Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(8):1915-1922. doi:10.7150/jca.27053 This issue Cite
Research Paper
Cyclin-Dependent Kinase Inhibitor 3 Promoted Cell Proliferation by Driving Cell Cycle from G1 to S Phase in Esophageal Squamous Cell Carcinoma
Juan Liu1, Li Min1, Shengtao Zhu1, Qingdong Guo1, Hengcun Li1, Zheng Zhang1, Yu Zhao1, Changqin Xu2 ![]() , Shutian Zhang1
, Shutian Zhang1 ![]()
1. Department of Gastroenterology, Beijing Friendship Hospital, Capital Medical University, National Clinical Research Center for Digestive Disease, Beijing Digestive Disease Center, Beijing Key Laboratory for Precancerous Lesion of Digestive Disease, Beijing, 100050, P. R. China
2. Shandong Provincial Hospital affiliated to Shandong university
Received 2018-5-3; Accepted 2019-1-12; Published 2019-4-21
Abstract
Background and aims. Cyclin-dependent kinase inhibitor 3 (CDKN3) has been found playing a varying role in carcinogenesis, but its biological function in esophageal squamous cell carcinoma (ESCC) is unclear. The aim of this study was to demonstrate the role of CDKN3 in ESCC.
Materials and Methods: Real-time PCR and Western blot was performed in 15 pairs of ESCC tissues and adjacent normal esophageal tissues. Then cell proliferation ability, cloning ability, cell cycle status and migration and invasion ability were explored in CDKN3 overexpressed TE1 cell line and CDKN3 siRNA transfected TE1 and KYSE70 cell lines. Finally, cell cycle related proteins CyclinD1, CDK4, pAKT, P53, P21, and P27 were tested by Western blot.
Results: mRNA level was higher in 11 ESCC tissues compared to adjacent normal tissues, and an increased protein expression was further detected in 8 of those 11 ESCC tissues. Functional assays showed that CDKN3 overexpression promoted ESCC cell proliferation, colony formation, migration and invasion, and facilitated G1/S transition. Opposite results were also got after transfected with CDKN3 siRNA. Cell cycle associated protein pAKT, CyclinD1, CDK4 and P27 were upregulated and P53, P21 and were downregulated under CDKN3 overexpression. All the protein levels were found changed in the opposite direction when CDKN3 expression was disturbed by siRNA.
Conclusions: Our study suggested that CDKN3 acted as an oncogene in human ESCC and may accelerate the G1/S transition by affecting CyclinD-CDK4 complex via regulating pAKT-p53-p21 axis and p27 independent of AKT.
Keywords: ESCC, CDKN3, cell cycle, G1/S transition, cyclinD-CDK4 complex
Introduction
Esophageal cancer is one of the most common cancers and Eastern Asia owns the highest incidence rate[1]. In developing countries such as China, major pathological type of esophageal cancers is squamous cell carcinomas (ESCC)[2], which ranked the third commonly diagnosed cancer among men and the fifth among women. Additionally, ESCC is also the fourth leading cause of cancer death among both men and women[3]. Although large efforts have been devoted into ESCC studies, the exact mechanism of ESCC is still unclear.
Cyclin-dependent kinase inhibitor 3 (CDKN3), which is also known as Cyclin-Dependent Kinase-Associated Protein Phosphatase (KAP), is the product of the CDKN3 gene[4]. CDKN3 is a dual specificity phosphatase and participates in the regulation of cell cycle by interacting with cyclin-dependent kinases which mainly include CDK1 and CDK2[5]. Activated CDK2-Cyclin A complex promotes the G1/S transition and cell proliferation. Since Thr160 phosphorylation is necessary for activating CKD2, CDKN3 can dephosphorylate CKD2 at Thr160 when CDK2 is degraded or dissociates from Cyclin A and then delay the G1/S transition. Besides, a study had shown that CDKN3 dephosphorylated CDK1 at Thr161 in late mitosis and loss of CDKN3 can induce abnormal mitosis and supernumerary centrosomes[6]. Another research identified that CDKN3 can maintain a proper number of centrosomes by forming a self-regulated feedback loop with Mps1 and controls mitosis[7].
Disorder of cell replication or proliferation is an important mechanism of tumorigenesis and progression. As a cell cycle regulator, CDKN3 has been investigated in different cancers. In glioblastoma, hepatocellular cancer and myelogenous leukemia, CDKN3 acts as tumor suppressor depending on its function of dephosphorylation of CDK1 or CDK2 [8-11]. However, in ovarian cancer, cervical cancer, colorectal cancer and breast cancer, it works in an opposite way[12-15]. Clinical data showed that overexpression of CDKN3 indicated poor cancer prognosis[13, 16]. It had been documented that CDKN3 is upregulated in esophageal cancer[17], but advanced research on the detailed biological function is still absent.
In this study, we aimed to assess the role of CDKN3 in ESCC. We found that CDKN3 was overexpressed in ESCC tissues and could increase ESCC cells proliferation by accelerating G1/S transition, which suggested that CDKN3 acted an oncogene in human ESCC.
Materials and Methods
Cell lines and culture
ESCC cell lines TE1, TE10 (purchased from RIKEN BioResource Center, Japan) and KYSE70 (purchased from DSMZ company, German) were cultured in RPMI 1640 medium (Gibco, American) mixed with 10 % fetal bovine serum (FBS) at 37 °C with humidified 5% CO2.
ESCC clinical samples
From May to October 2013, 15 pairs ESCC tissues and adjacent normal esophageal tissue were obtained from patients who underwent esophagectomy for esophageal cancer at Beijing Friendship Hospital. All the samples were frozen in liquid nitrogen and stored at -80°C.
The study was approved by the Clinical Research Ethics Committee of Beijing Friendship Hospital.
Western blotting
Proteins measured in this study was extracted by protein lysis buffer and quantified by Pierce™ BCA Protein Assay Kit (Thermo Scientific, American). Proteins were separated by 12% polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to PVDF. After blocking by 5% defatted milk for 1 hour, the membrane was incubated with a primary antibody overnight at 4℃. Primary antibody mainly included CDKN3 (LifeSpan BioSciences, 1:1000), Flag (Sigma, 1:1000), AKT (Invitrogen, 1:1000), pAKT (Life Technologies, 1:1000), P53 (Cell Signaling Technology, 1:1000), P27 (Cell Signaling Technology, 1:1000), P21 (Abcam, 1;1000), CyclinD1 (Abcam, 1:200), CDK4 (Proteintech, 1:1000), β-actin (Earthox, 1:1000). Then, the membrane was incubated with an appropriate secondary antibody for 1 hour at room temperature after washed by TBS-T (tris buffered saline-0.1% tween-20) for six times. At last, the blot was colored and observed by SuperSignal® West Dura Extended Duration Substrate (Thermo Fisher Scientific, USA).
Real-time quantitative PCR and PCR
RNA was extracted from cell or tissues by Trizol Reagent (Invitrogen, USA). High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, American) was used to accomplish reverse transcription in a 20μl reaction volume, which contains 10μl master mix and 2μg (10μl) RNA. The 10μl master mix was composed of 2μl 10×RT Buffer, 0.8μl dNTP mix, 2.0μl 10×RT random primers, 1.0μl MultiScribe Reverse Transcriptase and 4.2μl Nuclease-free water. The reaction was carried out at 25℃ for 10 minutes, 37℃ for 120 minutes and 85℃ for 5 minutes. The obtained cDNA was quantified by real-time PCR. The reaction mixture contained 6μl Fast SYBR™ Green Master Mix (Thermo Fisher Scientific, American), 2.5μl sense/antisense primer separately (Table S1), and 1μl cDNA. The reaction was incubated at 25℃ for 10 minutes, 37℃ for 120 minutes and 85℃ for 5 minutes. The relative expression level of CDKN3 was calculated by the comparative △CT (2-△△Ct) method.
siRNA transfection and Construction of CDKN3 overexpression cells
Depending on principles for siRNA designing, we synthesized four kinds of CDKN3 special siRNA (Table S2) and chose the most effective one which was verified by western blot in TE1 cell line (Figure S1A) to complete the study.
To construct CDKN3 expression vector, we firstly got amplified CDKN3 cDNA by PCR (primer was listed in table 1), then the cDNA was purified by 1% agarose gel electrophoresis and linked to pCMV-HA to form pCMV-HA-CDKN3. Secondly, target plasmid was extracted after pCMV-HA-CDKN3 transfecting competent E.coli DH-5α (TIANGEN Biotech Co.,Ltd., China) and was sequenced to confirm the CDKN3 gene sequence. Next, lentiviral vector Ubi-MCS-3FLAG-SV40-EGFP-IRES-puromycinco contained pCMV-HA-CDKN3 was constructed. Finally, lentiviral vector and vector with CDKN3 infected the TE1 cell line stably (abbreviated as TE1-vector and TE1-CDKN3) (Figure S1B) and confirmed by western blot (Figure S1C). Puromycin was selected to screen and maintain transfected cell and the MOI was 50. (TE1-CDKN3) or empty vector (TE1-vector).
Correlation between CDKN3 expression and clinicopathologic parameters of ESCC tissues
| CDKN3 mRNA level | |||
|---|---|---|---|
| High (n=11) | Normal (n=4) | P values* | |
| Gender (M/F) | 10/1 | 2/2 | 0.15 |
| Age(y) | 0.10 | ||
| ≤60 | 5 | 4 | |
| >60 | 6 | 0 | |
| Tumor location | 0.71 | ||
| Cervical segment | 1 | 0 | |
| Thoracic segment | 4 | 1 | |
| Abdominal segment | 6 | 3 | |
| Tumor size (cm) | 0.56 | ||
| ≤5cm | 8 | 2 | |
| >5cm | 3 | 2 | |
| Tumor invasion | 0.56 | ||
| T2 | 3 | 2 | |
| T3 | 8 | 2 | |
| Lymph node metasasis | 0.24 | ||
| Positive | 8 | 1 | |
| Negative | 3 | 3 | |
| Tumor differentiation | 0.08 | ||
| Moderate | 2 | 3 | |
| poor | 9 | 1 | |
ESCC, esophageal squamous cell carcinoma. * Fisher's exact test
Cell proliferation assay
Cell proliferation assay was measured by MTS. Cells were seeded in a 96-well plate at a density of 3× 103 cells / well, each type cell created 6 wells. Viability was measured at12, 24, 48, 72 hours described as day 0, 1, 2, 3. Before the test, 20ul MTS solution (CellTiter 96® AQueous One Solution Cell Proliferation Assay, Promega Corporation, USA) was added to each well and cultured at 37 °C for 2 hours. Then OD of each well was read at 490 nm. Each experiment was repeated three times. Data was addressed by the following method: firstly, excluded the maximum and minimum value. Secondly, got the mean OD value of day 0. Lastly, the mean OD value was divided by every OD value to get the fold change of OD value.
Cell colony formation assay
TE1 cells were seeded in 6-well plate at 1000 cells / well and KYSE70 cells were at 500 cells / well and cultured with RPIM 1640 with FBS for 10-14 days until colonies appeared. Then removed the solution and used phosphate buffered saline (PBS) to wash the plates and 1ml/well methanol was inserted for 15 minutes until PBS dried. Finally, cells were stained with 0.1% crystal violet for 10 minutes and the number of colonies was counted artificially.
Cell cycle analysis
Cells were resuspended in 75% ethyl alcohol which was pre-cooled in ice then kept at 4 ℃ for 12 hours. After washed the cells by PBS twice, 250μl PI/RNase Staining Buffer (BD Pharmingen™, USA) was added and incubated in the dark place for 30 minutes. Then samples were tested by flow cytometer (BD FACSVerse™, USA) and flow cytometric data were analyzed by ModFit LT (Version 3.2). Each cell line tested three samples and experiment was repeated three times.
Transwell cell invasion and migration assay
Cell invasion ability was evaluated by Transwell cell invasion assay. 12 hours before the experiment, cells were cultured by medium without FBS, then transwell with Matrigel (Corning, USA) on the bottom was hydrated by 500μl RPIM 1640 in the upper and lower chamber which should be placed in 24-well plate at 37 °C incubator for 2 hours. Then transwell was put in another well of 24-well plate with 750μl RPIM 1640 and 10% FBS, after that 1*105 cells which were resuspended by 500μl RPIM 1640 were added to the upper chamber of transwell and incubated at 37 °C incubator for 36 hours. Next, the membrane was fixed by methanol for 5 minutes and placed on slides after cutting off. Finally, cells which stained by DAPI staining solution were observed under a microscope. Transwell without Matrigel was used to perform migration assay in the same way with invasion assay except for hydration. Choosing three fields to count the cell number and accomplished data analysis.
Statistical analysis
The statistical data were analyzed by SPSS 21.0 (SPSS Inc., Chicago, IL, USA). Data were described as the mean ± SE and calculated with unpaired, two-tailed Student's t-test. Enumeration data were compared using the χ2 test or the Fisher's exact test. The correlation between CDKN3 expression and esophageal cancer patient survival span was analyzed using the chi-square tests. P < 0.05 was considered statistically with significant differences.
Results
Expression of CDKN3 was increased in ESCC tissues
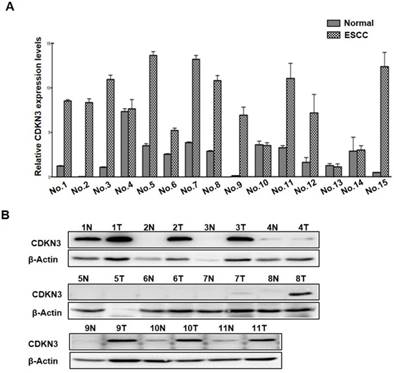
To investigate the expression levels of CDNK3 in ESCC tissues, we examined the mRNA levels of CDKN3 in 15 pairs ESCC tissues and adjacent normal esophageal tissue. The result showed that CDKN3 mRNA level was increased in 11 ESCC tissue samples compared to the relevant normal esophageal tissue, and the other 4 pairs were with subequal results (Figure 1A). Association between CDKN3 mRNA level and clinicopathologic parameters of these cases were analyzed, but no statistical difference was detected in gender, age, tumor location and size, invasion depth, lymph node metastasis and cell differentiation (table 1). Protein expression status of the 11 paired samples was further investigated by Western blot, of which 1 pair was failed for histolysis. The results indicated that CDKN3 expression was higher in cancer tissues in 8 pair samples, the other 2 pairs got similar bands (Figure 1B).
Expression of CDKN3 was increased in ESCC tissues. (A) mRNA levels of CDKN3 in 15 paired ESCC tissue and adjacent normal tissue. (B) Western blot result of CDKN3 expression level in 11 matched tissues. N is short for normal tissue and T is tumor tissue.
CDKN3 expression indicated poor survival
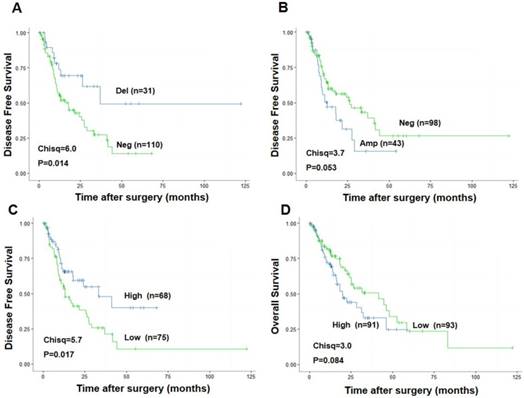
To explore the association between CDKN3 expression and ESCC patients survival, we performed the survival analysis of data from The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/). Patients with CDKN3 copy number variation (CNV) deletion (n=31) had a significant higher Disease-Free Survival (DFS) after surgery (Chisq=6.0, P=0.014) than patients without CNV deletion (n=110) (Figure 2A). Correspondingly, DFS of patients with CNV amplification (n=43) was much lower compared to the ones without CNV amplification (n=98), with a strong tendency towards statistical significance (Chisq=3.7, P=0.053) (Figure 2B). Higher CDKN3 expression level (n=68) was also related with longer DFS (Chisq=3.7, P=0.017) (Figure 2C) but a little lower overall survival rate (Chisq=3.0, P=0.084) (Figure 2D) compared with low CDKN3 expression level.
Association between CDKN3 expression and survival rate in patients with esophageal cancer. The data used was from the Cancer Genome Atlas (TCGA). (A) CDKN3 copy number variation (CNV) deletion was associated with higher disease free survival rate. (B) CDKN3 copy number variation (CNV) amplification was associated with poor disease free survival rate. (C) High CDKN3 expression level was related to higher disease free survival rate but (D)lower overall survival rate.
CDKN3 promoted ESCC cell proliferation and colony formation
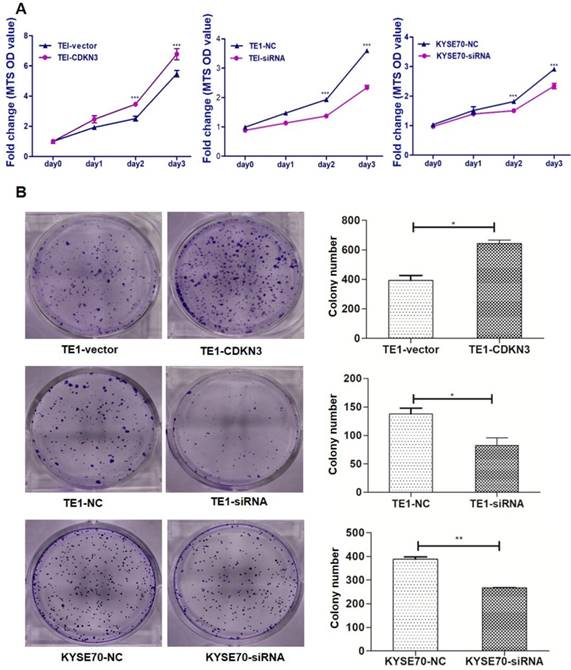
Cell proliferation assay and colony formation were carried out to evaluate the effect of CDKN3 on cell growth. MTS assays showed that CDKN3 overexpression promoted TE1 cells proliferating and CDKN3 knockdown decreased the proliferative ability of TE1 and KYSE70 cells (Fig. 3A). Colony formation assays got the similar results. Colony count was more in CDKN3 overexpression cells than those transfected with empty vector and was lower in CDKN3 siRNA interfering cell lines (Fig. 3B).
CDKN3 promoted ESCC cell proliferation and colony formation. (A) CDKN3 overexpression promoted cell proliferation in TE1 cell line (left), but TE1 and KYSE70 cells transfected with CDKN3 specific siRNA showed decreased cell proliferation ability (middle and right). (B) CDKN3 overexpression increased colony formation in TE1 cells (up), siRNA expressing decreased colony formation ability in TE1 (middle) and KYSE70 cell line (down). Columns, mean; bars, SE. *P<0.05; **P<0.01; ***P<0.001.
CDKN3 facilitated G1/S transition
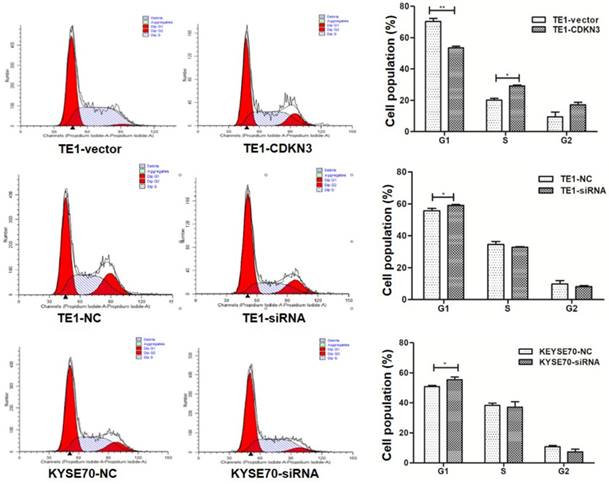
To detect the influence of CDKN3 on cell cycle, flow cytometer was conducted and the results revealed that G1 phase cell percentage in CDKN3 overexpression TE1 cells was less than the vector group (P<0.01) and the S phase cell percentage changed in the opposite (P<0.05). After the CDKN3 expression was disturbed by siRNA, G1 phase cell percentage was increased in both TE1 and KYSE70 cells(P<0.05) (Fig. 4). These outcomes identified that CDKN3 could accelerate the G1/S transition in both TE1 and KYSE70 cell lines.
CDKN3 facilitated G1/S transition. Cell cycle was measured by flow cytometer. In TE1 cell line, CDKN3 overexpression decreased the G1 phase cell percentage (up). When transfected with CDKN3 special siRNA, G1 phase cell percentage was increased in both TE1 (middle) and KYSE70 cell lines (down). Columns, mean; bars, SE. *P<0.05; **P<0.01; ***P<0.001
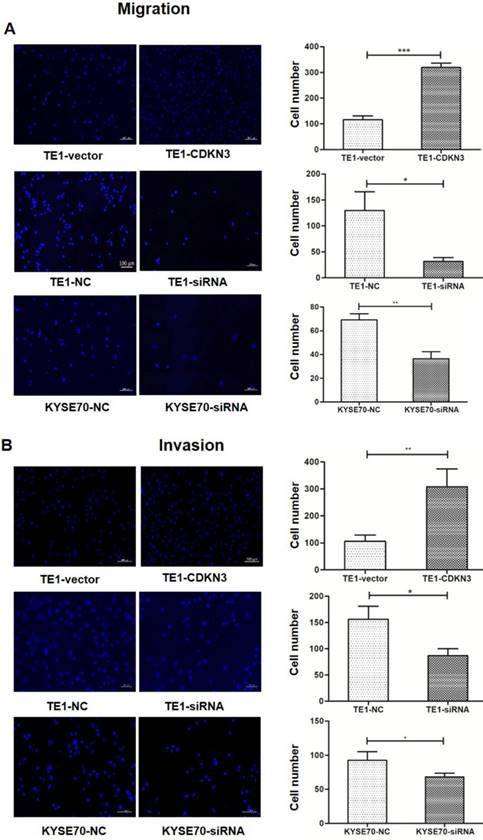
CDKN3 promoted ESCC cell migration and invasion
To investigate whether CKDN3 influence the migration and invasion of ESCC cells, transwell cell assays with or without Matrigel was performed. The result showed that both migration and invasion were increased in TE1 cell with overexpressed CDKN3 than control. Conversely, downregulated CDKN3 inhibit cell migration and invasion in both TE1 and KYSE70 cells (Fig 5).
CDKN3 increased ESCC cell migration and invasion. (A) Migration ability was evaluated by Transwell cell assay without Matrigel. Fluorescence microscope result showed that migration was enhanced in CDKN3 overexpressed TE1 cells (up) and was downregulated in TE1 (middle) and KYSE70 (down) cells which were disturbed by CDKN3 siRNA. (B) Invasion ability was evaluated by Transwell cell assay without Matrigel. Fluorescence microscope result showed that invasion was upregulated in CDKN3 overexpressed TE1 cells (up) and was downregulated in TE1 (middle) and KYSE70 (down) cells which were disturbed by CDKN3 siRNA. Columns, mean; bars, SE. *P<0.05; **P<0.01; ***P<0.001
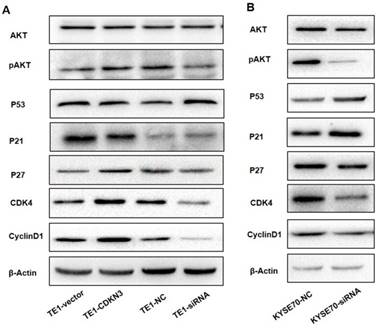
CDKN3 may regulate cell cycle by working on CyclinD-CDK4 complex via AKT-p53-p21 axis and p27 independent of AKT
AKT signal pathway played a key role in the regulation of cell cycle[18], so that we detected the AKT-related protein expression to investigate whether CDKN3 accelerate G1/S transition through this pathway. The result demonstrated that the total AKT protein level was stable, but pAKT level was upregulated when CDKN3 was overexpressed (Fig 6A). Overexpression of CDKN3 in TE1 cells decreased the expression of P53 and P21 which acted as a tumor suppressor, but increased CyclinD1, CDK4, and p27 protein level. Consistently, in CDKN3 siRNA transfected TE1 cells, all the detected protein level changed in the reverse direction (Fig 6. A). The variation tendency of these proteins in siRNA transfected KYSE70 cells was in keeping with TE1 cells (Fig 6.B).
CDKN3 regulate cell cycle associated protein expression which was included in AKT signal pathway. Western blot showed the expression level of AKT, pAKT, P53, P21, P27, CyclinD1 and CDK4. (A) Protein expression level in TE1 cell line, included both CDKN3 overexpressed cells and disturbed cells. (B) Protein expression level in CDKN3 special siRNA transfected KYSE70 cell line.
Discussion
Cancer is characterized by abnormal proliferation which was mainly due to the aberrant cell cycle activity, so that proteins associated with cell cycle are studied frequently. CDKN3, as a kind of dual specificity phosphatase, participated in the regulation of cell cycle and had been studied in several types of cancer except ESCC. In this study, we found that CDKN3 expression was upregulated in ESCC tissues. Further biological function research in two different ESCC cell lines indicated that overexpression of CDKN3 promoted cell proliferation, migration, and invasion. However, after its expression was disturbed by special siRNA, the tumor activity was suppressed. Thus, we concluded that CDKN3 acted as an oncogene in ESCC.
Several studies showed that CDKN3 acted as a tumor suppressor [8-11], others also presented that CDKN3 promoted cancer cell proliferation [12-15, 19-23], so the exact mechanism of CDKN3 in cancer was still unclear. Evidence supported CDKN3 as tumor suppressor gene was that with the progression of cancer, wild-type CDKN3 expression decreased and aberrant splicing transcription increased[24] which owned a negative dominant and inhibited the dephosphorylation process of CDK2 completed by wild CDKN3 and then forced the G1/S transition[8, 24]. However, when we overexpressed the complete coding sequence of CDKN3 exogenously in this study, the proliferation ability was enhanced in ESCC cell lines, so the tumorigenesis role of CDKN3 in esophageal cancer cannot be explained by aberrant splicing transcription theory. For CDKN3 was a cell cycle associated gene and do accelerate G1/S transition in this study, we suggested that it promoted cell proliferation by targeting other cell cycle related proteins.
CyclinD1-CDK4 complex played a key role in driving cell cycle from G1 to S phase[25]. It has been proved that CyclinD1-CDK4 complex participated in tumor cell proliferation and could be inhibited by p21 and p27[26, 27]. Latest research showed that CDKN3 facilitates the cell cycle progression by interacting with MdM2-P53 and then inhibiting the function of p21 in pancreatic ductal adenocarcinoma (PDAC)[28]. Studies in prostate and ovarian cancer revealed that the function of CDKN3 in accelerating cell cycle was associated with proliferating cell nuclear antigen (PCNA) [12, 22], which was required to stabilize the inhibitory function of p21 within CyclinD-CDK4 complex[29]. So, we speculated that the CyclinD1-CDK4 complex was furtherly affected by CDKN3 via AKT signal pathway. Here we detected those abovementioned proteins by Western blot and the result showed that pAKT, CyclinD1, and CDK4 protein level were all upregulated when CDKN3 was overexpressed and p21and p53 altered in the opposite direction. Thus we concluded that CDKN3 could work on the CyclinD1-CDK4 complex through the pAKT-p53-p21 axis, but further study was needed to explore the direct substrate of CDKN3.
However, p27 expression was positively correlated with CDKN3 in our experiment and was not consistent with the previous result in nasopharyngeal carcinoma which demonstrated CDKN3 may act as an oncogene by suppressing p27[23]. p27 was necessary for maintaining the stable formation of CyclinD1-CDK4 dimer, but it played different roles in different conformation. Phosphorylated p27 enabled the CyclinD1-CDK4 to phosphorylate downstream substrates and accelerated cell cycle, while non-phosphorylated p27 inhibited the function of the CyclinD1-CDK4 dimer[30, 31]. Thus we suggested that CDKN3 would affect the function of the CyclinD1-CDK4 complex by increasing the expression of active p27 independent of AKT. Additional work should be done to confirm the hypothesis and find the exact mechanism.
In this study, we preliminarily documented that CDKN3 expression was upregulated in ESCC tissues compared with normal esophageal tissue, and higher CDKN3 level suggested a worse prognosis. However, considering the limited case number of the study, current conclusion and the potential role of CDKN3 in the ESCC diagnosis and prognosis should be further validated by an independent cohort in our future work.
In conclusion, our data demonstrated that CDKN3 acted as an oncogene in ESCC, and could increase cell proliferation by accelerating the G1/S transition. CDKN3 could drive cell cycle by affecting CyclinD-CDK4 complex via regulating AKT-p53-p21 axis and p27 independent of AKT.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 81302160 and 81272447).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL. et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
2. Tran GD, Sun XD, Abnet CC. et al. Prospective study of risk factors for esophageal and gastric cancers in the Linxian general population trial cohort in China. Int J Cancer. 2005;113:456-63
3. Chen W, Zheng R, Baade PD. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
4. Maak S, Jaesert S, Neumann K. et al. Rapid communication: nucleotide sequence and physical mapping of the porcine cyclin-dependent kinase inhibitor 3 (CDKN3) gene. J Anim Sci. 2002;80:1698-9
5. Hannon GJ, Casso D, Beach D. KAP: A dual specificity phosphatase that interacts with cyclin-dependent kinases. Proc Natl Acad Sci U S A. 1994;91:1731-5
6. Nalepa G, Barnholtz-Sloan J, Enzor R. et al. The tumor suppressor CDKN3 controls mitosis. J Cell Biol. 2013;201:997-1012
7. Srinivas V, Kitagawa M, Wong J. et al. The tumor suppressor CDKN3 is required for maintaining the proper number of centrosomes by regulating the centrosomal stability of Mps1. Cell Rep. 2015;13:1569-77
8. Yu Y, Jiang X, Schoch BS. et al. Aberrant splicing of cyclin-dependent kinase-associated protein phosphatase KAP increases proliferation and migration in glioblastoma. Cancer Res. 2007;67:130-8
9. Dai W, Miao H, Fang S. et al. CDKN3 expression is negatively associated with pathological tumor stage and CDKN3 inhibition promotes cell survival in hepatocellular carcinoma. Mol Med Rep. 2016;14:1509-14
10. Chen Q, Chen K, Guo G. et al. A critical role of CDKN3 in Bcr-Abl-mediated tumorigenesis. PLoS One. 2014;9:e111611
11. Li H, Jiang X, Yu Y. et al. KAP regulates ROCK2 and Cdk2 in an RNA-activated glioblastoma invasion pathway. Oncogene. 2015;34:1432-41
12. Zhang LP, Li WJ, Zhu YF. et al. CDKN3 knockdown reduces cell proliferation, invasion and promotes apoptosis in human ovarian cancer. Int J Clin Exp Pathol. 2015;8:4535-44
13. Barrón EV, Roman-Bassaure E, Sánchez-Sandoval AL. et al. CDKN3 mRNA as a biomarker for survival and therapeutic target in cervical cancer. Plos One. 2015;10:e0137397
14. Yang C, Sun JJ. Mechanistic studies of cyclin-dependent kinase inhibitor 3 (CDKN3) in colorectal cancer. Asian Pac J Cancer Prev. 2015;16:965-70
15. Deng M, Wang J, Chen Y. et al. Silencing cyclin-dependent kinase inhibitor 3 inhibits the migration of breast cancer cell lines. Mol Med Rep. 2016;14:1523-30
16. Zang X, Chen M, Zhou Y. et al. Identifying CDKN3 gene expression as a prognostic biomarker in lung adenocarcinoma via meta-analysis. Cancer Inform. 2015;14:183-91
17. Su P, Wen S, Zhang Y. et al. Identification of the Key Genes and Pathways in Esophageal Carcinoma. Gastroenterol Res Pract. 2016;2016:2968106
18. Arcaro A, Guerreiro AS. The Phosphoinositide 3-Kinase Pathway in Human Cancer: Genetic Alterations and Therapeutic Implications. Current Genomics. 2007;8:271-306
19. Li Y, Ji S, Fu LY. et al. Knockdown of cyclin-dependent kinase inhibitor 3 inhibits proliferation and invasion in human gastric cancer cells. Oncol Res. 2017;25:721-31
20. Lai MW, Chen TC, Pang ST. et al. Overexpression of cyclin-dependent kinase-associated protein phosphatase enhances cell proliferation in renal cancer cells. Urol Oncol. 2012;30:871-8
21. Xing C, Xie H, Zhou L. et al. Cyclin-dependent kinase inhibitor 3 is overexpressed in hepatocellular carcinoma and promotes tumor cell proliferation. Biochem Biophys Res Commun. 2012;420:29-35
22. Yu C, Cao H, He X. et al. Cyclin-dependent kinase inhibitor 3 (CDKN3) plays a critical role in prostate cancer via regulating cell cycle and DNA replication signaling. Biomed Pharmacother. 2017;96:1109-18
23. Wang H, Chen H, Zhou H. et al. Cyclin-Dependent Kinase Inhibitor 3 Promotes Cancer Cell Proliferation and Tumorigenesis in Nasopharyngeal Carcinoma by Targeting p27. Oncol Res. 2017;25:1431-40
24. Yeh CT, Lu SC, Chen TC. et al. Aberrant transcripts of the cyclin-dependent kinase-associated protein phosphatase in hepatocellular carcinoma. Cancer Res. 2000;60:4697-700
25. Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17:93-115
26. Ye D, Luo H, Lai Z. et al. ClC-3 Chloride Channel Proteins Regulate the Cell Cycle by Up-regulating cyclin D1-CDK4/6 through Suppressing p21/p27 Expression in Nasopharyngeal Carcinoma Cells. Sci Rep. 2016;6:30276
27. Patel P, Tsiperson V, Gottesman SRS. et al. Dual Inhibition of CDK4 and CDK2 via Targeting p27 Tyrosine Phosphorylation Induces a Potent and Durable Response in Breast Cancer Cells. Mol Cancer Res. 2018;16:361-77
28. Liu D, Zhang J, Wu Y. et al. YY1 suppresses proliferation and migration of pancreatic ductal adenocarcinoma by regulating the CDKN3/MdM2/P53/P21 signaling pathway. Int J Cancer. 2018;142:1392-404
29. He G, Kuang J, Koomen J. et al. Recruitment of trimeric proliferating cell nuclear antigen by G1-phase cyclin-dependent kinases following DNA damage with platinum-based antitumour agents. Br J Cancer. 2013;109:2378-88
30. James MK, Ray A, Leznova D. et al. Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol Cell Biol. 2008;28:498-510
31. Ray A, James MK, Larochelle S. et al. p27Kip1 inhibits cyclin D-cyclin-dependent kinase 4 by two independent modes. Mol Cell Biol. 2009;29:986-99
Author contact
![]() Corresponding authors: Prof. Shutian Zhang, MD, PhD: Department of Gastroenterology, Beijing Friendship Hospital, Capital Medical University, National Clinical Research Center for Digestive Disease, Beijing Digestive Disease Center, Beijing Key Laboratory for Precancerous Lesion of Digestive Disease, Beijing, 100050, P. R. China, E-mail: zhangshutianedu.cn and Changqin Xu, MD: Shandong Provincial Hospital affiliated to Shandong university. E-mail: 64791275com.
Corresponding authors: Prof. Shutian Zhang, MD, PhD: Department of Gastroenterology, Beijing Friendship Hospital, Capital Medical University, National Clinical Research Center for Digestive Disease, Beijing Digestive Disease Center, Beijing Key Laboratory for Precancerous Lesion of Digestive Disease, Beijing, 100050, P. R. China, E-mail: zhangshutianedu.cn and Changqin Xu, MD: Shandong Provincial Hospital affiliated to Shandong university. E-mail: 64791275com.