Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(9):2063-2073. doi:10.7150/jca.29327 This issue Cite
Research Paper
Simultaneous Inhibition of EGFR and HER2 via Afatinib Augments the Radiosensitivity of Nasopharyngeal Carcinoma Cells
Fangling Huang1,2, Xujun Liang1, Xiaoli Min1, Ye Zhang1, Guoqiang Wang1, Zhengrong Peng2, Fang Peng1, Maoyu Li1, Lin Chen1, Yongheng Chen1 ![]()
1. Key Laboratory of Cancer Proteomics of Chinese Ministry of Health, XiangYa Hospital, Central South University, Changsha, Hunan 410008, China
2. Department of Hyperbaric Oxygen, Xiangya Hospital, Central South University, Changsha 410008, China
Received 2018-8-18; Accepted 2019-4-13; Published 2019-5-12
Abstract
Ionizing radiation (IR) is the central component of the therapeutic scheme for nasopharyngeal carcinoma (NPC) at present. Previous studies show that inhibition of epidermal growth factor receptor (EGFR) enhances the radiosensitivity of NPC; however the effects of EGFR-targeted agents are limited. In this study, we observed that simultaneously inhibition of EGFR and HER2 by afatinib could augment the radiosensitivity of NPC cells; this approach has an advantage over erlotinib-mediated inhibition of EGFR alone. The afatinib-induced augmentation of NPC cell radiosensitivity was associated with increases in apoptosis and accumulation of DNA damage that were induced by radiation. In addition, the crosstalk between radiation-induced activities and EGFR-, and HER2-related downstream pathways may contribute to the enhancement of radiosensitivity. Our findings indicate the potential of repositioning afatinib or other ERBB-family-targeted agents for improving radiation response in NPC cells.
Keywords: nasopharyngeal carcinoma, EGFR, HER2, radiosensitivity
Introduction
Currently, ionizing radiation (IR) is the foundation and central component of curative treatment regimens for nasopharyngeal carcinoma (NPC). Improving radiosensitivity is crucial for the treatment of locally advanced NPC. Epidermal growth factor receptor (EGFR), an important member of the ERBB family of receptors, is expressed at elevated levels in most malignancies of epithelial origin. EGFR has diverse roles in promoting cellular proliferation, mediating differentiation and protecting cells from apoptosis [1, 2]. Increasing evidence supports the concept that EGFR is also correlated with tumour responses to ionizing radiation (IR) [3-5]. Overexpression and activation of EGFR frequently result in tumour resistance to radiation and poor prognosis [6]. Several therapies have been investigated in the past decade that indicate that EGFR inhibition treatment could modulate cellular resistance to radiation and enhance tumour response to IR in vitro and in vivo [7-9].
Although investigators have confirmed the efficacy of EGFR monoclonal antibodies (cetuximab and nimotuzumab) and EGFR tyrosine kinase inhibitors (TKIs) in optimizing radiotherapy effect in preclinical and clinical research [10-12], definitive benefit in local control and survival remains unsatisfied for a fraction of patients [11]. Therefore, rational integration of molecular targeted agents with radiotherapy is still needed for improved treatment of NPC.
Recent studies have identified HER2 activation as a potential mechanism compromising the effectiveness of radiation [13, 14]. HER2, as another member of the ERBB family of receptors without a known ligand, could heterodimerize with EGFR and transduce the subsequent signalling pathway. Meanwhile, interreceptor kinase activation and transmodulation of EGFR and HER2 are observed as the results of heterodimerization [15, 16]. Moreover, HER2 amplification, overexpression and heterodimerization obviate the need for activated EGFR to initiate downstream signal pathways [17-19].
For the purpose of seeking more effective and rational approaches to drug radiotherapy combinations, we hypothesized that excessive activation of HER2 suppresses the ability of anti-EGFR therapies to regulate the biological effect of radiation in NPC cells. Afatinib is a novel TKI for both EGFR and HER2. The traditional EGFR TKIs, like erlotinib, bind in a reversible fashion to ATP banding site of EGFR. However, afatinib, a second generation of EGFR TKIs, combines with EGFR, HER2 and other ERBB family members via covalent bond, which irreversibly and more potently inhibit ERBB signalling pathways. In 2013, afatinib was approved by the U.S. Food and Drug Administration as a new first-line treatment for patients with metastatic non-small lung cancer. With the goal of further identifying mechanisms of radiation resistance in NPC, we performed a detailed analysis of the synergistic effect between radiation and pharmacological inhibition of both EGFR and HER2 by afatinib. Our data showed that pharmacological inhibition of both EGFR and HER2 by afatinib exhibited more improved radiosensitizing activity than inhibition of EGFR alone by erlotinib. Mechanisms of radiosensitizing activity of afatinib were associated with the potent suppression of radiation-induced activation of ERBB-family-related signalling pathways to regulate proliferation, apoptosis and radiation-induced DNA repair. Our results support the use of afatinib as a reliable radiation sensitizer and, provide the direction for optimizing the radiation response of advanced NPC.
Materials and methods
Cells culture
Human NPC cells (5-8F, 6-10B, HNE2 and HNE3) were cultured in RPMI-1640 (GIBCO) supplemented with 10% foetal bovine serum (GIBCO), and penicillin-streptomycin (GIBCO) to final concentrations of 100 U/ml and 100 μg/ml, respectively. An immortalized nasopharyngeal epithelial cell line, NP69, was cultured in Keratinocyte-SFM medium (GIBCO) with 0.2 μg/ml EGF (GIBCO) and 30 μg/ml Bovine Pituitary Extract (GIBCO). All cell lines were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air, as we have previously reported [20].
Chemical reagents and antibodies
Afatinib and erlotinib were purchased from Selleck Chemicals. For in vitro experiments, afatinib and erlotinib stock solutions were prepared in DMSO and both compounds were diluted in culture medium before dosing. The following antibodies were obtained from Cell Signaling Technology: phospho-Erk1/2 Thr202/Tyr204 (#4370, 1:2000 dilution), phospho-Akt Ser473 (#4060, 1:2000 dilution), Erk1/2 (#4695, 1:1000 dilution), Akt (#4691, 1:1000 dilution), cleaved PARP Asp214 (#5625, 1:1000 dilution). The following antibodies were purchased from Abcam: phosphor-EGFR Tyr1068 (#ab32430, 1:5000 dilution), phospho-HER2 Tyr1139 (#ab53290, 1:1000 dilution), EGFR (#ab52849, 1:5000 dilution), HER2 (#ab134182, 1:10000 dilution), ϒH2AX phospho-Ser139 (#ab26350, 1:1000 dilution). The Caspase 3 (#19677-1-AP, 1:1000 dilution) antibody and Beta actin (#60008-1-Ig, 1:10000 dilution) antibody were purchased from Proteintech.
Cell antiproliferation assay
The human nasopharyngeal carcinoma cells were seeded at 2000 cells/well in 96-well plates. After overnight incubation, the cells were treated with afatinib or erlotinib (0, 0.625, 1.25, 2.5, 5, 7.5, or 10 μM). After 72 hours of treatment, growth inhibition was assessed through the MTT assay, which was performed according to previously established methods [21]. The absorbance of the converted dye was measured at 570 nm using a microplate reader (Bio-Tek ELX800, USA). All experiments were repeated at least three times. The curves were fitted using a non-linear regression model with a sigmoidal dose response.
Clonogenic assays
5-8F and HNE2 human nasopharyngeal carcinoma cells were cultured in 6-well plates and were exposed to treatment the following day. The cells were treated with different doses (0Gy, 2Gy, 4Gy, 6Gy, and 8Gy) of radiation following a 1-hour pretreatment with afatinib, erlotinib or vehicle on day 1. After culturing for 10 days, the cells were fixed in ice-cold methanol and were strained with crystal violet. Colonies with more than 50 cells were counted. At each concentration, the surviving fraction was determined by dividing the total number of colonies after irradiation by the number of colonies without irradiation.
Determination of apoptosis with flow cytometry
5-8F cells and HNE2 cells were pretreated for half an hour with vehicle or 2 μM afatinib before irradiation (4Gy) and were incubated for 48 hours. Next, the apoptotic cells were analysed via flow cytometry using the Annexin V-FITC Detection Kit I (BD Pharmingen, San Diego, CA) according to the manufacturer's instructions. Flow cytometry data were plotted and analysed via the fluorescence activated cell-sorting (FACS-Vantage) system using the Cell quest software (Becton-Dickinson, San Jose, CA) within 1 hour of staining. The percentages of differently labelled cells were calculated using the FlowJo software (Tree Star).
Ionizing irradiation treatment of cells
5-8F cells and HNE2 cells in flask or plates were exposed to X-rays (2-8 Gy, 4Gy per minute), using a 6-MV photon beam from a linear accelerator (Varian Medical Systems, Inc) at the Department of Clinical Oncology of Xiangya Hospital (Changsha, China). The plates were covered with a tissue-equivalent gel to promote dose build-up.
Immunofluorescence
Cells were seeded on a glass slide, incubated overnight and pretreated with either vehicle or 2 μM EGFR- tyrosine kinase inhibitors (TKIs), afatinib or erlotinib, half an hour before IR (4 Gy). The number of ϒ-H2AX foci was determined at half an hour after IR. The cells were treated as described previously [22] and incubated with a primary antibody against γ-H2AX (Abcam). Next, the primary antibody was washed off, and a secondary antibody conjugated to FITC was applied to the slides. DNA damage was visualized using laser scanning confocal microscopy (Leica, SP8). For each group, the ϒ-H2AX foci were counted in at least 50 cells. The captured images were processed using Image-Pro Plus software (Media Cybernetics, Rockville, Maryland, USA) and were displayed using Adobe Photoshop CS6.
Western blot analysis
Cells were lysed in lysis buffer (50 mM Tris pH = 6.8, 2% SDS, 10% glycerol, 5% β-mercaptoethanol and 1% protease inhibitor cocktail, with or without 1% phosphatase inhibitor cocktail). An equal amount of protein from each cell extract was separated via SDS-PAGE gel (8-12% polyacrylamide), and the separated proteins were transferred to PVDF membranes and immunoblotted with various antibodies, as we have previously reported [23]. The proteins of interest were visualized via enhanced chemiluminescence.
Data presentation and statistical analysis
Numerical results were expressed as the means ± standard deviation and were graphically displayed using GraphPad Prism version 7.0 for Windows, (GraphPad Software; www.graphpad.com). Each figure represents results from three independent experiments. Statistical significance was evaluated using one-way ANOVA for normally distributed and homoscedasic data or Kruskal-Wallis H tests for data which is non-normally distributed or with different variances. P values less than 0.05 compared with the control were considered statistically significant.
Result
Afatinib inhibits NPC cell proliferation by blocking EGFR and HER2 signalling
Since phosphorylations activate the EGFR and HER2 signal pathways, we first examined the phosphorylation levels and the total expression of EGFR and HER2 in four NPC cell lines (5-8F, 6-10B, HNE2, and HNE3) and an immortalized nasopharyngeal epithelial cell line (NP69). Western blotting showed that the expression and phosphorylation levels of EGFR and HER2 vary in all these cell lines (Fig. 1A). Compared to immortalized nasopharyngeal epithelial cell line NP69, HNE2 cells exhibited equivalent expression of phospho-EGFR, but only weak expression (p=0.000) of phospho-HER2 (Fig. 1B). However, the expression levels of phosphorylated EGFR and HER2 in 5-8F cells are contrast to those in HNE2 cells. We chose 5-8F cells and HNE2 cells in the following experiments to investigate the effect of afatinib in NPC cells.
Afatinib block EGFR and HER2 signalling to inhibit NPC cell proliferation. (A) Lysates of the immortalized nasopharyngeal epithelial cell line NP69 and 4 different cell lines (5-8F, 6-10B, HNE3, HNE2) were analysed via immunoblotting to detect phosphorylated EGFR (p- EGFR), total EGFR, phosphorylated HER2 (p-HER2), total HER2 and ACTB. (B) Phosphorylated protein levels were quantified by densitometry and normalized by total protein levels. Compared with the control group. *P< 0.05, ** P< 0.01, *** P< 0.001. (C) Lysates of 5-8F and HNE2 were treated for 30minutes with increasing concentrations of the EGFR TKI afatinib (0, 0.125, 0.25, 0.5, 1 and 2μM) and were analysed via immunoblotting to detect p-EGFR, total EGFR, p-HER2, total HER2, p-Erk1/2, total Erk1/2, p-Akt, total Akt and ACTB. D. Viability curve describing the viability of 5-8F and HNE2 NPC cells that were treated with vehicle or the indicated concentrations of afatinib or erlotinib for 72 hours. Each data point represents the average value from 3 samples, and is expressed as a percentage of surviving cells relative to untreated controls. Compared with the control group. *P< 0.05.
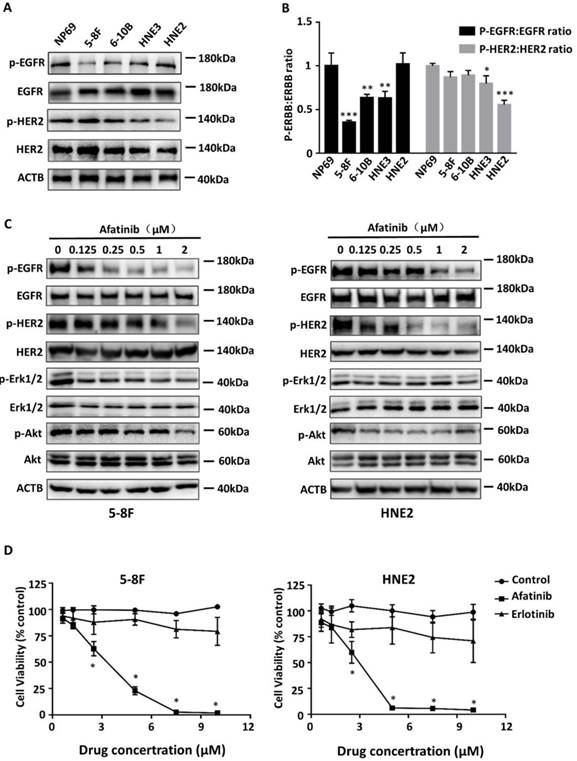
To address whether afatinib is a potent inhibitor, we evaluated its ability to reduce autophosphorylation of EGFR, HER2 and their downstream signalling properties (Erk1/2 and Akt) in 5-8F cells and HNE2 cells without ligand stimulation (Fig. 1C). The expression levels of phospho-EGFR, phospho-HER2, phospho-Erk1/2 and phospho-Akt were generally reduced by afatinib in a dose-dependent manner (Supplemental Fig. 1). Furthermore, to determine whether the inhibition of both EGFR and HER2 phosphorylation could more effectively inhibit cell proliferation, we treated 5-8F cells and HNE2 cells with different doses of afatinib or with a standard EGRF-TKI (erlotinib). Consistent with our signalling data, proliferation of NPC cells declined sharply with increasing dose of afatinib (P=0.034 and p=0.022 in 5-8F and HNE2 cell lines with 2.5μM afatinib, respectively. Fig. 1D). Compared to erlotinib, afatinib had a much better capacity to decrease the viability of NPC cells, with an IC50 of approximately 5 μM in both NPC cell lines. These results demonstrate that afatinib effectively inhibit proliferation of NPC cells by blocking the EGFR and HER2 signalling pathways.
Simultaneous blockade of EGFR and HER2 tyrosine kinase activities significantly radiosensitizes nasopharyngeal carcinoma cells
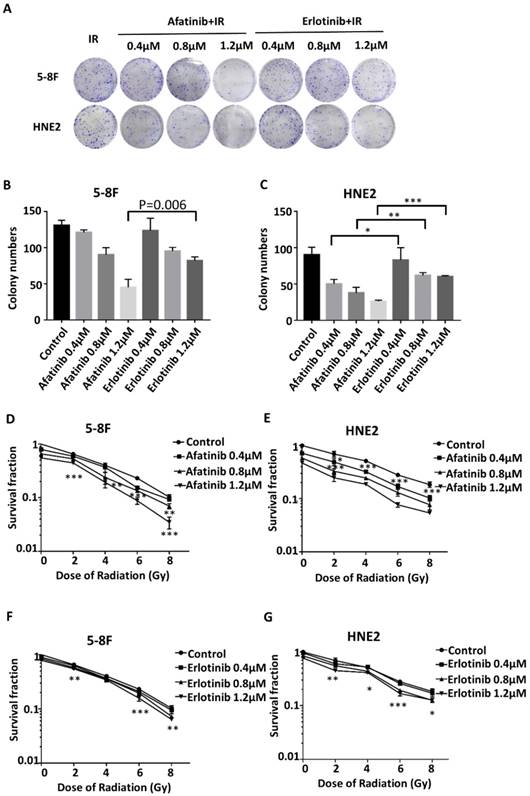
To substantiate the statement that combined inhibition of EGFR and HER2 tyrosine kinase could increase the radiosensitivity of NPC cells; the clonogenic survival assay was performed to estimate the colony formation ability of 5-8F and HNE2 NPC cells after treatment with 0-8 Gy irradiation with or without afatinib compared with that with erlotinib. As described in Fig. 2A, the colony formation assay demonstrated that afatinib and erlotinib dose-dependently reduced the survival fraction of 5-8F cells and HNE2 cells following 8 Gy irradiation. Moreover, afatinib was more successful in reducing the NPC colony numbers than erlotinib at the same drug dose; the difference became obvious and significant with the increasing dose (P=0.006 for 1.2μM drugs in 5-8F cells and p=0.013, p= 0.008 and p=0.000 for 0.4 μM, 0.8 μM and 1.2μM drugs in HNE2 cells, respectively.) of afatinib and erlotinib (Fig. 2B and 2C). We further described the potent inhibition ability of afatinib against colony formation with different doses of radiation (Fig. 2D-2G). As shown in Fig. 2F and 2G, the clonogenic survival rate of 5-8F cells and HNE2 cells were mildly reduced with the addition of erlotinib. However, afatinib sharply reduced the clonogenic survival rate with increase in drug dose (Fig. 2D and 2E). Our data strongly suggests that afatinib is a satisfying radiosensitizing agent for NPC cells. Moreover, the ability of increasing radiosensitivity of afatinib is equal in 5-8F cells and HNE2 cells, indicating that afatinib could radiosensitize NPC cells regardless of the expression levels of phosphorylated EGFR and HER2 in NPC cells before irradiation.
Afatinib significantly radiosensitizes nasopharyngeal carcinoma cells. (A) 5-8F and HNE2 cells were seeded at 2000 cells/plate, and were treated with either IR at 8Gy alone or the combination of 0.4μM, 0.8μM, or 1.2μM of afatinib or erlotinib with IR at 8Gy. Following treatment, the plates were fixed and stained with crystal violet. All experiments were performed in triplicate. The representative plates are shown. (B and C) Colony numbers of 5-8F (B) and HNE2 (C) cells were counted following the treatment described in Fig.2A. Data from three independent experiments was averaged. (bar graph). *p<0.05, **p<0.01, ***p<0.001. D-G. 5-8F (D, F) and HNE2 (E,G) cells either treated with IR (2Gy, 4 Gy, 6 Gy, or 8 Gy) alone or the combination of 0.4μM, 0.8μM, and 1.2μM of afatinib (D, E) or erlotinib (F, G) with IR. Colony- forming efficiency was determined, and survival curves were generated approximated 10 days later. Data from three independent experiments was averaged. *p<0.05, **p<0.01, ***p<0.001.
Combined EGFR and HER2 tyrosine kinase inhibition promotes radiation-induced apoptosis
To further characterize the availability of afatinib as a radiation sensitizer in NPC cells, we examined its pro-apoptosis effect in 5-8F cells and HNE2 cells via flow cytometry using Annexin V/propidium iodide (PI) staining (Fig. 3A and 3B). The population of Annexin-V-positive cells was significantly increased (P=0.047 and p=0.000 in 5-8F and HNE2 cell lines, respectively.) after pretreatment with afatinib half an hour before IR in both NPC cell lines, especially in the HNE2 cell line (Fig. 3C and 3D). Furthermore, the increase in Annexin-V-positive cells treated with IR alone mainly counted on the contribution of early apoptotic cells (Annexin-V-FITC-positive and PI-negative), whereas afatinib equally promoted IR-induced early apoptosis and advanced apoptosis (Annexin-V-FITC-positive and PI-positive). These results show that afatinib is capable of promoting radiation-induced apoptosis.
Afatinib promotes the pro-apoptosis ability of IR. (A and B) Flow cytometry was performed to analysis cell apoptosis of 5-8F cells (A) and HNE2 cells (B). Cells were labelled with Annexin V conjugated to FITC and propidium iodide (PI) 48 hours after IR at 4Gy or control treatment, with or without addition of 2μM afatinib as indicated. (C and D) Percent of apoptosis cells in 5-8F cells (C) and HNE2 cells (D) that were treated as described in Figure 3A-B were quantified following flow cytometry. (E) 5-8F cells and HNE2 cells were treated with IR at 4Gy and 2μM afatinib for half an hour before IR as indicated. Lysates from 5-8F and HNE2 cells were analysed via immunoblotting to detect caspase-3, cleaved caspase-3 and ACTB 72 hours later. (F) Lysates from 5-8F and HNE2 cells that were treated as described in Figure 3E were analysed via immunoblotting to detect cleaved PARP and ACTB. (G) Expression levels of caspase-3 (upper panel), Cleaved Caspase-3 (middle panel) and Cleaved PARP (bottom panel) were quantified by densitometry and normalized by ACTB levels.
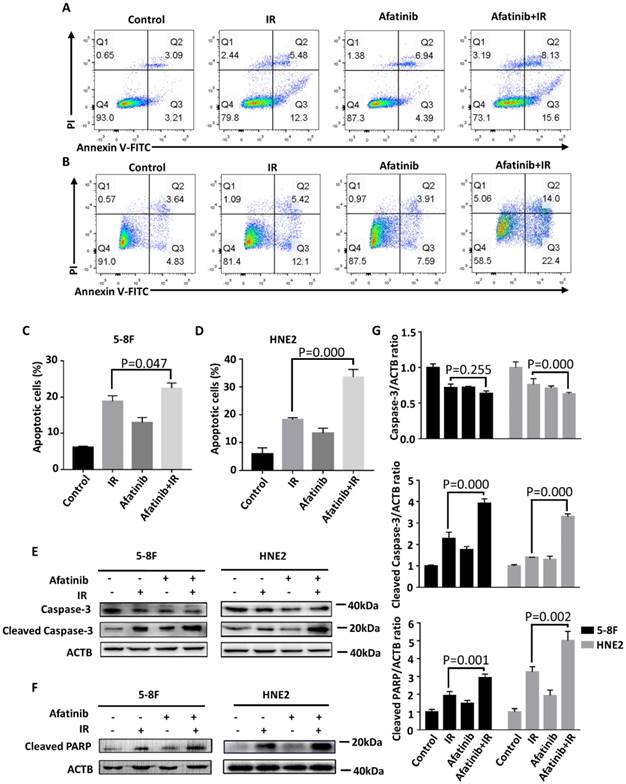
We further assayed the expression levels of apoptosis-related proteins at 72 hours after treatment with IR and afatinib. In line with the flow cytometry results, pretreatment with afatinib significantly increased radiation-induced cleaved caspase-3 (P=0.000 in both 5-8F and HNE2 cell lines. Fig. 3E and 3G) and poly ADP-ribose polymerase (PARP) (P=0.001 and p=0.002 in 5-8F and HNE2 cell lines, respectively. Fig. 3F and 3G). Caspase-3 preforms different degrees of reduction owing to increasing of cleaved caspase-3 in 5-8F cells and HNE2 cells. Caspase-3 plays a crucial role in the execution-phase of cell apoptosis and is activated through both extrinsic and intrinsic pathways [24]. PARP, the most important substrate of caspase-3, is the symbol of caspase-3 activation. These data indicate that the promotion of radiation-induced caspase-dependent apoptosis is one of reasons that afatinib radiosensitizes NPC cells.
Combined EGFR and HER2 tyrosine kinase inhibition enhances radiation-induced DNA damage
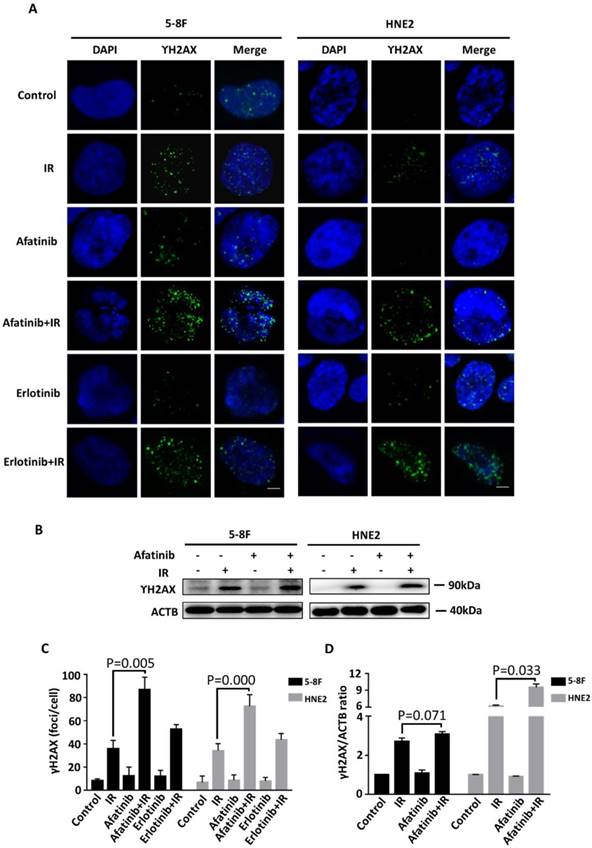
Effective DNA damage, especially unrepaired DNA double strand break (DSB) is the main mechanism that underlies the elimination of carcinoma cells via irradiation. Next, we investigated whether afatinib modulated the level of phosphorylated histone H2AX (γ-H2AX), which formed foci at DNA double-strand break areas. We first assessed the quantity of γ-H2AX using the immunofluorescence technique. As shown in Fig.4A, treatment with afatinib or erlotinib alone exhibited little effect on the γ-H2AX foci. However irradiation led to a significant increase in the quantity of γ-H2AX foci. Half an hour of pretreatment with afatinib before irradiation made further effort (P=0.005 and p=0.000 in 5-8F and HNE2 cell lines, respectively.) on DNA damage on the basis of irradiation in both 5-8F and HNE2 cells (Fig. 4C) whereas erlotinib had less effect on it. Our data certify that, compared with EGFR alone, combined EGFR and HER2 tyrosine kinase inhibition enhances radiation-induced DNA damage to improve radiation response. Next, we examined the protein expression level of γ-H2AX in both 5-8F and HNE2 cells. In accordance with immunofluorescence results, afatinib augmented the DNA damage triggered by IR (P=0.071 and p=0.033 in 5-8F and HNE2 cell lines, respectively. Fig. 4B and 4D) especially in HNE2 cells. Because DNA damage in cells treated with afatinib or erlotinib alone was considerably less than that in cells treated with IR, our results suggest that the increase in DNA damage may be due to the inhibition of DNA damage repair instead of enhancement in cytotoxicity caused by EGFR and HER2 inhibitors.
Afatinib enhances radiation-induced DNA damage. (A) Confocal microscopy was used to obtain images of 5-8F cells and HNE2 cells that were treated with IR at 4 Gy and /or 2 μM afatinib as indicated. Cells were fixed 30 minutes following the treatments, and the cell nuclei were stained with γ-H2AX (green) and DAPI (blue). The scale bar represents 5μm. (B) Lysates of 5-8F and HNE2 cells that were treated as described in Figure 4A were analysed via immunoblotting to detect γ-H2AX and ACTB. (C) γ-H2AX foci in the nuclei of 5-8F cells and HNE2 cells that were treated as described in Figure 4A were quantified. For each group, at least 50 cells were counted, and the data are expressed as the means ± standard deviation. (D) Expression levels of γ-H2AX were quantified by densitometry and normalized by ACTB levels.
Concomitant EGFR and HER2 tyrosine kinase inhibition significantly suppresses radiation-activated signalling pathways
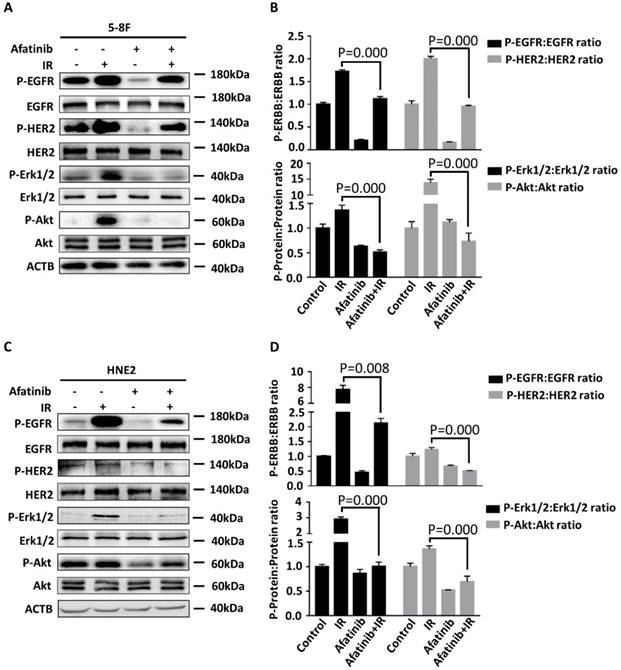
We sought to determine whether afatinib is an effective therapeutic strategy for the inhibition of IR-activated EGFR and HER2 signalling pathways. We treated 5-8F cells and HNE2 cells with the vehicle or afatinib 30 minutes before IR and assessed the effects of these treatments on the auto-phosphorylation of EGFR and HER2 and their downstream signalling properties (such as phospho-Erk 1/2, phospho-Akt). Hyperp-hosphorylation of HER2 (P=0.000 and p=0.017 in 5-8F and HNE2 cells, respectively.) was observed in both NPC cell lines following IR, which suggested that radiation induced hyperactivation of the HER2 pathway and that it may led to the failure of EGFR-targeted therapy (Fig 5A and 5C). Meanwhile, IR damage conspicuously increased the activation of EGFR, HER2, and all of their downstream signals in 5-8F and HNE2 cells as previously reported (Fig 5A and 5C) [25]. Afatinib is a potent inhibitor of IR-induced EGFR and HER2 autophosphorylation. As expected, activation of downstream Erk 1/2 and Akt signalling were both inhibited after drug treatment. We conclude that afatinib can significantly suppress EGFR, HER2 and their downstream signalling properties that were activated by IR and improve the radiosensitivity of NPC cells.
Afatinib inhibits radiation-activated EGFR and HER2 signalling. 5-8F (A) and HNE2 (C) were treated with IR at 4 Gy and /or 2 μM afatinib as indicated. Lysates of 5- 8F cells and HNE2 cells were prepared 2 hours following treatments and were analysed via immunoblotting to detect p-EGFR, p-HER2, and their downstream signalling phosphorylated proteins p-Erk1/2, p-Akt and ACTB. Phosphorylated protein levels of 5-8F (B) and HNE2 (D) were quantified by densitometry and normalized by total protein levels.
Discussion
Here, we provided compelling evidence that demonstrated that blockage of HER2 augments the radiotherapy sensitization ability of anti-EGFR therapies through suppressing tumour proliferation, improving apoptosis and increasing DNA damage induced by irradiation.
The role of EGFR in mediating the effect of radiotherapy is partly investigated. PI3K-Akt-mTOR signalling is the most important downstream pathway for EGFR to sensitize NPC cells to radiotherapy. PI3K-Akt-mTOR signalling is involved in elevating DNA-damage repair capability through the activation DNA-dependent protein kinase (DNA-PK) [26]. DNA-PK is a crucial protein kinase in DNA non-homologous end joining (NHEJ) repair and recombination for IR-induced DNA double strand breaks, and it mediates radiosensitivity [27, 28]. In addition, PI3K-Akt-mTOR signalling mediates inhibition of Nuclear factor-κB( NF-κB) kinase α phosphorylation and activation of NF-κB, in an Akt-dependent or Akt-independent manner [29, 30]. In addition to regulating the induction of inflammatory cytokines and molecules via radiation exposure [31], activated NF-κB upregulates the expression and activation of some oncogenes, such as MET, which promote IR-induced cell invasion, increase proliferation and angiogenesis and protect cells from apoptosis [32-34]. In our study, PI3K-Akt-mTOR signalling is the most dramatically inhibited downstream pathway by afatinib following IR. The results demonstrate that afatinib is a potent potential therapeutic agent to inhibit IR-induced EGFR signalling activation and enhance the toxic effect of IR.
The mechanisms for tumour resistance to anti-EGFR therapy are not completely understood; however, one reason behind this may be the upregulation, coexpression and heterodimerization of other EGFR family members, such as HER2. This is similar to the contribution of increased expression of EGFR to treatment resistance in HER-2-positive breast cancer [35]. In cells co-expressing EGFR and HER2, ligands of EGFR prefer to stimulate the formation of EGFR/HER2 heterodimers via cross-phosphorylation [15, 36]. More importantly, reciprocal activation between ligand-independent tyrosine protein kinase in c-Src and ERBB families contributes to ligand-independent transmodulation of EGFR and HER2 [16]. Transactivation of EGFR and HER2 suggests that single-target inhibitors or antibodies may be insufficient to block EGFR signal transduction.
In addition to drug efflux, populations of “cancer stem cells”, cellular metabolism [37] and reversible “drug-tolerant persisters” [38], epithelial-mesenchymal transition (EMT) may be an underlying mechanism of EGFR-TKI drug resistance [39-41]. Compared with the bulk tumour compartment of epithelial cells, mesenchymal cells tend to be more resistant to EGFR inhibitors and antibodies. Previous studies have revealed that activation of Src/focal adhesion kinase (FAK) signalling was crucial in maintaining the mesenchymal status, and it partly generated EMT-associated drug resistance [39]. HER2 promotes the expression levels of proteins associated with EMT via the activation of FAK [42]. Consequently, combination inhibition of HER2 in mesenchymal cells may be a useful strategy to overcome certain types of anti-EGFR drug resistance.
In our study, EGFR, HER2 and their vital downstream proteins were all hyperactivated following irradiation, which is tremendously abrogated by pretreatment with afatinib, regardless of their initial expression. Additionally, blocking of the EGFR and HER2 signalling pathways that were hyperactivated by irradiation was associated with improved sensitivity of NPC cells to IR, compared to blocking EGFR alone. We further demonstrated that the improvement of radiosensitivity was related to the promotion of cell apoptosis and the inhibition of DNA damage repair. Our data explain the resistant mechanism of EGFR antibodies or EGFR TKIs as radiosensitizer, and state the superiority of afatinib in augmenting radiation response of NPC cells. Moreover, our research establishes foundation for selecting treatment agents to promote the sensitivity of malignant tumours to IR. Further study will be necessary to fully elucidate the mechanisms of the effect of HER2 on the EGFR pathway and radiosensitivity in NPC.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 81372904 and 81570537) awarded to Yongheng Chen.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Herchenhorn D, Ferreira CG. Targeting epidermal growth factor receptor to optimize chemoradiotherapy in locally advanced head and neck cancer: has biology been taken into account? Journal of clinical oncology: official journal of the American Society of Clini cal Oncology. 2011;29:e283-4 author reply e5-7
2. Casalini P, Iorio MV, Galmozzi E, Menard S. Role of HER receptors family in development and differentiation. Journal of cellular physiology. 2004;200:343-50
3. Wang M, Kern AM, Hulskotter M, Greninger P, Singh A, Pan Y. et al. EGFR-mediated chromatin condensation protects KRAS-mutant cancer cells against ionizing radiation. Cancer research. 2014;74:2825-34
4. Bentzen SM, Atasoy BM, Daley FM, Dische S, Richman PI, Saunders MI. et al. Epidermal growth factor receptor expression in pretreatment biopsies from head and neck squamous cell carcinoma as a predictive factor for a benefit from accelerated radiation therapy in a randomized controlled trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2005;23:5560-7
5. El Bezawy R, Cominetti D, Fenderico N, Zuco V, Beretta GL, Dugo M. et al. miR-875-5p counteracts epithelial-to-mesenchymal transition and enhances radiation response in prostate cancer through repression of the EGFR-ZEB1 axis. Cancer letters. 2017;395:53-62
6. Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF. et al. Stem cell-related "self-renewal" signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:3015-24
7. Morris ZS, Harari PM. Interaction of radiation therapy with molecular targeted agents. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014;32:2886-93
8. Lu H, Liang K, Lu Y, Fan Z. The anti-EGFR antibody cetuximab sensitizes human head and neck squamous cell carcinoma cells to radiation in part through inhibiting radiation-induced upregulation of HIF-1α. Cancer letters. 2012;322:78-85
9. Zhao L, He L-R, Xi M, Cai M-Y, Shen J-X, Li Q-Q. et al. Nimotuzumab promotes radiosensitivity of EGFR-overexpression esophageal squamous cell carcinoma cells by upregulating IGFBP-3. Journal of translational medicine. 2012;10:249
10. You R, Hua YJ, Liu YP, Yang Q, Zhang YN, Li JB. et al. Concurrent Chemoradiotherapy with or without Anti-EGFR-Targeted Treatment for Stage II-IVb Nasopharyngeal Carcinoma: Retrospective Analysis with a Large Cohort and Long Follow-up. Theranostics. 2017;7:2314-24
11. Herchenhorn D, Ferreira CG. Targeting epidermal growth factor receptor to optimize chemoradiotherapy in locally advanced head and neck cancer: has biology been taken into account? Journal of Clinical Oncology. 2011;29:e283-e4
12. Lin M, You R, Liu Y-P, Zhang Y-N, Zhang H-J, Zou X. et al. Beneficial effects of anti-EGFR agents, Cetuximab or Nimotuzumab, in combination with concurrent chemoradiotherapy in advanced nasopharyngeal carcinoma. Oral oncology. 2018;80:1-8
13. Duru N, Fan M, Candas D, Menaa C, Liu HC, Nantajit D. et al. HER2-associated radioresistance of breast cancer stem cells isolated from HER2-negative breast cancer cells. Clinical cancer research: an official journal of the American Association for Cancer Research. 2012;18:6634-47
14. Cao N, Li S, Wang Z, Ahmed KM, Degnan ME, Fan M. et al. NF-kappaB-mediated HER2 overexpression in radiation-adaptive resistance. Radiation research. 2009;171:9-21
15. Qian X, LeVea CM, Freeman JK, Dougall WC, Greene MI. Heterodimerization of epidermal growth factor receptor and wild-type or kinase-deficient Neu: a mechanism of interreceptor kinase activation and transphosphorylation. Proceedings of the National Academy of Sciences. 1994;91:1500-4
16. Garcia-Recio S, Pastor-Arroyo EM, Marin-Aguilera M, Almendro V, Gascon P. The Transmodulation of HER2 and EGFR by Substance P in Breast Cancer Cells Requires c-Src and Metalloproteinase Activation. PloS one. 2015;10:e0129661
17. Takezawa K, Pirazzoli V, Arcila ME, Nebhan CA, Song X, de Stanchina E. et al. HER2 amplification: a potential mechanism of acquired resistance to EGFR inhibition in EGFR-mutant lung cancers that lack the second-site EGFRT790M mutation. Cancer discovery. 2012;2:922-33
18. Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C. et al. A molecularly annotated platform of patient-derived xenografts ("xenopatients") identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer discovery. 2011;1:508-23
19. Ciardiello F, Normanno N. HER2 signaling and resistance to the anti-EGFR monoclonal antibody cetuximab: a further step toward personalized medicine for patients with colorectal cancer. Cancer discovery. 2011;1:472-4
20. Liu J, Zhan X, Li M, Li G, Zhang P, Xiao Z. et al. Mitochondrial proteomics of nasopharyngeal carcinoma metastasis. BMC medical genomics. 2012;5:62
21. Li X, Wang Z, Liu J, Tang C, Duan C, Li C. Proteomic analysis of differentially expressed proteins in normal human thyroid cells transfected with PPFP. Endocrine-related cancer. 2012;19:681-94
22. Han X, Zhang J, Peng Y, Peng M, Chen X, Chen H. et al. Unexpected role for p19INK4d in posttranscriptional regulation of GATA1 and modulation of human terminal erythropoiesis. Blood. 2017;129:226-37
23. Liu Y, Li G, Liu C, Tang Y, Zhang S. RSF1 regulates the proliferation and paclitaxel resistance via modulating NF-κB signaling pathway in nasopharyngeal carcinoma. Journal of Cancer. 2017;8:354
24. Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, Eshraghi M. et al. Apoptosis and cancer: mutations within caspase genes. Journal of Medical Genetics. 2009;46:497-510
25. Zhang T, Zhang L, Zhang T, Fan J, Wu K, Guan Z. et al. Metformin sensitizes prostate cancer cells to radiation through EGFR/p-DNA-PKCS in vitro and in vivo. Radiation research. 2014;181:641-9
26. Horn D, Hess J, Freier K, Hoffmann J, Freudlsperger C. Targeting EGFR-PI3K-AKT-mTOR signaling enhances radiosensitivity in head and neck squamous cell carcinoma. Expert opinion on therapeutic targets. 2015;19:795-805
27. Jette N, Lees-Miller SP. The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Progress in biophysics and molecular biology. 2015;117:194-205
28. Lu J, Tang M, Li H, Xu Z, Weng X, Li J. et al. EBV-LMP1 suppresses the DNA damage response through DNA-PK/AMPK signaling to promote radioresistance in nasopharyngeal carcinoma. Cancer letters. 2016;380:191-200
29. Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82-5
30. Tanaka K, Babic I, Nathanson D, Akhavan D, Guo D, Gini B. et al. Oncogenic EGFR signaling activates an mTORC2-NF-kappaB pathway that promotes chemotherapy resistance. Cancer discovery. 2011;1:524-38
31. Criswell T, Leskov K, Miyamoto S, Luo G, Boothman DA. Transcription factors activated in mammalian cells after clinically relevant doses of ionizing radiation. Oncogene. 2003;22:5813-27
32. De Bacco F, Luraghi P, Medico E, Reato G, Girolami F, Perera T. et al. Induction of MET by ionizing radiation and its role in radioresistance and invasive growth of cancer. Journal of the National Cancer Institute. 2011;103:645-61
33. Sau A, Lau R, Cabrita MA, Nolan E, Crooks PA, Visvader JE. et al. Persistent Activation of NF-kappaB in BRCA1-Deficient Mammary Progenitors Drives Aberrant Proliferation and Accumulation of DNA Damage. Cell stem cell. 2016;19:52-65
34. Zheng Z, Qu J-Q, Yi H-M, Ye X, Huang W, Xiao T. et al. MiR-125b regulates proliferation and apoptosis of nasopharyngeal carcinoma by targeting A20/NF-κB signaling pathway. Cell death & disease. 2017;8:e2855
35. Razumienko EJ, Chen JC, Cai Z, Chan C, Reilly RM. Dual-Receptor-Targeted Radioimmunotherapy of Human Breast Cancer Xenografts in Athymic Mice Coexpressing HER2 and EGFR Using 177Lu- or 111In-Labeled Bispecific Radioimmunoconjugates. Journal of Nuclear Medicine. 2015;57:444-52
36. Landgraf R. HER2 therapy. HER2 (ERBB2): functional diversity from structurally conserved building blocks. Breast Cancer Research. 2007;9:202
37. Xiao L, Hu Z, Dong X, Tan Z, Li W, Tang M. et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene. 2014;33:4568
38. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69-80
39. Wilson C, Nicholes K, Bustos D, Lin E, Song Q, Stephan J-P. et al. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget. 2014;5:7328
40. Izumchenko E, Chang X, Michailidi C, Kagohara L, Ravi R, Paz K. et al. The TGFbeta-miR200-MIG6 pathway orchestrates the EMT-associated kinase switch that induces resistance to EGFR inhibitors. Cancer research. 2014;74:3995-4005
41. Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T. et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nature genetics. 2012;44:852-60
42. Hou J, Zhou Z, Chen X, Zhao R, Yang Z, Wei N. et al. HER2 reduces breast cancer radiosensitivity by activating focal adhesion kinase in vitro and in vivo. Oncotarget. 2016;7:45186-98
Author contact
![]() Corresponding author: Prof. Yongheng Chen, Key Laboratory of Cancer Proteomics of Chinese Ministry of Health, XiangYa Hospital, Central South University, 110 Xiangya Road, Changsha, Hunan 410078, P.R. China. E-mail:yonghenccom.
Corresponding author: Prof. Yongheng Chen, Key Laboratory of Cancer Proteomics of Chinese Ministry of Health, XiangYa Hospital, Central South University, 110 Xiangya Road, Changsha, Hunan 410078, P.R. China. E-mail:yonghenccom.