Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(9):2128-2138. doi:10.7150/jca.30235 This issue Cite
Research Paper
PSME3 Promotes TGFB1 Secretion by Pancreatic Cancer Cells to Induce Pancreatic Stellate Cell Proliferation
Lianyuan Yu1, Jun-Jie Li1, Xiao-Long Liang2, Huanwen Wu1, Zhiyong Liang1 ![]()
1. Molecular Pathology Research Center, Department of Pathology, Peking Union Medical College Hospital, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing.
2. Department of Pathology, Chao-Yang Hospital, Beijing.
Received 2018-9-27; Accepted 2019-4-23; Published 2019-5-16
Abstract
Pancreatic cancer is a highly malignant disease that is associated with poor prognosis. One hallmark of pancreatic cancer is excessive desmoplasia, characterized by fibrous or connective tissue growth and altered tumor stroma. Pancreatic stellate cells (PSCs) comprise a mesenchymal cell type that contributes to pancreas fibrosis and cancer progression. PSME3 is a regulatory subunit of the proteasome that is expressed in various cancers such as breast, ovarian, and pancreatic. Notably, PSME3 modulates lactate secretion in pancreatic cancer, suggesting a potential function in regulating pancreas fibrosis. However, the role of PSME3 in pancreatic cancer cell (PCC)-PSC interactions remains unclear. The current study, for the first time, explored the mechanism involved in PSME3-mediated PCC-PSC interactions. IHC showed that PSME3 is highly expressed in PCCs, and this was found to correlate with tumor differentiation. RNA interference (RNAi) indicated that PSME3 is involved in PCC apoptosis. PCR array and cell co-culture experiments suggested that conditioned culture medium (CM) from PSME3-knockdown PCCs could suppress PSC proliferation by down-regulating TGFB1 secretion. Transcription factor (TF) activation assays showed that PSME3 regulates TGFB1 production by inhibiting activation protein-1 (AP-1). Together, these data demonstrate that PSME3 interacts with AP-1 to regulate TGFB1 secretion in PCCs and promote PSC proliferation. Our results indicate a novel PSME3-regulated association between PSCs and PCCs and provide a promising therapeutic strategy for this malignancy.
Keywords: PSME3, TGFB1, Pancreatic Stellate Cell
Introduction
Pancreatic cancer remains the fourth leading cause of death in the United States. Further, most cases are diagnosed with distant metastasis and are often resistant to chemical treatment [1]. One major characteristic of pancreatic cancer is the abundance of tumor stroma referred to as desmoplasia, which is correlated with poor disease prognosis [2]. Approaches targeting desmoplasia are associated with poor responses, which implies that there is another predominant desmoplastic regulatory pathway [3]. Recent studies have considered cancer-associated fibroblasts, namely PSCs (pancreatic stellate cells), as a major contributor to the induction of pancreatic desmoplasia and cancer progression [4]. Our previous study also indicated that pancreatic cancer cells (PCCs) are capable of interacting with PSCs in a paracrine manner [5-7]. Thus, to provide novel therapeutic and diagnostic strategies, exploration of interaction patterns between PSCs and PCCs is important.
The proteasome is a specific protein complex that regulates protein degradation in mammalian cells. The activated proteasome is composed of a 20S core particle with either a 19S or 11S regulatory particle [8]. PSME3 (also called REGγ), one of the subunits of the 11S family, forms a homodimer and is predominantly located in the nucleus [9]. Unlike other 11S subunits, the functions of PSME3 are correlated with multiple biological behaviors, many of which are still under investigation. Many studies have demonstrated an aberrant expression of PSME3 in human malignant tumors such as thyroid and colorectal cancers [10, 11]. PSME3 promotes cell growth by mediating the degradation of the cell cycle inhibitor p21 [12]. It also acts as a regulator by targeting the MDM2/P53 complex [13]. A recent report showed that PSME3 acts as a transcriptional regulator to mediate the secretion of TNFα and IL1β [14]. Additionally, PSME3 facilities lactate secretion by targeting c-myc in pancreatic cancer [15]. Together, these studies suggest that there is a potential PSME3-mediated cell cross-talk mechanism. However, whether a PSME3-mediated cell interaction pathway exists between PCCs and PSCs has not been reported.
In the present study, our aim was to clarify the potential role of PSME3 in PCC-PSC interactions. Accordingly, we demonstrated proliferation-inducing effects on PSCs that occur via AP-1-mediated TGFB1 secretion by PCCs. We also show that PSME3 is highly expressed in PCCs and is involved in cancer cell apoptosis.
Materials and Methods
Immunohistochemical (IHC) staining
Forty-eight surgically resected pancreatic cancer FFPE specimens were obtained from the Department of Pathology, Peking Union Medical College Hospital, from January 2015 to May 2016. Patient clinical information was obtained from medical records. All human samples were obtained in accordance with the policies and practices of the Institutional Review Board of Peking Union Medical College Hospital. FFPE slides were treated with xylene dewaxing followed by gradient alcohol hydration. Citric acid repair solution (pH 6.0) was used for antigen retrieval for 4 minutes. After cooling to room temperature, the slides were treated with 3% hydrogen peroxide for 10 min to block endogenous peroxidase activity. The slides were then incubated with an anti-PSME3 primary antibody (Santa Cruz, USA) at 1:200 at 4 °C overnight. The next day, the slides were incubated with secondary antibody (ZSBIO, Beijing, China) for 30 min at room temperature and stained with DAB before observation. All IHC slides were assessed by two experienced pathologists. The staining intensity was assessed using the following scale: 0, negative; 1, weak; 2, moderate; and 3, strong. Staining quantity was quantified using the following scale: 0, < 1% positive; 1, 1-10% positive; 2, 10-50% positive; 3, 50-75% positive; 4, > 75%. The two scores were multiplied to achieve a final IHC staining score as follows:0 was considered not detected; 1-4, was considered weak; 5-8, was considered medium; 9 or more was considered high.
Cell culture and transfection
Bxpc-3 and Panc-1 pancreatic cancer cell lines were purchased from PUMC Cell Resource Center (Beijing, China). Panc-1 cells were cultured in DMEM (Gibco, Waltham, MA, USA) with 10% FBS (Thermo Scientific; Waltham, MA, USA) and penicillin/streptomycin (100 U/mL; PUMC Cell Resource Center). Bxpc-3 cells were cultured with RPMI1640 (Gibco, Waltham, MA, USA) with 10% FBS and penicillin/streptomycin.PSCs were isolated from fresh human pancreatic surgical tissue and cultured in DMEM/F-12 (1:1) (Gibco, Waltham, MA, USA) supplemented with 10% FBS and 100 U/mL penicillin/streptomycin as previously described. The clinicopathological data of the PSC donors are presented in Table 2. All cells were incubated at 37 °C with 5% CO2.
The clinicopathological data of the three PSCs donors with surgically resected PDAC.
| Number | Gender | Age | Differentiation |
|---|---|---|---|
| PSC1 | Female | 67 | Moderate |
| PSC2 | Female | 71 | Moderate |
| PSC3 | Female | 61 | Poor |
Three unique human 27-mer siRNA duplexes targeting PSME3 (SR306885) were obtained from OriGene Technologies, Inc, USA. Lipofectamine® RNAiMAX Transfection Reagent was purchased from Thermo Scientific (Waltham, MA, USA) and used in accordance with the manufacturer's protocols.
Western blot analysis
Total cell protein was obtained using Minute™ Protein Extraction Kits (Invent Biotech, USA) following the manufacturer's instructions. Protein levels were quantified using a BSA kit (Thermo Scientific). TGX FastCast premixed acrylamide solutions (Bio-rad, USA) were used for electrophoretic protein separation. After blocking with 5% Difco skim milk (BD, USA) for 1 h, proteins were then transferred to 0.45-μm PVDF membranes (Merck Millipore, USA). The membranes were then incubated with primary antibodies (1:1000) overnight at 4 °C. HRP-conjugated secondary antibody (1:1000) was added the next day and incubated for 1 h at room temperature. After washing three times with TBST, the membranes were stained with a chemiluminescence kit (Merck Millipore, USA) and detected with a ChemiDoc Touch Imaging System (Bio-rad). The results were analyzed using Quantity One Software (Bio-rad).
The PSME3 primary antibody (Cat.No2412) and Pro-apoptosis primary antibodies kit (Cat.No98322) were purchased from Cell Signal Technology (Danvers, MA). TGFB1 (Cat.NoYM0616) and AP-1 primary antibodies (Cat.NoYT0249) were purchased from ImmunoWay Biotechnology Company (TX, USA).
Real-time PCR and PCR array
Total cell RNA was isolated using the RNeasy Mini Kit (Qiagen, Hamburg, Germany) in accordance with the manufacturer's protocols. The RT2 First Strand Kit (Qiagen) was used to generate the first-strand cDNA template. Real-time PCR was conducted using PowerUp SYBR Green Master Mix (Applied Biosystems, Thermo Fisher Scientific, USA) as per the manufacturer's instructions. Results were analyzed using ABI 7500 Software v2.0.6. GAPDH was used as an endogenous reference gene. Primers utilized for real-time PCR were as follows: PSME3, forward primer: 5′-CCAGACCTAAGCTGCCTTCT-3′, reverse primer: 5′-GATAGCAGCCTCTACTGGCA-3′; BAK, forward primer: 5′-GTTTTCCGCAGCTACGTTTTT-3′, reverse primer: 5′-GCAGAGGTAAGGTGACCATCTC-3′; BIK, forward primer: 5′-GACCTGGACCCTATGGAGGAC-3′, reverse primer: 5′-CCTCAGTCTGGTCGTAGATGA-3′; TGFB1, forward primer: 5′-GGATACCAACTATTGCTTCAGCTCC-3′, reverse primer: 5′-AGGCTCCAAATATAGGGGCAGGGTC-3′; FOS, forward primer: 5′-CACTCCAAGCGGAGACAGAC-3′, reverse primer: 5′-AGGTCATCAGGGATCTTGCAG-3′; JUN, forward primer: 5′-AACAGGTGGCACAGCTTAAAC-3′, reverse primer: 5′-CAACTGCTGCGTTAGCATGAG-3′; GAPDH, forward primer: 5′-TCCAGAAACTAATGGCAGATCCC-3′, reverse primer: 5′-AATTCCCTACGCTTTGGGTTTT-3′.
RT2 Profiler PCR Arrays (Qiagen) were used to identify differentially expressed extracellular matrix and adhesion molecules according to the manufacturer's description. The results were analyzed using the Qiagen data analysis center (www.qiagen.com/cn/shop/genes-and-pathways/data-analysis-center).
Cell proliferation assays
For this, 5 × 103 Panc-1 or Bxpc3 cells were plated in 96-well plates and incubated at 37 °C with 5% CO2. After cell attachment, a CCK8-kit (DOJINDO laboratory, Kumamoto Techno Research Park, Japan) was added to the cells, which were analyzed using an EPOCH2T microplate reader (Biotek instruments Inc, Highland Park Winooski, USA) at an absorbance of 450 nm at day 1, 2 and 3 after transfection with PSME3 siRNAs. Four wells were tested for each sample at every time point.
Cell apoptosis analysis
Hoechst 33258 (Beyotime Biotechnology, Shanghai, China) and an Annexin V Apoptosis Detection Kit APC (Thermo Fisher Scientific, USA) were used to evaluate cell apoptosis. Briefly, 5 × 105 Panc-1 or Bxpc3 cells were plated in 6-well plates. Cells were harvested at day 3 after transfection with PSME3 siRNAs. Cells were fixed with neutral formalin and stained with Hoechst 33258 in accordance with the manufacturer's instructions.
For Annexin V detection, 1 × 106 cells and culture supernatant were harvested and centrifuged at 1000 × g fo 5 min to collect the cells. After incubation with an Annexin V detection kit, the cells were then subjected to flow cytometric (Accrui C6, BD, USA) analysis.
Cell cycle analysis
A cell cycle analysis PI kit (Beyotime Biotechnology, Shanghai, China) was used to evaluate cell cycle. Briefly, 1 × 106 Panc-1 or Bxpc3 cells were harvested at day 3 after transfection with PSME3 siRNA and fixed in 70% ethanol overnight. Then, the cells were incubated with propidium staining in accordance with the manufacturer's instructions and subjected to flow cytometric (Accrui C6, BD, USA) detection. The results were analyzed using ModFitLT v3.1 (Verity Software House, Inc).
Cell co-culture assays
For co-culture, 0.4-μm cell culture inserts (Costar, Corning, NY, USA) were used to determine PSC proliferation; 1 × 105 Panc-1 or Bxpc3 cells were plated in lower chambers after transfection them with siRNA, and 1 × 103 PSCs were plated in the upper chamber. After 3 days of co-culture, cells were treated with 0.1% crystal violet stain solution for cell counts. All cells were cultured with FBS-free culture medium at 37 °C with 5% CO2. Three wells were tested for each sample. A phase contrast microscope (CKX41; Olympus) and image acquisition software (Bio-Life DP, version 4.8) were used to capture pictures. The results were analyzed using Photoshop CS5. Cell viability of PSCs was measured by performing a CCK-8 assay at day 3 after co-culture with either PSME3-silenced PCCs or control PCCs.
ELISA assays
For this, 1 × 106 Panc-1 or Bxpc3 cells were plated in 10-mm plates and transfected with PSME3 siRNA for 3 days. Cell culture media were then collected and TGFB1 levels were detected using the Human TGFB1 ELISA Kit (Proteintech, USA) in accordance with the manufacturer's protocols.
Transcription factor (TF) activation profiling
A TF Activation Profiling Plate Array Kit (Signosis, Inc., USA) was adopted to identify differentially expressed TFs between the negative control group and PSME3 RNAi group, in accordance with the manufacturer's instructions. Briefly, a 10-mm plate with approximately 1 × 107 cells was harvested for nuclear protein isolation using a Nuclear Extraction Kit (Signosis Inc.) after transfection with siRNA for 3 days. Next, approximately 10 μg of nuclear protein was incubated with biotin-labeled probes mixed to form TF/probe complexes. After purification and washing to remove free probes, the complexes were hybridized with a plate pre-coated with complementary sequences. The captured DNA probes were further detected via streptavidin-HRP luminescence using a chemiluminescence plate reader (Turner microplate multimode, Turner BioSystems, United States).
Statistical analysis
Statistical analysis was carried out using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA). P-values < 0.05 were considered statistically significant.
Results
PSME3 is highly expressed in pancreatic cancer
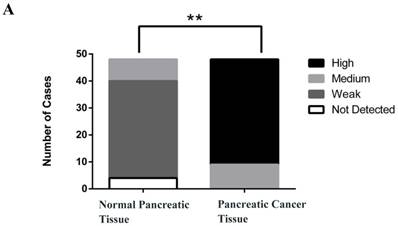
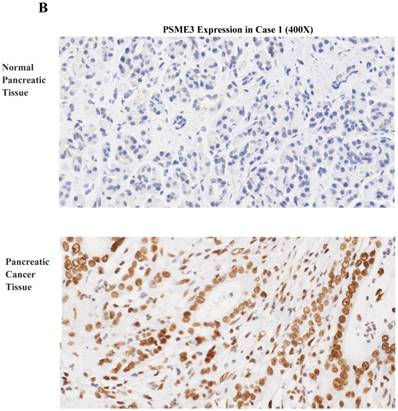
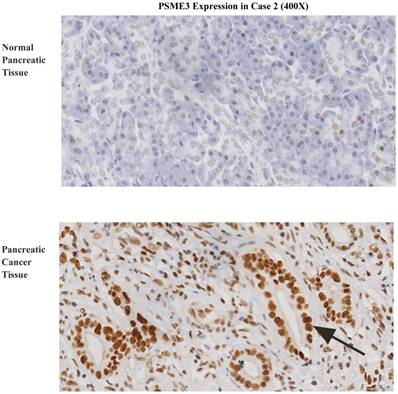
To investigate the expression pattern of PSME3 in pancreatic cancer tissue, IHC staining was conducted. Forty-eight surgically-resected pancreatic cancer FFPE specimens were tested. Associations between PSME3 expression and clinicopathological parameters were also studied. PSME3 was mainly located in the nuclei of PCCs. 48 pancreatic cancer tissues were all stained, of which no weakly stained was observed; 9 were moderately stained (18.75%); 39 were highly stained (81.25%); Correspondingly, in 48 normal pancreatic adjacent tissues, 4 were not PSME3 detected, 36 were weakly stained (75%); 8 were moderately stained (16.67%), and no highly stained was observed. As shown in Fig 1, the expression level of PSME3 in pancreatic cancer tissue was stronger than that in normal pancreatic tissue (P<0.01). Notably, the expression of PSME3 was positively associated with pancreatic cancer differentiation (p < 0.01), but not with sex, age, margins, lymph node metastasis, tumor size and recurrence (Table 1). These results indicate that PSME3 is a potential therapeutic and diagnostic marker in pancreatic cancer.
PSME3 is highly expressed in pancreatic cancer tissue and is positively associated with the cancer differentiation. (A) The protein expression level was calculated and shown at right panel (P < 0.01 ). (B) Representative IHC staining images showing PSME3 expression in pancreatic cancer tissue and in normal adjacent pancreatic tissue. Black Arrow indicates that PSME3 is mainly located in the nuclei of pancreatic cancer cells.
Clinical and pathological characteristics of PCC patients (n=48)
| Clinicopathological Features | Total Cases | PSME3 expression | P value | ||
|---|---|---|---|---|---|
| medium | high | ||||
| Gender | Male | 26 | 4 | 22 | 0.713 |
| Female | 22 | 5 | 17 | ||
| Age (years) | ≥60 | 29 | 4 | 25 | 0.451 |
| <60 | 19 | 5 | 14 | ||
| Differentiation | Well | 3 | 3 | 0 | 0.004 * |
| Moderate/Poor | 42 | 5 | 37 | ||
| Margins | Positive | 11 | 1 | 10 | 0.657 |
| Negative | 34 | 7 | 27 | ||
| Nodal metastasis | Yes | 28 | 6 | 22 | 0.716 |
| No | 20 | 3 | 17 | ||
| Tumor size (cm) | ≤4 | 34 | 16 | 18 | 0.321 |
| >4 | 12 | 3 | 9 | ||
| Recurrence | Yes | 12 | 3 | 9 | 0.671 |
| No | 36 | 6 | 30 | ||
PSME3 knockdown suppresses PCC proliferation and induces apoptosis
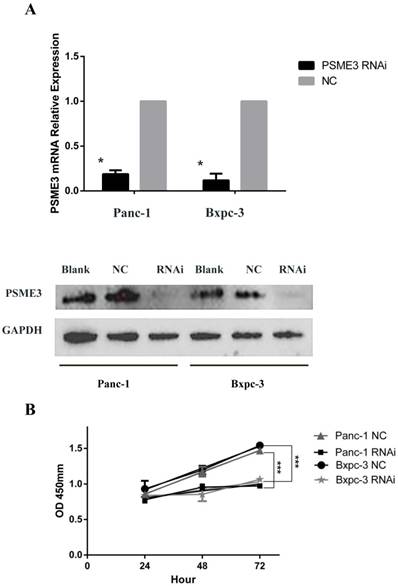
Since PSME3 was found to be highly expressed in PCCs, RNAi was used to verify the molecular function of this marker. As shown in Fig 2, the expression of PSME3 was effectively inhibited in Panc-1 and Bxpc-3 cell lines after transfection with siRNA at both the mRNA and protein level. Cell proliferation declined significantly after siRNA treatment in both pancreatic cell lines. Additionally, flow cytometry and Hoechst staining showed that apoptosis was dramatically increased after PSME3 ablation. These results suggest that PSME3 can modulate pancreatic cell proliferation and apoptosis, which is consistent with previous reports [13].
PSME3-RNAi silencing suppresses PCC proliferation and induces PCC apoptosis. (A) The PSME3-RNAi silencing efficiency in Panc-1 and Bxpc-3 cells is detected by Real-time PCR and Western Blot. (B) Cell proliferation curves of Panc-1 and Bxpc-3 cells. (C) Cell apoptosis of Panc-1 and Bxpc-3 cells is detected via flow cytomerty. PI signal was detected in FL2-H, Annexin V signal was detected in FL4-H. (D) Apoptotic cells were examined by Hoechst 33258 staining, and the counts of apoptotic cells are statistically analyzed. Data represent mean ± SD. *P < 0.05, **P < 0.01 versus control group.
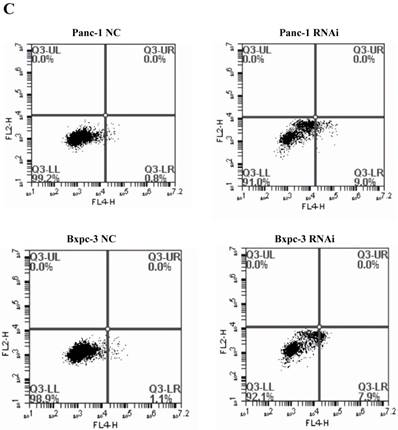
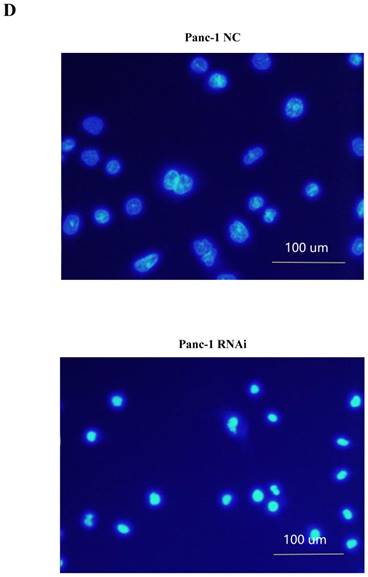
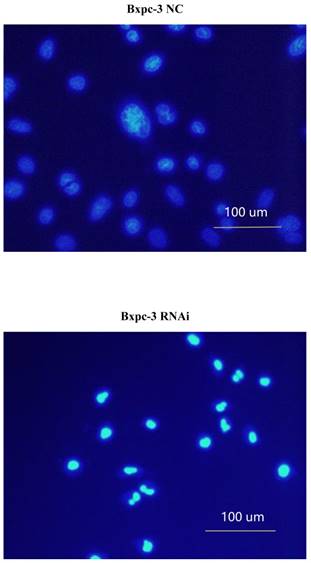
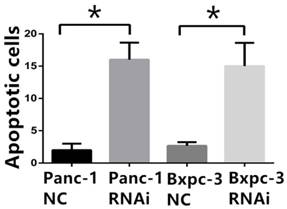
Pro-apoptotic BCL-2 family members are associated with PSME3-mediated PCC apoptosis
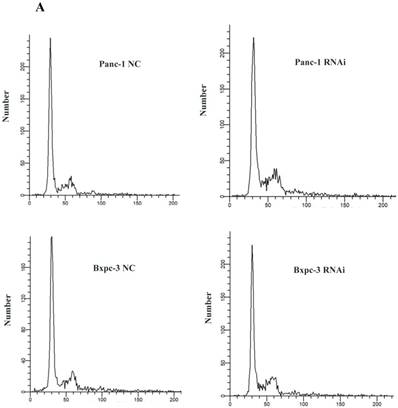
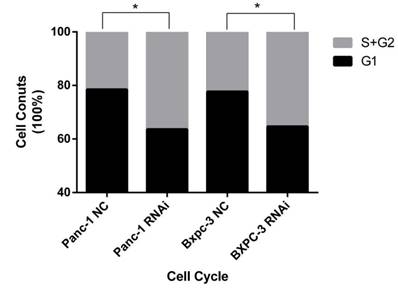
To clarify the mechanism related to PSME3-knockdown-induced suppression of cell proliferation, cell cycle analysis was performed. Flow cytometry indicated G1/S phase transition after treatment with PSME3 RNAi in both Panc-1 and Bxpc-3 cell lines, suggesting that this molecule affects cell proliferation by modulating cell cycle progression (Fig. 3A).
BCL-2 family pro-apoptosis members correlate with PSME3-mediated PCC apoptosis. (A) Cell cycle of Panc-1 and Bxpc-3 cells is measured by flow cytomerty. (B) Apoptosis PCR array showing significant mRNA profiling distinction of BCL-2 family pro-apoptosis genes BIK and BAK in Panc-1 cells. The effect of PSME3-RNAi silencing on BIK and BAK is measured using Real-time PCR and western blot analysisin Panc-1 and Bxpc-3 cells. Data represent mean ± SD. *P < 0.05, **P < 0.01 versus control group.
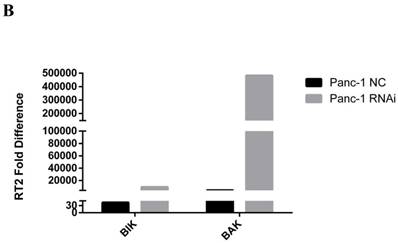
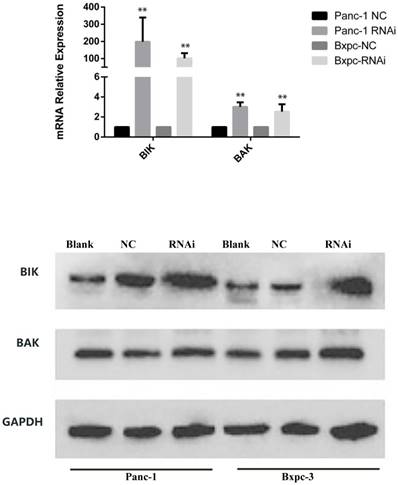
To demonstrate the mechanism associated with PSME3-mediated apoptosis, an apoptosis PCR array was used. Briefly, RNA was extracted from PSME3-knockdown or negative control Panc-1 cells and then incubated with a PCR array containing apoptosis-related genes. Two BCL-2 family pro-apoptosis genes, namely BIK, and BAK were found to be dramatically increased with PSME3 RNAi. Real-time PCR was performed to confirm the PCR array results using both Panc-1 and Bxpc-3 cell lines. Result showed that the mRNA expression of BIK and BAK was increased in both cell lines, which was consistent with PCR array data. Western blotting also confirmed these results in Panc-1 and Bxpc-3 cells. These results indicate that PSME3 mediates PCC apoptosis by regulating the expression of BCL-2 family pro-apoptosis members BIK and BAK (Fig. 3B).
PSC proliferation is inhibited by conditional medium (CM) from PSME3-knockdown PCCs
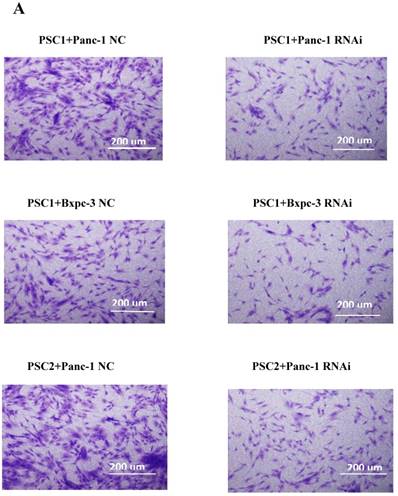
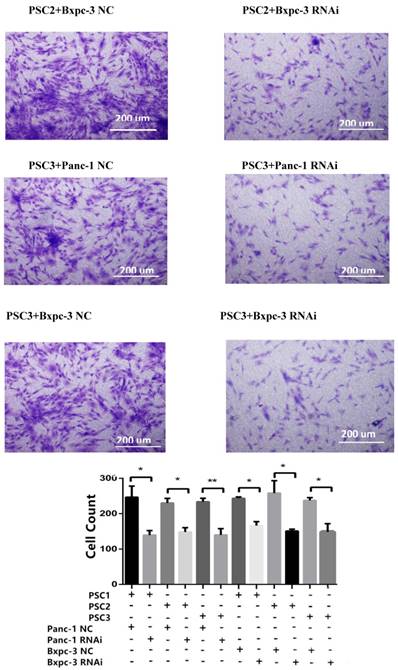
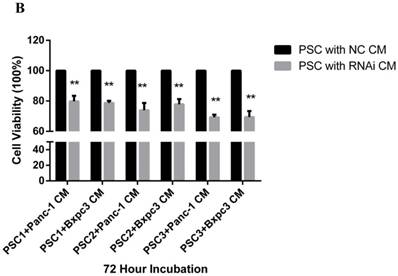
To explore the effect of PSME3 RNAi in PCCs on PSCs, co-culture and CCK8 assays were conducted. PSCs from three different patients (PSC1, PSC2, PSC3) were seeded in the upper layer of 0.4-μm transwell chambers, and PSME3-knockdown Panc-1 or Bxpc-3 cells were seeded in the lower layer. After 72 hours' co-culture, PSCs were treated with 0.1% crystal violet stain solution for cell counts. Additionally, The proliferation of PSCs was also assessed by CCK8 assays after 72 hours of treatment with PSME3-knockdown Panc-1 or Bxpc-3 CM (Conditional medium). As shown in Fig. 4, the PSC number was significantly decreased after co-culture with PSME3-knockdown Panc-1 or Bxpc-3 and CM. These results indicate that PSC cell growth is inhibited by PSME3-knockdown PCCs CM.
PSC proliferation is inhibited by PSME3 RNAi PCC CM. (A) Representative images showing that PSCs co-culture with either PSME3-RNAi silencing PCC or control PCC in 0.4um transwell membrane. The counts of PSCs are also statistically analyzed. (B) Cell viability of PSCs are measured using CCK-8 assay at day 3 after co-culture with either PSME3-RNAi silencing PCC or control PCC CM. Data represent mean ± SD. *P < 0.05, **P < 0.01 versus control group.
CM from PSME3-knockdown PCCs inhibits PSC growth by suppressing TGFB1 secretion
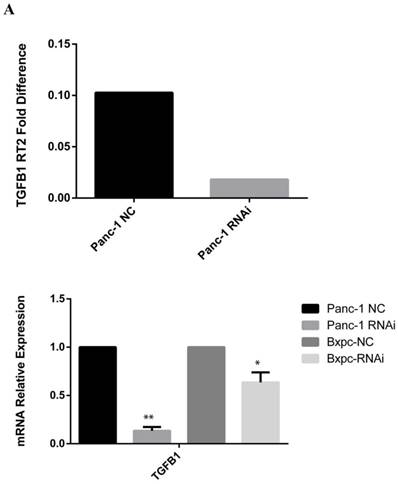
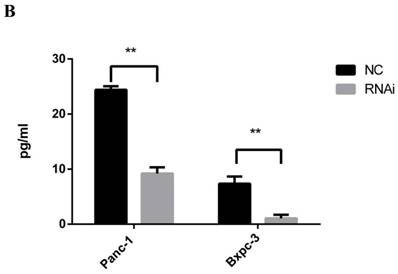
A PCR array to assess extracellular matrix and adhesion molecules was performed to explore the mechanism responsible for the growth inhibitory effect of PSME3-knockdown CM from PCCs on PSCs. In brief, RNA was extracted from PSME3 RNAi-treated or negative control Panc-1 cells and then incubated with a PCR array containing cell interaction-associated genes. From these results, the secreted protein transforming growth factor beta 1 (TGFB1) was dramatically decreased after knockdown. Real-time PCR was used to confirm the PCR array results using both Panc-1 and Bxpc-3 cell lines. Results showed that the mRNA expression of TGFB1 was decreased in both cell lines, which was consistent with PCR array results. An ELISA was also performed to detect secreted TGFB1 levels. Results showed that secreted TGFB1 was decreased in both Panc-1 and Bxpc-3 cells after PSME3 RNAi treatment. These results suggest that the growth inhibitory effect of PSME3 suppression in PCCs on PSCs is through the regulation of TGFB1 secretion (Fig. 5).
PSME3-RNAi PCC CM inhibits PSC growth via mediating TGFB1 secretion. (A) ECM PCR array showing significant mRNA profiling distinction of TGFB1 in Panc-1 cells. The effect of PSME3-RNAi silencing on TGFB1 mRNA is measured using Real-time PCR in Panc-1 and Bxpc-3 cells. (B)The effect of PSME3-RNAi silencing on TGFB1 secretion is detected via ELISA analysis in Panc-1 and Bxpc-3 cells. Data represent mean ± SD. *P < 0.05, **P < 0.01 versus control group.
PSME3 regulates TGFB1 secretion in PCCs by mediating AP-1 protein expression
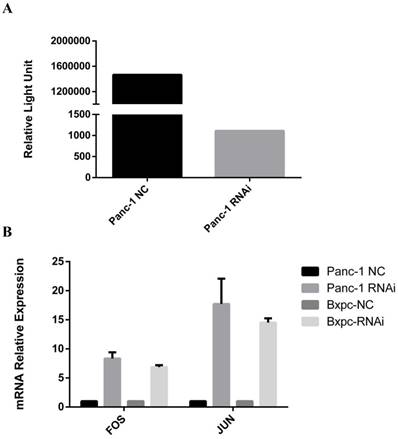
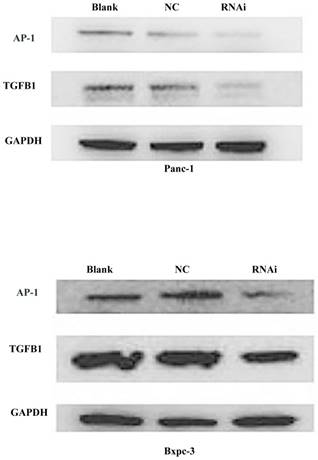
To further demonstrate the mechanism through which PSME3 regulates TGFB1 secretion in PCCs, a TF activation assay was performed. Briefly, Panc-1 nuclear proteins were extracted and then subjected to TF activity testing. As shown in Fig 6, the transcription factor activation protein 1 (AP-1) was significantly decreased after PSME3 RNAi. Reports have shown that Ap-1 is a potent TGFB1 transcriptional activator, and this TF consists of Fos and Jun subunits. Accordingly, real-time PCR and western blotting were used to confirm the aforementioned results in both Panc-1 and Bxpc-3 cell lines. Results showed that AP-1 was decreased in both cell lines at the protein level, whereas mRNA expression (Fos/Jun) dramatically increased. The above results suggest that PSME3 regulates TGFB1 secretion in PCCs by repressing AP-1 transcriptional activity at the protein level but that it does not affect its mRNA expression.
PSME3 regulates PCC TGFB1 secretion via mediating AP-1 protein expression. (A) Transcription factor activation assay showing dramatic Ap-1 TF activity differentiation in panc-1 cells. (B) The effect of PSME3-RNAi silencing on FOS/JUN and TGFB1 is detected via Real-time and weatern blots in Panc-1 and Bxpc-3 cells. Data represent mean ± SD. *P < 0.05, **P < 0.01 versus control group.
Discussion
In the present study, we show that PSME3 is highly expressed in PCCs and is involved in modulating cancer cell apoptosis and proliferation. Inhibiting PSME3 expression in PCCs dramatically suppressed TGFB1 secretion by down-regulating AP-1. TGFB1 is a potent growth factor for PSCs and has important roles in the development of pancreatic fibrosis [16]. Together, these data demonstrate the proliferative effect of PCCs on PSC, which occurs in an AP-1/TGFB1-dependent paracrine manner.
PSCs have been characterized as cancer-associated fibroblasts. In normal pancreatic tissue, these cells remain in a quiescent state. However, when stimulated by extracellular factors, they become activated and are a major source of pancreatic cancer tumor desmoplasia [17]. Many studies have verified the correlation between PSCs and pancreatic tumor progression such as through limiting drug delivery and immunosuppression [18]. Therefore, therapies targeting only PCCs are insufficient. Thus, the vital function of PSCs in pancreatic cancer has forced us to explore novel therapeutic approaches to improve the poor survival associated with pancreatic cancer.
Tumor cell-derived signals have long been identified as a major mechanism involved in PSC activation [19]. The TGFB family best represents these instructive soluble regulators [20]. Studies have shown that treating a mouse model of pancreatitis with TGFB1 results in the development of significant fibrosis [21]. Additionally, the proliferation of PSCs is dramatically increased after incubating these cells with TGFB1 [22]. The promoter regions of the human TGFB1 gene contain consensus sequences for AP-1, which is a transcriptional factor that is highly conserved in humans [23]. c-Jun, with other binding partners such as c-Fos, forms the dimer structure of Ap-1 [24, 25]. Studies have confirmed that Ap-1, as a transcription factor, up-regulates the transcription of TGFB1 [26]. These studies indicate that the AP-1/TGFB1 axis has a vital role in the regulation of PSC growth.
The proteasome pathway is critical to mediate cellular functions and maintain homeostasis. Aberrant activation of this pathway is associated with many diseases including cancer [27]. Proteasome inhibitors have been identified as inducers of apoptosis in leukemia. Further investigations have demonstrated the anti-proliferative role of proteasome inhibitors in solid tumor [28-30]. PSME3 is a proteasome component that has a unique expression pattern in many types of solid tumors including pancreatic cancer [15]. Multiple functions of PSME3 such as the regulation of cell cycle, chromosome stability, apoptosis, and immune defects have been reported [31-33]. The aforementioned data suggest that PSME3 could be considered a potential therapeutic target. One recent study demonstrated that PSME3 participates in the release of soluble inflammatory factors [14], which suggests an uncharacterized role for this protein in mediating cell interactions. However, the mechanism involved in this effect has not been investigated.
In the current study, our results showed that PSME3 is highly expressed in PCCs and is correlated with cancer differentiation. Inhibition of PSME3 expression was found to induce apoptosis in PCC lines by up-regulating BCL-2 pro-apoptotic factors. In co-cultures with CM from PSME3-knockdown PCCs, a dramatic suppression of PSC proliferation was observed. A PCR array and ELISA indicated the down-regulation of TGFB1 secretion in PCC CM. Moreover, a TF activation assay suggested the inhibition of AP-1 transcriptional activity in PSME3-RNAi-treated PCCs. Further investigation showed that PSME3 mainly affects AP-1 at the protein level, whereas the mRNA level of AP-1 (Fos/ Jun) was up-regulated. Together, these results indicate that PSME3-RNAi indeed inhibits the proliferation of PSCs via AP-1/TGFB1 secretion. As a regulatory subunit of the proteasome, PSME3 might regulate the protein levels of Ap-1 or the binding of Ap-1 subunits by degrading key regulatory factors. The corresponding increase in mRNA (Fos/ Jun) could be due to feedback-induced transcriptional activation caused by excessive Ap-1 protein complex degradation. The stimulation of mRNA expression also suggests that PSME3 does not affect FOS/JUN gene transcription. To demonstrate the key regulatory factors and feedback pathways involved in the PSME3-AP1 interactions, further fundamental studies such as immunoprecipitation experiments will be required.
In conclusion, PCCs exert proliferative effects on PSCs through the PSME3-mediated AP-1/TGFB1 secretion pathway. PSME3 is mainly involved in AP-1 down-regulation at the protein level, but not at the mRNA level. This critical role of PSME3 suggests a novel PCC-PSC interaction pattern that is regulated by the proteasome. This new association could become a promising therapeutic target for pancreatic cancer.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31471366, 81602162, 81502625), Chinese Academy of Medical Sciences (CAMS) Initiative for Innovative Medicine (2016-I2M-1-002), Center for molecular pathology, Chinese Academy of Medicine Science & Peking Union Medical College, Beijing, China (2017PT31008).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67(1):7-30
2. Watanabe I, Hasebe T, Sasaki S, Konishi M, Inoue K, Nakagohri T. et al. Advanced pancreatic ductal cancer: fibrotic focus and beta-catenin expression correlate with outcome. Pancreas. 2003;26(4):326-333
3. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA. et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25(6):735-747
4. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A. et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68(3):918-926
5. Wang L, Wu H, Wang L, Zhang H, Lu J, Liang Z. et al. Asporin promotes pancreatic cancer cell invasion and migration by regulating the epithelial-to-mesenchymal transition (EMT) through both autocrine and paracrine mechanisms. Cancer Lett. 2017;398:24-36
6. Zhang H, Wu H, Guan J, Wang L, Ren X, Shi X. Paracrine SDF-1alpha signaling mediates the effects of PSCs on GEM chemoresistance through an IL-6 autocrine loop in pancreatic cancer cells. Oncotarget. 2015;6(5):3085-3097
7. Wu H, Liang Z, Shi X, Ren X, Wang K, Liu T. Intrinsic chemoresistance to gemcitabine is associated with constitutive and laminin-induced phosphorylation of FAK in pancreatic cancer cell lines. Mol Cancer. 2009;8:125
8. Peters JM, Franke WW, Kleinschmidt JA. Distinct 19 S and 20 S subcomplexes of the 26 S proteasome and their distribution in the nucleus and the cytoplasm. J Biol Chem. 1994;269(10):7709-7718
9. Masson P, Andersson O, Petersen UM, Young P. Identification and characterization of a Drosophila nuclear proteasome regulator. A homolog of human 11 S REGgamma (PA28gamma ). J Biol Chem. 2001;276(2):1383-1390
10. Okamura T, Taniguchi S, Ohkura T, Yoshida A, Shimizu H, Sakai M. et al. Abnormally high expression of proteasome activator-gamma in thyroid neoplasm. J Clin Endocrinol Metab. 2003;88(3):1374-1383
11. Roessler M, Rollinger W, Mantovani-Endl L, Hagmann ML, Palme S, Berndt P. et al. Identification of PSME3 as a novel serum tumor marker for colorectal cancer by combining two-dimensional polyacrylamide gel electrophoresis with a strictly mass spectrometry-based approach for data analysis. Mol Cell Proteomics. 2006;5(11):2092-2101
12. Li X, Amazit L, Long W, Lonard DM, Monaco JJ, O'Malley BW. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGgamma-proteasome pathway. Mol Cell. 2007;26(6):831-842
13. Zhang Z, Zhang R. Proteasome activator PA28 gamma regulates p53 by enhancing its MDM2-mediated degradation. EMBO J. 2008;27(6):852-864
14. Sun J, Luan Y, Xiang D, Tan X, Chen H, Deng Q. et al. The 11S Proteasome Subunit PSME3 Is a Positive Feedforward Regulator of NF-kappaB and Important for Host Defense against Bacterial Pathogens. Cell Rep. 2016;14(4):737-749
15. Guo J, Hao J, Jiang H, Jin J, Wu H, Jin Z. et al. Proteasome activator subunit 3 promotes pancreatic cancer growth via c-Myc-glycolysis signaling axis. Cancer Lett. 2017;386:161-167
16. Ebert M, Guo F, Herrera P, Friess H, Buechler M, Malfertheiner P. Overexpression of transforming growth factor beta-1 (TGF-B1) is associated with reduced pten expression in pancreatic fibrosis and cancer. Gastroenterology. 2000;118(4):A650
17. Apte M, Pirola RC, Wilson JS. Pancreatic stellate cell: physiologic role, role in fibrosis and cancer. Curr Opin Gastroenterol. 2015;31(5):416-423
18. Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC. et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145(5):1121-1132
19. Korc M. Pancreatic cancer-associated stroma production. Am J Surg. 2007;194(Suppl 4):S84-S86
20. Friess H, Lu Z, Riesle E, Uhl W, Brundler AM, Horvath L. et al. Enhanced expression of TGF-betas and their receptors in human acute pancreatitis. Ann Surg. 1998;227(1):95-104
21. van Laethem JL, Deviere J, Resibois A, Rickaert F, Vertongen P, Ohtani H. et al. Localization of transforming growth factor beta 1 and its latent binding protein in human chronic pancreatitis. Gastroenterology. 1995;108(6):1873-1881
22. Schneider E, Schmid-Kotsas A, Zhao J, Weidenbach H, Schmid RM, Menke A. et al. Identification of mediators stimulating proliferation and matrix synthesis of rat pancreatic stellate cells. Am J Physiol Cell Physiol. 2001;281(2):C532-C543
23. Kim SJ, Glick A, Sporn MB, Roberts AB. Characterization of the promoter region of the human transforming growth factor-beta 1 gene. J Biol Chem. 1989;264(1):402-408
24. Hess J, Angel P, Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci. 2004;117(Pt 25):5965-5973
25. Halazonetis TD, Georgopoulos K, Greenberg ME, Leder P. c-Jun dimerizes with itself and with c-Fos, forming complexes of different DNA binding affinities. Cell. 1988;55(5):917-924
26. Schneider E, Schmid-Kotsas A, Zhao J, Weidenbach H, Schmid RM, Menke A. et al. TNF-alpha induces TGF-beta1 expression in lung fibroblasts at the transcriptional level via AP-1 activation. J Cell Mol Med. 2009;13(8B):1866-1876
27. Crawford LJ, Walker B, Irvine AE. Proteasome inhibitors in cancer therapy. J Cell Commun Signal. 2011;5(2):101-110
28. Imajoh-Ohmi S, Kawaguchi T, Sugiyama S, Tanaka K, Omura S, Kikuchi H. Lactacystin, a specific inhibitor of the proteasome, induces apoptosis in human monoblast U937 cells. Biochem Biophys Res Commun. 1995;217(3):1070-1077
29. Shinohara K, Tomioka M, Nakano H, Tone S, Ito H, Kawashima S. Apoptosis induction resulting from proteasome inhibition. Biochem J. 1996;317( Pt 2):385-388
30. Drexler HC. Activation of the cell death program by inhibition of proteasome function. Proc Natl Acad Sci U S A. 1997;94(3):855-860
31. Murata S, Kawahara H, Tohma S, Yamamoto K, Kasahara M, Nabeshima Y. et al. Growth retardation in mice lacking the proteasome activator PA28gamma. J Biol Chem. 1999;274(53):38211-38215
32. Barton LF, Runnels HA, Schell TD, Cho Y, Gibbons R. Immune defects in 28-kDa proteasome activator gamma-deficient mice. J Immunol. 2004;172(6):3948-3954
33. Zannini L, Lecis D, Buscemi G, Carlessi L, Gasparini P. et al. REGgamma proteasome activator is involved in the maintenance of chromosomal stability. Cell Cycle. 2008;7(4):504-512
Author contact
![]() Corresponding author: Liang Zhiyong, MD, PhD, Molecular Pathology Research Center, Department of Pathology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100730, People's Republic of China. Tel/Fax: 86 10 6529 5522. Email: liangzhiyong1220com.
Corresponding author: Liang Zhiyong, MD, PhD, Molecular Pathology Research Center, Department of Pathology, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100730, People's Republic of China. Tel/Fax: 86 10 6529 5522. Email: liangzhiyong1220com.