecular mechanisms and identify novel gene targets of HCC.
Cell division cycle associated 5 (CDCA5, also known as sororin) is located at 11q13.1. It is expressed in the nucleus in interphase cells, disperses from the chromatin in mitosis, and interacts with the cohesin complex [3]. In Xenopus embryos, CDCA5 overexpression leads to disorder in segregating sister chromatids in mitosis and increases the combination between cohesin and chromosomes. In cultured cells, CDCA5 knockdown leads to mitotic arrest and sister chromatid cohesion failure [4]. Thus, CDCA5 plays an essential role as a cell cycle-dependent mediator of sister chromatid cohesion. As reported, CDCA5 is overexpressed in gastric cancer[5], urothelial carcinoma [6], malignant pleural mesothelioma [7], oral squamous cell carcinoma [8], and acral melanoma [9]. Meanwhile, CDCA5 may be a poor prognostic factor in patients with these cancers. The expression and role of CDCA5 in HCC still need to be further explored.
The E2F transcription factors are conserved among species and control a series of cell behaviors [10-12]. As a key member of the E2F family, E2F1 is overexpressed in many cancers including HCC. As reported, E2F1 overexpression triggers apoptosis subsequent to S phase entry of quiescent cells [13]. The effects of E2F1 on apoptosis can be regulated by p53. Meanwhile, E2F1 sustains abnormal c-Myc-driven cell growth via suppression of c-Myc-induced apoptosis [14]. However, the interaction between E2F1 and CDCA5 is poorly investigated.
At present, the expression of CDCA5 was upregulated in HCC samples and patients with higher CDCA5 expression suffered poorer survival. Silencing of CDCA5 suppressed cancer cell proliferation and tumorigenesis in vivo and in vitro, indicating the oncogenic role of CDCA5 in HCC progression. Luciferase reporter assay and chromatin immunoprecipitation (ChIP) identified that CDCA5 was transcribed by E2F1. Furthermore, CDCA5 interrupted cell behavior via AKT phosphorylation at Ser473. In summary, our work revealed the functional role of CDCA5 in HCC progression and its underlying mechanism. All of these results may lead to a better understanding of HCC and provide a novel target for HCC treatment.
Materials and Methods
Tissue samples
Tumor tissues and matched normal tissues of 201 HCC cases were collected at The First Affiliated Hospital, School of Medical, Zhejiang University, from 2008 to 2015. The patients were followed up and their clinicopathological features were collected. All samples were stored at -80°C for further research.
RNA isolation and reverse-transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted by TRIzol Reagent (Invitrogen, USA) and reverse-transcribed using PrimeScript RT Reagent Kit with gDNA Eraser (Takara, Japan) under conditions provided by the supplier. PCR was performed with FastStart Universal SYBR Green Master (ROX) (Roche, German) using the ABI 7900 System (Applied Biosystems, USA) according to the manufacturer's instructions. GAPDH was used as an internal control. Primers used for RT-qPCR are listed below: CDCA5 forward: CCACCACCCGAGAAACAGAA, reverse: GGTGGCTATGTACAGGACAGG; GAPDH forward: AATGGGCAGCCGTTAGGAAA, reverse: GCGCCCAATACGACCAAATC.
Immunohistochemistry (IHC)
Paired paraffin-embedded HCC tissues were cut into 4 μm, deparaffinized, and rehydrated. Antigen retrieval was performed using 10 mmol/L citric acid buffer (pH 6.0) at 100°C for 15 minutes. After incubation with anti-CDCA5 antibody (1:200) at 4°C overnight, the slides were rinsed in PBS and incubated with secondary antibody for 30 minutes. The slides were rinsed in PBS, incubated with DAB for 2 minutes, then rinsed, and stained with hematoxylin.
Western blot
Tissues were homogenized and cells were lysed in RIPA buffer and quantified by the bicinchoninic acid assay kit (Pierce, USA). Total protein was separated by sodium dodecyl-sulfate polyacrylamide gel electrophoresis under denaturing conditions and transferred to polyvinylidene fluoride membranes. The membranes were blocked in 5% non-fat milk for 1 h and then incubated with primary antibodies overnight. On the next day, the membranes were incubated with secondary antibodies. Proteins were detected using enhanced chemiluminescence substrate (CST, USA). Antibodies used for western blot are listed below: CDCA5 (ab192237) was purchased from Abcam (UK). AKT (#4685), p-AKT (Ser473, #4060), NF-κB (#8242), Caspase-3 (#9662), and Cleaved caspase-3 (#9665) were purchased from CST (USA).
Cell lines
Two HCC cell lines (LM3 and HepG2) and 293T were purchased from the Shanghai cell bank at the Chinese Academy of Sciences and investigated in this study. Cells were cultured in alpha-Minimum Essential Medium (BI industry, Israel) supplemented with 10% fetal bovine serum (Gemini, USA).
Small interfering RNA (siRNA) transfection
siRNA transfections were performed according to the manufacturer's protocol. Cells were seeded in 6-well plates. Three different siRNA duplexes targeting CDCA5 were transfected using Lipofectamine 2000 reagent (Invitrogen, USA) after 24 h. The sequences of siRNA duplexes are listed below: 1#: GCCAGAGACTTGGAAATGTCT, 2#: CGAGAAACAGAAACGTAAGAA, 3#: CCAAAGTACCATAGCCAGTTT.
Cell viability assay
Cells were seeded in 96-well plates. Cell Counting Kit (CCK)-8 assay was performed at 24, 48, 72, and 96 h according to the manufacturer's instructions. The resulting colored solution was quantified using an ELx800 absorbance microplate reader (BioTek, USA) at 450 nm.
Cell apoptosis assay
Cell apoptosis was analyzed using Annexin V, FITC Apoptosis Detection Kit (Dojindo, Japan) according to the manufacturer's instructions. Cells were trypsinized and incubated with annexin V and propidium iodide at room temperature for 15 min. The cells were then evaluated by flow cytometry and analyzed using FlowJo (Tree Star).
Lentiviral infection
CDCA5 lentivirus and vector were purchased from Genecopoeia. LM3 cells were infected with lentivirus or vector with 8 μg/mL polybrene (Sigma, USA). CDCA5 knockdown efficiency was determined by immunoblotting. The short hairpin RNA (shRNA) sequences are listed below: 1#: CCCATCGTCTTAAAGAGGATC, 2#: GCGGAAATCAGGCTCTGAACT.
Xenograft model in vivo
18 Six-week-old male nude mice (Shanghai SLAC Laboratory Animal Co., Ltd, China) were purchased for the xenograft assay. LM3 cells with stable expression of control shRNA or two different CDCA5 shRNAs were used (6 mice in each group). These stable cells were trypsinized and suspended in phosphate-buffered saline (PBS). A total volume of 0.1 mL of PBS containing 1 × 106 LM3 cells was injected subcutaneously into the mouse flank. Approximately seven days later, tumors were observed and measured by a caliper. Tumor volumes were calculated by the formula V = 1/2 × ab2, where 'a' represents the longest dimension and 'b' represents the shortest dimension. The animal study protocol was reviewed and approved by the Animal Care Committee of Zhejiang University and designed in accordance with the Interdisciplinary Principles and Guidelines for the Use of Animals in Research, Testing, and Education by the New York Academy of Sciences, Ad Hoc Animal Research Committee.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
Apoptosis of xenograft tumors was detected using the One Step TUNEL Apoptosis Assay Kit (Beyotime, China) according to the manufacturer's instructions. Apoptosis of tumor tissues was measured by fluorescence microscopy.
Reporter assay
The ~2000-bp promoter region of CDCA5 was cloned into pGL3 luciferase reporter vectors. 293T cells were seeded in a 24-well plate. Reporter plasmid was transfected alone or with E2F1 plasmid. After 48 h, cells were collected for reporter assays using a dual luciferase reporter assay system (Promega, USA) according to the manufacturer's instructions. Renilla and firefly luciferase activities were measured by a Fluoroskan Ascent FL fluorometer (Thermo Fisher Scientific, USA).
ChIP assay
293T cells were seeded in a 24-well plate and transfected with E2F1-Flag or vector plasmid. After 48 h, cells were collected for ChIP. ChIP assay (Qiagen, German) was conducted according to the manufacturer's instructions. Flag antibody (Sigma, USA) or IgG antibody (CST, USA) was used for immunoprecipitation. DNA was quantified using PCR. Primers are listed below: ChIP-CDCA5-1: 5'-CACCACTACGCTCTCACCAG-3', 5'-CAGGAATCTGAGATCGGGGC-3'; ChIP-CDCA5-2:5'-GGAAAGGGGCATGAGTTGGTA-3', 5'-GGCCACCAGAACCTGACTTC-3'; ChIP-CDCA5-3: 5'-GACCCGATGTTCGAAGCAGA-3', 5'-CAACTCATGCCCCTTTCCCA-3'; ChIP-CDCA5-4: 5'-CACCTTGGCGTGATTGGCTA-3', 5'-TACCAACTCATGCCCCTTTCC-3'.
Statistical analysis
Values were presented as the mean ± SD. The comparison of CDCA5 levels were performed using student's t test in tumor and normal tissues. The overall survival of patients was assessed by Kaplan- Meier survival analysis. GraphPad PRISM 6.01 software (San Diego, CA, US) was used to analyze experimental data. P < 0.05 indicates a statistically significant difference. * represents P < 0.05, ** represents P < 0.01, and *** represents P < 0.001.
Results
The expression of CDCA5 and its clinical significance in HCC
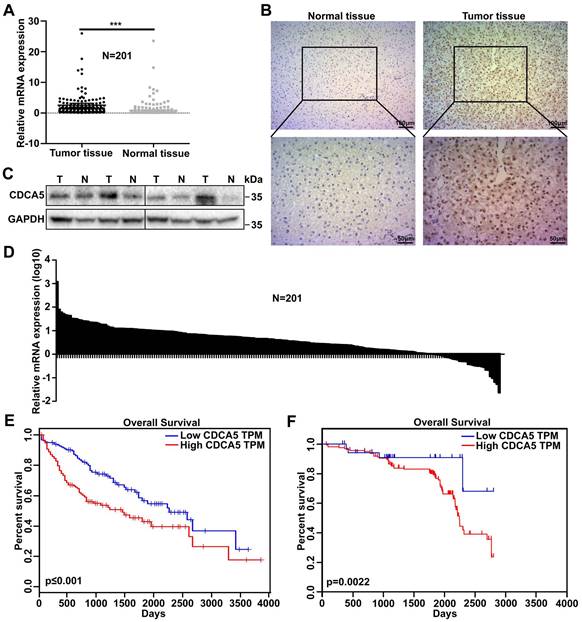
To investigate the possible role of CDCA5 in HCC progression, messenger RNA (mRNA) expression levels of CDCA5 were analyzed in 201 pairs of tumor tissues and matched normal tissues from HCC patients by RT-qPCR. Overall, the expression of CDCA5 was much higher in tumor tissues than in normal tissues (Figure 1A and 1B). The protein levels of CDCA5 in paired tissues were measured by western blot and IHC and the results showed that CDCA5 overexpressed in cell nucleus of HCC tissues (Figure 1C and 1D). The relationship between CDCA5 expression and prognosis was analyzed by Oncolnc, which indicated that patients with higher CDCA5 expression were correlated with poorer survival (Figure 1E). In order to confirm this relationship, we analyzed the CDCA5 mRNA data with the patients' clinical features and prognosis. Similarly, the results indicated that patients with higher CDCA5 expression were correlated with poorer overall survival (Figure 1F). Meanwhile, CDCA5 expression was related to tumor size and number (Table 1). Collectively, these results indicated that higher CDCA5 expression is associated with poorer prognosis and worse malignancy in HCC.
CDCA5 overexpressed in hepatocellular carcinoma. A. CDCA5 overexpressed in HCC tissue compared to paired normal liver tissue (n=201) by RT-qPCR. B. Relative CDCA5 mRNA expression in HCC tissue compared to paired normal liver tissue (n=201). C. CDCA5 protein expression in HCC tissue and paired normal liver tissue (n=4). D. Representative IHC images of CDCA5 in HCC tissue and paired normal liver tissue. E. Data from Oncolnc indicated Patients with higher CDCA5 expression (n=180) had poorer overall survival compared to those with lower CDCA5 expression (n=180). F. Kaplan-Meier analysis demonstrated that CDCA5 expression had a negative correlation with patient survival (n=201).
Relationship between CDCA5 expression and clinicopathological feature in 201 HCC cases
| Characteristic | CDCA5 Expression Level | P-Value | |
|---|---|---|---|
| Low | High | ||
| Mean age | 58.11±9.38 | 53.80±9.09 | 0.572 |
| AFP | 0.290 | ||
| <400 | 22 | 108 | |
| ≥400 | 15 | 56 | |
| Tumor grade | 0.161 | ||
| Low and moderate | 22 | 80 | |
| High | 15 | 84 | |
| Size(cm) | 0.004** | ||
| <5 | 19 | 44 | |
| ≥5 | 18 | 120 | |
| Tumor number | 0.019*; ※ | ||
| 1 | 34 | 124 | |
| ≥2 | 3 | 40 | |
| PVTT | 0.568 | ||
| Absent | 31 | 144 | |
| Present | 6 | 20 | |
| Child-Pugh class | 0.264 | ||
| A | 27 | 108 | |
| B or C | 10 | 56 | |
| BCLC stage | 0.518 | ||
| 0 | 2 | 3 | |
| A | 12 | 52 | |
| B | 17 | 71 | |
| C | 6 | 38 | |
*(p<0.05); ** (p<0.01); ※(continuity correction); BCLC, BCLC staging system.
CDCA5 promoted liver cancer cell proliferation and inhibited cell apoptosis in vitro
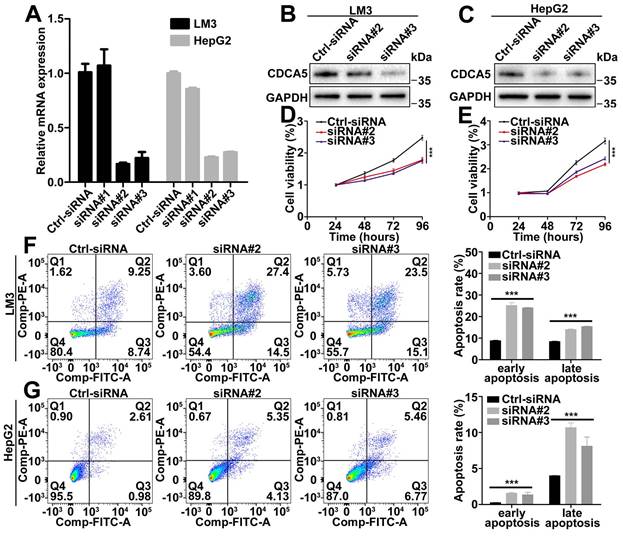
Based on the CDCA5 mRNA levels in liver cancer cell lines, LM3 and HepG2 were selected to detect the functional role of CDCA5. The efficacy of siRNA was detected by RT-qPCR and western blot (Figure 2A, 2B, and 2C). Depletion of CDCA5 with siRNA in LM3 and HepG2 cells led to reduced cell proliferation (Figure 2D and 2E). Meanwhile, downregulation of CDCA5 induced both early and late cell apoptosis (Figure 2F and 2G). However, no cell cycle alteration was observed in CDCA5-downregulated cells (data not shown).
CDCA5 promoted liver cancer cell proliferation and inhibited cell apoptosis in vitro. A. Evaluation of CDCA5 knockdown in LM3 and HepG2 cells after transfection with CDCA5 siRNA by RT-qPCR. B, C. Evaluation of CDCA5 knockdown in LM3 and HepG2 cells after transfection with CDCA5 siRNA by western blot. D, E. Knockdown of CDCA5 inhibited cell viability in LM3 and HepG2 cells. F, G. Knockdown of CDCA5 promoted cell apoptosis in LM3 and HepG2 cells.
CDCA5 induced HCC tumorigenicity in vivo
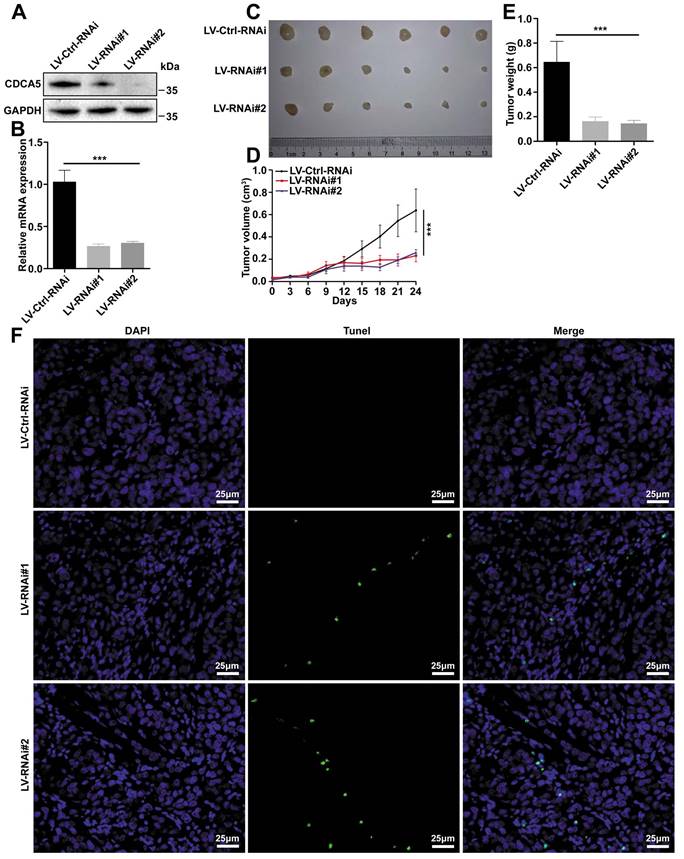
To confirm the results observed in vitro, a xenograft model was built using the LM3 cell line. In order to knockdown the expression of CDCA5, LM3 cells were transfected with lentivirus and filtered by puromycin. The silence efficiency was assessed by western blot and RT-qPCR (Figure 3A and 3B). After transfection, cells were injected into nude mice for the subcutaneous xenograft model. The tumors were harvested and imaged at 31 days (Figure 3C). Both tumor size and tumor weight demonstrated that knockdown of CDCA5 led to tumor growth inhibition (Figure 3D and 3E). Apoptosis was induced after CDCA5 depletion, which was detected by TUNEL assay (Figure 3F).
CDCA5 promoted tumorigenicity in vivo. A. Evaluation of CDCA5 knockdown in LM3 cells after transfection with CDCA5 shRNA by western blot. B. Evaluation of CDCA5 knockdown in LM3 cells after transfection with CDCA5 shRNA by RT-qPCR. C. Representative images of xenografts from CDCA5-knockdown group and control group. D. Tumor growth curve of CDCA5-knockdown LM3 cells in nude mice compared to control group. E. Tumor weight of CDCA5-knockdown xenografts decreased significantly compared to control group. F. More apoptotic cells were observed in CDCA5-knockdown xenografts compared to control group detected by TUNEL assays (400 ×).
CDCA5 was a direct downstream target of E2F1
As the mRNA level of CDCA5 was upregulated in tumor tissues, we doubted that transcription factors (TFs) involved in mRNA transcription may also regulate the function of CDCA5. A series of TFs were predicted, among which E2F1 was highly expressed in HCC tissues. We analyzed the relationship between E2F1 and prognosis in patients by Gene Expression Profiling Interactive Analysis (GEPIA) and found that patients with higher expression of E2F1 suffered worse overall and worse disease-free survival (Figure 4A and 4B). Moreover, we found that CDCA5 was highly correlated with E2F1 in HCC by GEPIA (Figure 4C). Then E2F1 was overexpressed in LM3 and HepG2 cells, and RT-qQCR assay showed that CDCA5 was induced by E2F1 (Figure 4D).The luciferase reporter assay was then conducted. We cloned the promoter region of CDCA5 (~2000 to 0), inserted the fragment into the pGL3-basic luciferase vector, and transfected it into 293T cells. As a result, the luciferase activity of CDCA5 promoter can be activated by E2F1 (Figure 4E).
CDCA5 was a direct downstream target of E2F1. A, B. Data from Gepia indicated Patients with higher E2F1 expression (n=182) had worse overall and worse disease-free survival compared to those with lower CDCA5 expression (n=182). C. The expression of CDCA5 was highly correlated to the expression of E2F1 (Data from GEPIA). D. The expression of CDCA5 was induced by E2F1 in LM3 and HepG2 cells. E. E2F1 activated the transcription of CDCA5 by luciferase reporter assay in 293T cells. F. The locations of the primer pairs used in the ChIP assay. G. The promoter occupancy detected with four pairs of primers in E2F1-overexpressing 293T cells or control.
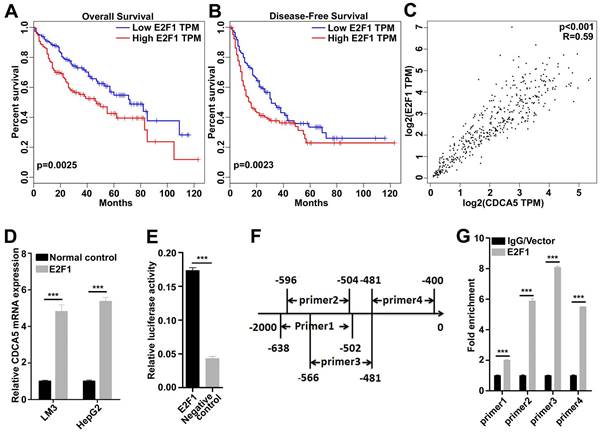
To determine whether E2F1 directly binds to the promoter region of CDCA5, we designed four pairs of E2F1 binding region-specific primers (Figure 4F) and performed ChIP assays using flag-specific antibodies. In 293T cells, the core promoter region was detected in the E2F1 co-precipitate complex (Figure 4G). Together, these findings suggested that the transcription of CDCA5 was regulated by E2F1.
CDCA5 regulated cell behavior via the AKT pathway
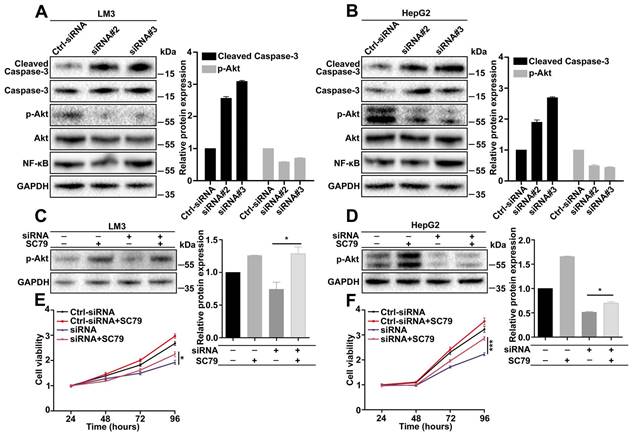
To explore the downstream signaling pathways regulated by CDCA5, a series of pathways were analyzed by western blot. According to the effects of CDCA5, we focused on apoptosis-associated pathways. The results showed that cleaved caspase-3 was increased and p-AKT (Ser473) was decreased in CDCA5-knockdown cells (Figure 5A and 5B). To confirm the effects of AKT, a rescue experiment was operated using SC79, an AKT-specific activator. Results showed that the expression of p-AKT can be partly rescued by SC79 (Figure 5C and 5D). Cell viability assay also showed that the suppression of cell proliferation by CDCA5 knockdown can be partly relieved by SC79 (Figure 5E and 5F). In a word, CDCA5 regulated cell behavior via the AKT pathway.
CDCA5 regulated cell behavior via the AKT pathway. A, B. Knockdown of CDCA5 significantly increased cleaved caspase-3 and decreased p-AKT (Ser473) by Western blot. C, D. SC79 restored the expression of p- AKT (Ser473) in CDCA5 silenced LM3 and HepG2 cells. E, F. SC79 restored cell viability in CDCA5 silenced LM3 and HepG2 cells.
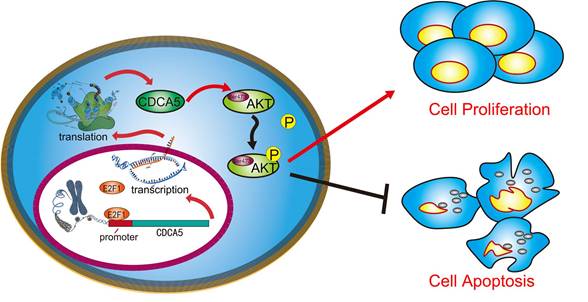
A schematic illustration showed that CDCA5, transcribed by E2F1, promotes oncogenesis by enhancing cell proliferation and inhibiting apoptosis via the AKT pathway
Discussion
The mammalian cell cycle is induced by the continuous activation or deactivation of proteins that regulate the various phases of the cell cycle [15]. Cell cycle arrests permit repair of mistakes that occurred during DNA replication in order to maintain an intact genome. Meanwhile, cell cycle arrests block the propagation of high-risk cells [16]. Loss of normal cell cycle is a hallmark of human cancers [17]. Accumulation of tumor cell mutations leads to genomic instability and unscheduled proliferation. Sister chromatid cohesion is vital for the proper co-segregation of newly replicated chromosomes at anaphase. CDCA5 is a critical regulator of DNA repair and chromosome segregation [16], which was degenerated by anaphase-promoting complex-dependent ubiquitination in the G1 phase [4]. It regulates sister chromatid cohesion by altering the interaction between cohesin and chromosomes. CDCA5 is abnormally expressed in several types of cancer, which makes it possible to be a significant prognostic indicator and a potential therapeutic target in tumor patients. However, few studies have investigated the expression and function of CDCA5 in HCC. In this study, we observed a significant upregulation of CDCA5 mRNA in clinical liver cancer tissues compared with adjacent paired tissue. Moreover, the mRNA level of CDCA5 in tumor tissues was tightly correlated with poor survival. Furthermore, depletion of CDCA5 dramatically inhibited cell growth and tumorigenesis both in vitro and in vivo. Thus, consistent with previous studies, our results supported the oncogenic role of CDCA5 in liver cancer progression and indicated the possibility of using CDCA5 as a therapeutic target for liver cancer.
In an effort to elucidate the upstream regulators, we identified E2F1 as the transcription factor that modulates CDCA5 expression. E2F1 is a key transcription factor in the control of proliferation [18, 19], differentiation [20], and apoptosis [21]. E2F1 is a well-known oncogene, which was upregulated in a series of tumors, including HCC [14, 22]. As reported, E2F1 may act as a vital anti-apoptotic factor in liver cancer for its ability to offset c-Myc-driven apoptosis. It was reported that E2F1 was upregulated at an early phase and then downregulated as Myc increased. Similarly, we found that E2F1 was overexpressed and negatively related with patient survival, which indicated the oncogenic function of E2F1 in HCC [23]. Meanwhile, the expression of E2F1 and CDCA5 were positively associated with. Dual luciferase reporter assay and ChIP supported the result that CDCA5 was transcribed by E2F1, implying that E2F1 is the upstream regulator of CDCA5 in HCC.
Moreover, we elucidated the signaling pathways medicated by CDCA5 and found that CDCA5 silencing induces suppression of AKT signaling pathway, which in turn lead to the activation of pro-apoptotic signaling pathways. AKT is a serine/threonine protein kinase that regulates cell growth, survival, and apoptosis [24, 25]. AKT is regulated by site-specific phosphorylation or dephosphorylation [26]. Once activated, AKT undergo phosphorylation, which regulates cellular function. Phosphorylation at four domains of AKT plays a vital function: Ser124, Thr308, Thr450, and Ser473. Ser124 and Thr450 are phosphorylated basally while Thr308 and Ser473 are phosphorylated conditionally [27, 28]. Phosphorylation of AKT at Ser473 indicates poor clinical outcome in many cancers [29-31]. CDCA5 knockdown repressed the levels of AKT phosphorylation at Ser473 sites and which induced the expression of cleaved caspase-3, subsequently leading to cell death. The activity of caspase-3 was correlated with the growth-inhibitory and apoptosis-inducing effects of CDCA5 knockdown. Collectively, these results indicated that CDCA5 is involved in the regulation of the AKT signaling pathway in HCC.
Conclusion
In conclusion, we identified a novel amplification of CDCA5 in HCC. CDCA5 played a pivotal oncogenic role in hepatocellular carcinogenesis and was transcribed by E2F1 by directly binding to its promoter. Meanwhile, CDCA5 knockdown can inhibit cell proliferation and induce apoptosis by repressing the AKT signaling pathway. All of these results provide a mechanistic explanation of the involvement of CDCA5 in hepatocellular carcinogenesis. CDCA5 expression may serve as an independent poor prognostic factor for HCC patients.
Acknowledgements
This research was supported by Innovative Research Groups of National Natural Science Foundation of China (No. 81721091), Major program of National Natural Science Foundation of China (No. 91542205), the National Natural Science Foundation of China (No. 81272675 and 81570575), National S&T Major Project (No. 2017ZX10203205), and Zhejiang International Science and Technology Cooperation Project (NO. 2016C04003).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29
2. Valery PC, Laversanne M, Clark PJ, Petrick JL, McGlynn KA, Bray F. Projections of primary liver cancer to 2030 in 30 countries worldwide. 2017.
3. Zhang N, Pati D. Sororin is a master regulator of sister chromatid cohesion and separation. Cell Cycle. 2012;11:2073-83
4. Rankin S, Ayad NG, Kirschner MW. Sororin, a substrate of the anaphase-promoting complex, is required for sister chromatid cohesion in vertebrates. Mol Cell. 2005;18:185-200
5. Zhang Z, Shen M, Zhou G. Upregulation of CDCA5 promotes gastric cancer malignant progression via influencing cyclin E1. Biochem Biophys Res Commun. 2018;496:482-9
6. Chang IW, Lin VC, He HL, Hsu CT, Li CC, Wu WJ. et al. CDCA5 overexpression is an indicator of poor prognosis in patients with urothelial carcinomas of the upper urinary tract and urinary bladder. Am J Transl Res. 2015;7:710-22
7. Kato T, Lee D, Wu L, Patel P, Young AJ, Wada H. et al. SORORIN and PLK1 as potential therapeutic targets in malignant pleural mesothelioma. Int J Oncol. 2016;49:2411-20
8. Tokuzen N, Nakashiro K, Tanaka H, Iwamoto K, Hamakawa H. Therapeutic potential of targeting cell division cycle associated 5 for oral squamous cell carcinoma. Oncotarget. 2016;7:2343-53
9. Xu T, Ma M, Dai J, Yu S, Wu X, Tang H. et al. Gene expression screening identifies CDCA5 as a potential therapeutic target in acral melanoma. Hum Pathol. 2018
10. Chen D, Pacal M, Wenzel P, Knoepfler PS, Leone G, Bremner R. Division and apoptosis of E2f-deficient retinal progenitors. Nature. 2009;462:925-9
11. Santoni-Rugiu E, Duro D, Farkas T, Mathiasen IS, Jaattela M, Bartek J. et al. E2F activity is essential for survival of Myc-overexpressing human cancer cells. Oncogene. 2002;21:6498-509
12. Miller TW, Balko JM, Fox EM, Ghazoui Z, Dunbier A, Anderson H. et al. ERalpha-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov. 2011;1:338-51
13. Qin XQ, Livingston DM, Kaelin WG Jr, Adams PD. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci U S A. 1994;91:10918-22
14. Ladu S, Calvisi DF, Conner EA, Farina M, Factor VM, Thorgeirsson SS. E2F1 inhibits c-Myc-driven apoptosis via PIK3CA/Akt/mTOR and COX-2 in a mouse model of human liver cancer. Gastroenterology. 2008;135:1322-32
15. Dominguez-Brauer C, Thu KL, Mason JM, Blaser H, Bray MR, Mak TW. Targeting Mitosis in Cancer: Emerging Strategies. Mol Cell. 2015;60:524-36
16. Huang CJ, Yuan YF, Wu D, Khan FA, Jiao XF, Huo LJ. The cohesion stabilizer sororin favors DNA repair and chromosome segregation during mouse oocyte meiosis. In Vitro Cell Dev Biol Anim. 2017;53:258-64
17. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
18. Huang Y, Tai AW, Tong S, Lok AS. HBV core promoter mutations promote cellular proliferation through E2F1-mediated upregulation of S-phase kinase-associated protein 2 transcription. J Hepatol. 2013;58:1068-73
19. Wang B, Hsu SH, Wang X, Kutay H, Bid HK, Yu J. et al. Reciprocal regulation of microRNA-122 and c-Myc in hepatocellular cancer: role of E2F1 and transcription factor dimerization partner 2. Hepatology. 2014;59:555-66
20. Xu S, Tao J, Yang L, Zhang E, Boriboun C, Zhou J. et al. E2F1 Suppresses Oxidative Metabolism and Endothelial Differentiation of Bone Marrow Progenitor Cells. Circ Res. 2018;122:701-11
21. Lu Z, Bauzon F, Fu H, Cui J, Zhao H, Nakayama K. et al. Skp2 suppresses apoptosis in Rb1-deficient tumours by limiting E2F1 activity. Nat Commun. 2014;5:3463
22. Chang L, Xi L, Liu Y, Liu R, Wu Z, Jian Z. SIRT5 promotes cell proliferation and invasion in hepatocellular carcinoma by targeting E2F1. Mol Med Rep. 2018;17:342-9
23. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017;45:W98-w102
24. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS. et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857-68
25. Zhou H, Li XM, Meinkoth J, Pittman RN. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol. 2000;151:483-94
26. Liao Y, Hung MC. Physiological regulation of Akt activity and stability. Am J Transl Res. 2010;2:19-42
27. Li J, Dokka S, Wang L, Shi X, Castranova V, Yan Y. et al. Activation of aPKC is required for vanadate-induced phosphorylation of protein kinase B (Akt), but not p70S6k in mouse epidermal JB6 cells. Mol Cell Biochem. 2004;255:217-25
28. Lee HC, Park IC, Park MJ, An S, Woo SH, Jin HO. et al. Sulindac and its metabolites inhibit invasion of glioblastoma cells via down-regulation of Akt/PKB and MMP-2. J Cell Biochem. 2005;94:597-610
29. Lin A, Piao HL, Zhuang L, Sarbassov dos D, Ma L, Gan B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacologic inhibition of the PI3K-AKT pathway. Cancer Res. 2014;74:1682-93
30. Yu Z, Weinberger PM, Sasaki C, Egleston BL, Speier WFt, Haffty B. et al. Phosphorylation of Akt (Ser473) predicts poor clinical outcome in oropharyngeal squamous cell cancer. Cancer Epidemiol Biomarkers Prev. 2007;16:553-8
31. Chua BT, Gallego-Ortega D, Ramirez de Molina A, Ullrich A, Lacal JC, Downward J. Regulation of Akt(ser473) phosphorylation by choline kinase in breast carcinoma cells. Mol Cancer. 2009;8:131
Author contact
![]() Corresponding authors: Prof. Shusen Zheng, E-mail: shusenzhengedu.cn; and Prof. Penghong Song, E-mail: songpenghongedu.cn.
Corresponding authors: Prof. Shusen Zheng, E-mail: shusenzhengedu.cn; and Prof. Penghong Song, E-mail: songpenghongedu.cn.