Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(10):2250-2260. doi:10.7150/jca.31156 This issue Cite
Research Paper
Identification of novel methylated DNA marker ZNF569 for head and neck squamous cell carcinoma
Xiangzhen Liu1# ![]() , Xinyuan Zhao2#, Chenyu Gou3
, Xinyuan Zhao2#, Chenyu Gou3
1. First Affiliated Hospital, Sun Yat-sen University, Guangzhou, China
2. Stomatological Hospital, Southern Medical University, Guangzhou, China
3. Sixth Affiliated Hospital, Sun Yat-sen University, Guangzhou, China
# Equal contribution
Received 2018-11-1; Accepted 2019-2-9; Published 2019-5-21
Abstract
Aberrant DNA methylation pattern plays an indispensable role in the initiation and development of head and neck squamous cell carcinoma (HNSCC). It is well recognized that lymph node metastasis is closely with unfavorable prognosis of HNSCC. Therefore, exploring the methylation events accounting for the lymph node metastasis of HNSCC is very important for improving the clinical outcome of HNSCC. Methylation data, RNA-seq data and clinical data were downloaded from The Cancer Genome Atlas (TCGA) and processed using the R package TCGA-Assembler. MethylMix was use for data analysis by integrating both methylation and gene expression data on HNSCC patients with lymph node metastasis and without lymph node metastasis. Pathway analysis was performed on significantly altered genes using ConsensusPathDB. The role of our interested gene zinc figure protein 569 (ZNF569) in HNSCC was further evaluated. Our results identified many novel hypermethylated/hypomethylated genes that might be closely associated with the lymph node metastasis of HNSCC. Pathway analysis revealed that increase in methylation of genes involved in generic transcription pathway including zinc figure proteins. ZNF569 was hypermethylated in HNSCC tissues especially those with lymph node metastasis. In addition, the expression levels of ZNF569 mRNA and protein were significantly lower in HNSCC tissues and cell lines compared to their respective controls. Moreover, overexpression of ZNF569 inhibited the proliferation, migration and invasion of HNSCC cells. HNSCC patients with lower ZNF569 expression suffered a significantly shorter overall survival than those with higher ZNF569 expression. In conclusion, we have identified many novel differentially methylated genes that might be important for the lymph node metastasis of HNSCC. In addition, ZNF569 might play a tumor suppressive role in carcinogenesis of HNSCC.
Keywords: Head and neck squamous cell carcinoma, Zinc figure protein 569, Methylation, Carcinogenesis
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common type of cancer [1-3]. A combination of genetic alterations, environmental risk factors and viral infection leads to the initiation and development of HNSCC. Despite the advances in surgery, radiation therapy and chemotherapy in the past few decades, there has been little change regarding the 5-year overall survival rate for HNSCC [4]. The poor prognosis of HNSCC is mainly due to disease detection at the advanced stages with lymph node metastasis. This highlights the importance of studying the underlying molecular events accounting for the carcinogenesis of HNSCC, which might help early detection of this malignancy.
DNA methylation is a heritable epigenetic mechanism that occurs by the addition of a methyl group onto the C5 position of the cytosine to form 5-methylcytosine [5]. DNA methylation plays critical roles in maintaining normal regulation of transcription, embryonic development, genomic imprinting, genome stability and chromatin structure [6]. Changes in DNA methylation pattern has been demonstrated to be a hallmark of cancer [7]. Methylation of p16, RARbeta, and MGMT was commonly detected in HNSCC cell lines and tumor specimens, indicating that downregulation of these genes might be closely associated with tumorigenesis of HNSCC [8-9]. Oral squamous cell carcinoma (OSCC) is the most common type of HNSCC. The promoter of SOX21-AS1was frequently hypermethylated in OSCC tissues. In addition, downregulation of SOX21-AS1 was associated with poor prognosis of OSCC, indicating SOX21-AS1 plays a tumor suppressive role in OSCC [10].
The Cancer Genome Atlas (TCGA) is a large-scale cancer genome project which provides researchers with multi-dimensional maps of the key genomic changes and clinicopathological information in 33 types of cancer (http://cancergenome.nih.gov/) [11]. The large number of tumor sample in TCGA datasets not only contributes to study the global methylation and gene expression patterns of HNSCC with increased statistical power, but also help screen novel biomarkers for early detection or/and predicting the prognosis of HNSCC. To the best of our knowledge, global methylation pattern analysis the genes associated with the lymph node metastasis of HNSCC is not available.
In this study, we first identified the differentially expressed hypermethylated / hypomethylated genes between HNSCC tumor samples with lymph node metastasis and those without lymph node metastasis based on TCGA HNSCC Methyl-seq and RNA-seq data. The role of our interested gene zinc finger protein 569 (ZNF569) in tumorigenesis of HNSCC was further explored.
Materials and Methods
Data acquisition and processing
The preprocessed level 3 RNA-seq data, methylation data and corresponding clinical information of HNSCC were downloaded from TCGA data portal. TCGA pipelines provide the methylation data in the form of beta values, which represent the ratio of methylated probe intensities to the total probe intensities. Probe-level data was condensed to a summary beta value for each gene using the Methylation 450_single_value function in TCGA-Assembler, which calculates the average methylation value for all CpG sites associated with a gene. For TCGA data, edgeR package was used for differentiated express genes (DEGs) screening. P<0.05 and absolute log2FC>1 were chosen as the cut-off criteria based on Benjamini& Hochberg (BH) procedure. The R package MethylMix was used for data analysis by integrating methylation data and RNA-seq data. Briefly, only methylation data highly correlated with gene expression data was selected for further analysis. Then a beta mixture model was used to define a DNA methylation state. Finally, a Wilcoxon rank sum test was employed to compare the methylation status in HNSCC samples with lymph node metastasis versus those without lymph node metastasis. A positive/negative differential methylation (DM) value indicated hypermethylation/hypomethylation respectively.
Pathway analysis
ConsensusPathDB was used for pathway analysis and the genes that were not statistically significant by MethylMix were filtered out. The following pathway databases including Inoh, Pid, Biocarta, Netpath, Humancyc, Kegg, Wikipathways, Smpdb, Pharmgkb, Ehmn, and Signalink were chosen for analysis. The cut-off P value was 0.01.
Tissue samples
This study was approved by the Institutional Research Ethics Committee at the First Affiliated Hospital of Sun Yat-sen University. Informed consent was obtained from all of patients or their relatives for the use of the tissues.
Cell culture
The HNSCC cell lines UM-SCC1 and UM-SCC23 were cultured in Dulbecco's modified eagle medium supplemented with 10% heat-inactivated fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 μg/mL). Normal human oral keratinocytes (NHOKs) were cultured in EpiLife media supplemented with the human keratinocyte growth supplement (Invitrogen, Carlsbad, CA, USA). All the cell lines were maintained in a 37°C humidified incubator with a mixture of 95% air and 5% CO2 and have been tested for mycoplasma contamination.
Lentivirus construction and infection
Full length ZNF569 was cloned into the pGCL-GFP vector. Recombinant lentiviral vectors combined with the packaging vectors were then transfected into 293T cells to generate lentiviruses. The supernatants were collected at 72 h after transfection and purified by ultracentrifugation. The empty vector was packaged as the negative control.
Real-time PCR
TRIzol (Takara, Dalian, China) was used to extract the total RNA from the cell lines and tissue samples based on the manufacturer's protocol. SuperScript III Reverse Transcriptase (Invitrogen) was used to synthesize the first-strand complementary DNA. The complementary DNA levels were amplified with Light Cycler 480® SYBR Green I MasterMix (Roche, Applied Science, Indianapolis, IN, USA) on an ABI 7500 real-time PCR system (Applied Biosystems, CA, USA). Human GAPDH was used as an endogenous control and the 2-ΔΔCt method was used for relative quantification.
Western blotting
The protein samples were loaded and separated on a 4-12% Bis-Tris NuPAGE gel (Invitrogen) and transferred onto a nitrocellulose membrane at 120V for 1.5 h. The membranes were blocked in TBST buffer containing 5% nonfat milk at room temperature for 1h, and incubated with rabbit antibody against human ZNF569 at a dilution of 1:150 (Abcam, Cambridge, UK) overnight in the cold room, followed by HRP conjugated goat anti-rabbit IgG (1:5000; Abcam) at room temperature for 1h. Signal detection was performed with the ECL-Plus Western blotting reagent kit (GE Healthcare, Piscataway, NJ, USA).
MTT assay
The ZNF569 overexpression cancer cells and the control cells were seeded into a 96-well plate at a density of 3000 cells/well. Twenty microliter of MTT solution (5 mg/ml in PBS) was added at each indicated time point and incubated for 4 h at 37 °C. The supernatant was then removed and 200 μl of dimethyl sulfoxide was added to dissolve the precipitate. The absorbance at 570 nm was measured using a microculture plate reader (Tecan, Mannedorf, Switzerland).
5-ethynyl-2′-deoxyuridine assay
The 5-ethynyl-2′-deoxyuridine (EdU) detection kit (Invitrogen) was used to evaluate the percentage of proliferating cells at the S phase. Briefly, 10 μmol/L EdU was added to the cells and cultured for 2 h at 37°C. The cells were fixed with 3.7% formaldehyde and washed with PBS. Then 0.5% Triton X-100 was added to the cells to increase the permeability of the cellular membrane. The cells protected from light were stained with 1 × Click-iT reaction cocktail for 30 min at room temperature. Hoechst 33342 dye was subsequently used to stain the cell nucleus following PBS washing. Images were captured under a confocal laser scanning microscope (Olympus, Center Valley, PA).
Wound healing assay
Briefly, cancer cells were seeded at a density of 5 × 105 cells/well into a 6-well plate and grown until monolayer confluence. Vertical scratch wounds were generated using 200μl micropipette tips. At least four random images per well were photographed (Eclipse TE2000, Nikon, Tokyo, Japan) at the beginning and after 24 h. The width of the wounds was calculated and analyzed by the NIH Image J software.
Transwell Matrigel invasion assay
Following 24 h serum starvation, trypsinized cells (5 × 105 cells /well) were resuspended in DMEM without FBS supplement and added to upper chamber of transwell matrigel invasion inserts (BD Biosciences. Bedford, MA, USA). The lower chamber was filled with complete culture medium. After 24 h, cells that had invaded through the membrane were fixed by 3.7% paraformaldehyde and stained with the crystal violet. At least four random images per insert were photographed and the NIH Image J software was used to count the average number of invaded cells per field.
Statistical analysis
The results were analyzed by the independent samples t-test using GraphPad Prism (version 7.0, GraphPad Software Inc., San Diego, CA, USA). X tile software (https://medicine.yale.edu/lab/rimm/research/software.aspx) was used to find out the optimum cutoff point to divide the HNSCC cases into high ZNF569 expression group and low ZNF569 expression group. Kaplan-Meier method in combination with log-rank test were used for the survival analysis, and P values < 0.05 was statistically significant.
Results
The hypermethylated/hypomethylated genes associated with lymph node metastasis of HNSCC
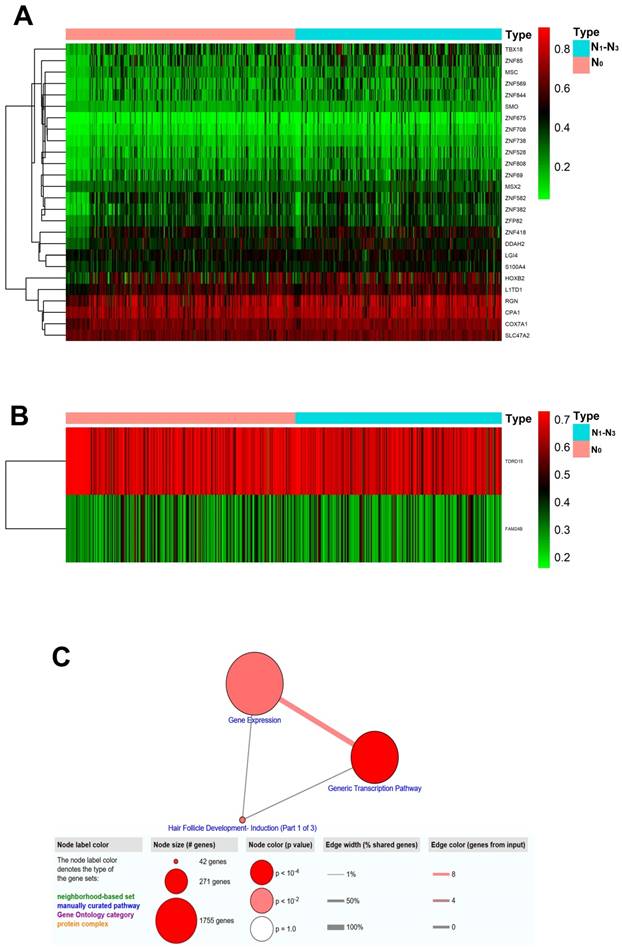
Figure 1 summarized the top hypermethylated or hypomethylated genes associated with lymph node metastasis of HNSCC. COX7A1, HOXB2, L1TD1, MSX2, RGN, SMO, TBX18, ZNF69, ZNF85, ZNF569 were the top ten hypermethylated genes in tumor samples with lymph node metastasis. However, only two hypomethylated genes namely FAM24B and TDRD15 were identified. The list of statistically different hypermethylated or hypomethylated genes were listed in Table 1 and Table 2 respectively. Figure 2A-2B revealed the heatmaps of the hypermethylated or hypomethylated genes. The pathway analysis of the hypermethylated genes showed that generic transcription pathway and gene expression were the top enriched pathways (Figure 2C).
The top methylated DNA markers (MDMs) associated with lymph node metastasis of HNSCC. (A) The top ten hypermethylated genes associated with lymph node metastasis of HNSCC; (B) The top hypomethylated genes associated with lymph node metastasis of HNSCC.
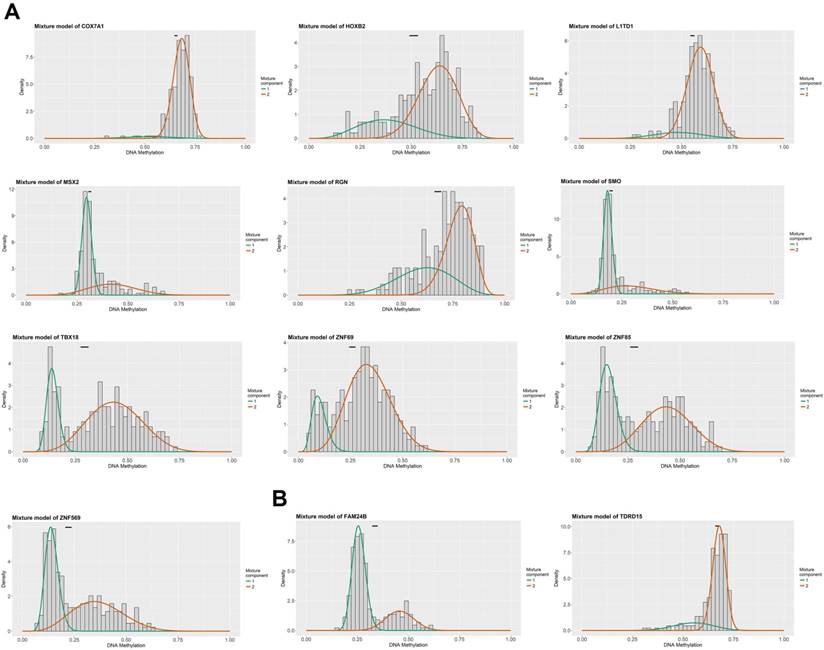
The hypermethylated genes associated with lymph node metastasis of HNSCC
| Gene | LN (-) mean | LN (+) mean | P value | Correlation |
|---|---|---|---|---|
| TBX18 | 0.294 | 0.363 | 0.000 | -0.632 |
| COX7A1 | 0.655 | 0.673 | 0.000 | -0.416 |
| ZNF69 | 0.259 | 0.301 | 0.000 | -0.475 |
| MSX2 | 0.312 | 0.342 | 0.001 | -0.347 |
| RGN | 0.678 | 0.713 | 0.001 | -0.389 |
| ZNF569 | 0.222 | 0.260 | 0.001 | -0.580 |
| L1TD1 | 0.553 | 0.579 | 0.002 | -0.403 |
| HOXB2 | 0.512 | 0.563 | 0.002 | -0.561 |
| ZNF85 | 0.280 | 0.329 | 0.002 | -0.425 |
| SMO | 0.198 | 0.217 | 0.003 | -0.368 |
| ZNF582 | 0.354 | 0.392 | 0.003 | -0.573 |
| DDAH2 | 0.432 | 0.455 | 0.003 | -0.418 |
| ZNF418 | 0.447 | 0.477 | 0.003 | -0.527 |
| ZNF382 | 0.307 | 0.343 | 0.003 | -0.382 |
| LGI4 | 0.466 | 0.481 | 0.004 | -0.436 |
| ZNF675 | 0.085 | 0.111 | 0.004 | -0.409 |
| S100A4 | 0.424 | 0.440 | 0.004 | -0.343 |
| MSC | 0.249 | 0.276 | 0.004 | -0.408 |
| ZNF844 | 0.233 | 0.265 | 0.004 | -0.621 |
| ZNF708 | 0.109 | 0.130 | 0.005 | -0.350 |
| ZNF528 | 0.192 | 0.226 | 0.005 | -0.546 |
| ZNF738 | 0.158 | 0.177 | 0.005 | -0.346 |
| ZNF808 | 0.214 | 0.236 | 0.006 | -0.544 |
| ZFP82 | 0.339 | 0.365 | 0.006 | -0.519 |
| SLC47A2 | 0.634 | 0.646 | 0.006 | -0.528 |
| CPA1 | 0.712 | 0.728 | 0.006 | -0.324 |
The hypermethylated genes associated with lymph node metastasis of HNSCC
| Gene | LN (-) mean | LN (+) mean | P value | Correlation |
|---|---|---|---|---|
| TDRD15 | 0.668 | 0.656 | 0.001 | -0.516 |
| FAM24B | 0.336 | 0.314 | 0.007 | -0.516 |
Heatmaps and pathway analysis of the altered MDMs. (A) Heatmap of hypermethylated genes associated with lymph node metastasis of HNSCC. The green/red color indicates relatively low/high methylation level. Each column represented an independent biological sample and each row indicates a gene; (B) Heatmap of hypomethylated genes associated with lymph node metastasis of HNSCC; (C) Pathway analysis of the hypermethylated genes.
ZNF569 was downregulated in HNSCC tissues and cell lines
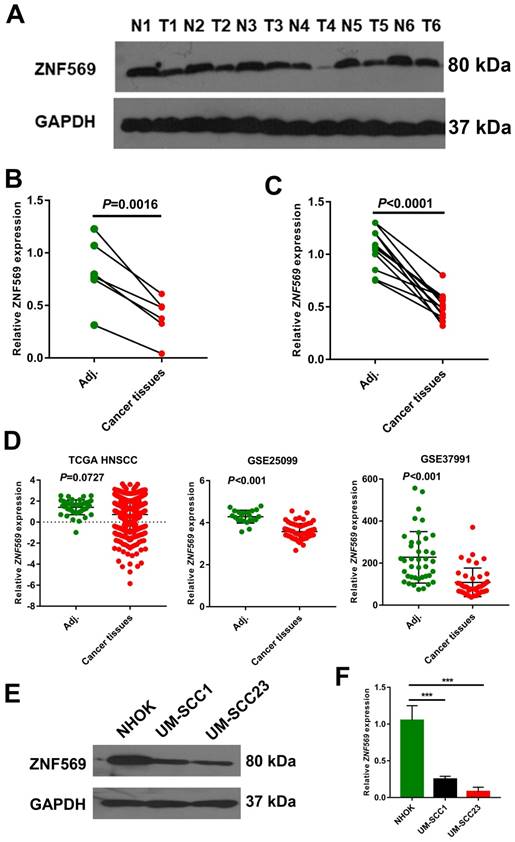
We have also calculated the beta value of ZNF569 in HNSCC tumor and normal samples from TCGA HNSCC cohort. The MethylMix analysis showed that ZNF569 was also highly methylated in the HNSCC tissues (beta value=0.258) compared to the normal tissues (beta value=0.118) (P<0.001) (Table 3). Then we compared ZNF569 level in six HNSCC tissues and paired adjacent normal tissues. Our western blotting result showed that ZNF569 level was significantly lower in HNSCC tissues compared to the normal tissues (P=0.0016) (Figure 3A-3B). Similarly, the expression level of ZNF569 mRNA was dramatically reduced in HNSCC tissues compared to the adjacent normal control tissues (P<0.001) (Figure 3C). Moreover, we have compared the expression level of ZNF-569 between HNSCC and normal control using the samples from TCGA database and GEO database. Our results showed that the expression levels of ZNF-569 were significantly downregulated in HNSCC tissues compared to the normal tissues in GSE25099 and GSE37991 (P<0.001). For the TCGA data, the ZNF-569 level was also found to be reduced in HNSCC samples compared to the adjacent normal tissues, although the P value approached marginal significance (P=0.0727) (Figure 3D). Furthermore, the expression levels of ZNF569 were remarkably lower in HNSCC cell lines (UM-SCC1 and UM-SCC23) compared with the normal oral epithelial cells (NHOK) (P<0.001) (Figure 3E-3F).
ZNF569 was hypermethylated in HNSCC tissues
| Gene | Normal mean | Tumor mean | P value | Correlation |
|---|---|---|---|---|
| ZNF569 | 0.118 | 0.258 | 0.000 | -0.603 |
ZNF569 was downregulated in HNSCC tissues and cell lines. (A-B) The expression level of ZNF569 protein was significantly downregulated in HNSCC tissues compared with the adjacent normal tissues (P=0.0016); (C) The expression level of ZNF569 mRNA was significantly downregulated in HNSCC tissues compared with the adjacent normal tissues (P<0.001); (D) Similarly, ZNF569 levels was found to be downregulated in cancer tissues compared to normal tissues in various independent cohort study such as GSE25099 (P<0.001), GSE37991 (P<0.001) and TCGA (P=0.0727). (E-F) The levels of ZNF569 mRNA and protein were remarkably lower in HNSCC cell lines compared to control cell line (P<0.001).
ZNF569 overexpression lentiviruses upregulated ZNF569 mRNA and protein levels
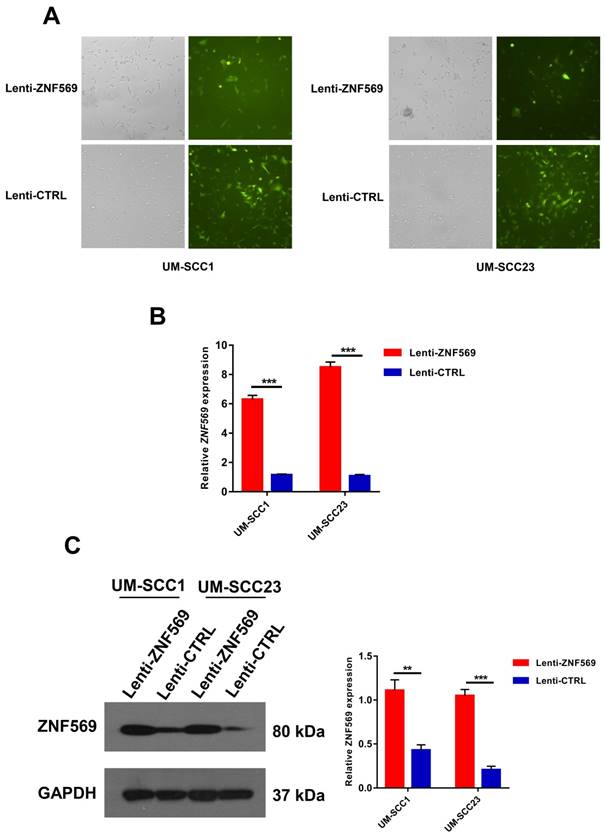
Both UM-SCC1 and UM-SCC23 cancer cells were successfully infected with ZNF569 overexpression and control lentiviruses. GFP was used to monitor the infection efficiency (Figure 4A). Real-time PCR and western blotting demonstrated the ZNF569 lentiviruses we constructed could efficiently upregulated ZNF569 at both mRNA and protein levels (**P<0.01, ***P<0.001) (Figure 4B-4C).
The construction of ZNF-569 overexpression lentiviruses. (A) The lentiviruses we constructed were able to infect HNSCC cell lines with high efficiency; (B-C) The expression levels of ZNF569 mRNA and protein were significantly higher in cancer cells that infected with ZNF-569 overexpression lentiviruses compared to those infected with the control lentiviruses (**P<0.01, ***P<0.001).
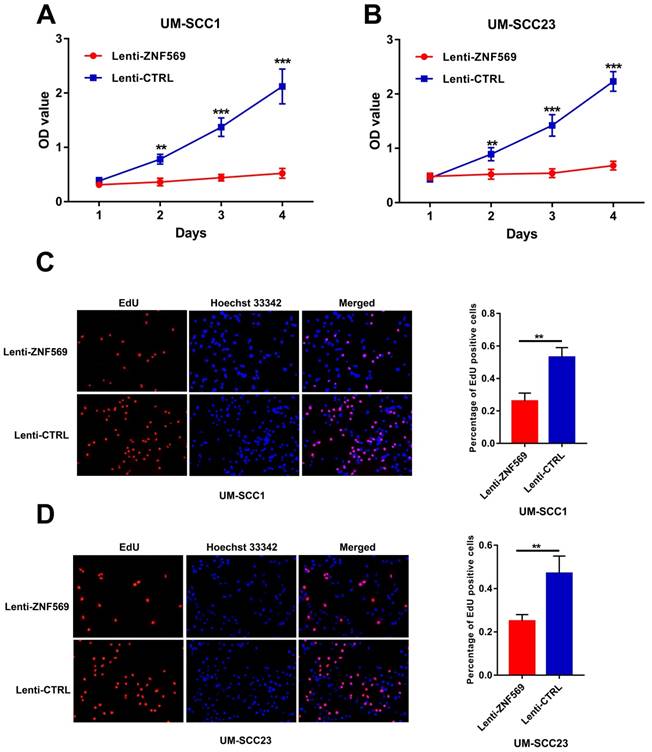
ZNF569 overexpression inhibited the proliferation of HNSCC cells
The MTT assay showed that the OD values were significantly lower in cancer cells that infected with ZNF569 lentiviruses compared to those infected with control lentiviruses in both UM-SCC1 and UM-SCC23 cell lines at all the time points except day 1 (**P<0.01, ***P<0.001) (Figure 5A-5B). In addition, the EdU assay demonstrated that the percentage of EdU positive UM-SCC1 cells was dramatically lower in the ZNF569 overexpression group than in the control group. Similar findings were observed in UM-SCC23 cells (P<0.01) (Figure 5C-5D).
ZNF569 overexpression inhibited the proliferation of HNSCC cell lines. (A-B) The OD values were significantly lower in ZNF569 overexpression cancer cells compared to the control cells (**P<0.01, ***P<0.001); (C-D) The percentage of EdU positive cells were lower in ZNF569 overexpression cancer cells compared to the control cells (**P<0.01).
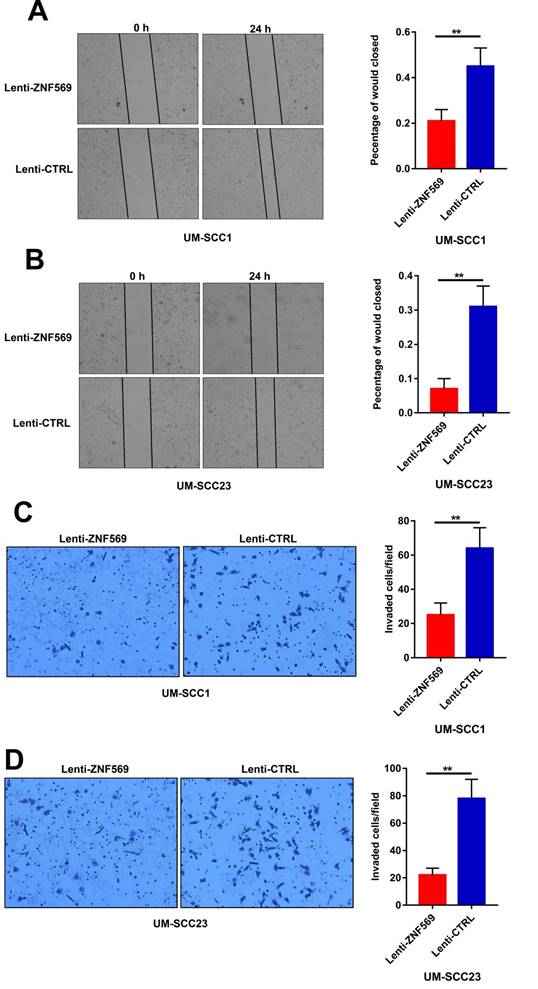
ZNF569 overexpression suppressed the migration and invasion capacity of HNSCC cells
The wound healing assay revealed that the cancer cells in the control group had a higher efficiency at closing the wound width than those in the ZNF569 overexpression group (P<0.01) (Figure 6A-6B). Similarly, the matrigel invasion assay showed that the number of cancer cells that invaded through the membrane was significantly lower in ZNF569 overexpression group in comparison with the control group (P<0.01) (Figure 6C-6D).
ZNF569 overexpression suppressed the migration and invasion of HNSCC cells. (A-B) The wound widths were larger in ZNF569 overexpression cancer cells compared to the control cells (**P<0.01); (C-D) The number of cancer cells that invaded through the membrane was lower in cancer cells from ZNF569 overexpression group (**P<0.01).
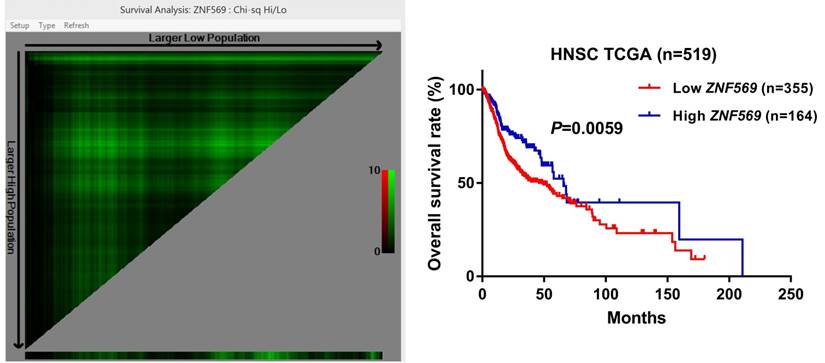
ZNF569 overexpression was associated with unfavorable clinical outcome of HNSCC
Our survival analysis showed that the HNSCC patients in the low ZNF569 expression group suffered a significantly shorter long-term overall survival than those in the high ZNF569 expression group (P=0.0059) (Figure 7), indicating ZNF569 might function as a tumor suppressor in HNSCC.
Low ZNF569 expression was associated with unfavorable overall survival. HNSCC patients with lower ZNF569 expression had a significantly worse long-term overall survival than those with higher ZNF569 expression (P=0.0059).
Discussion
In this study, we have identified many methylated DNA markers (MDMs) that were closely associated with the lymph node metastasis of HNSCC. The reasons we chose ZNF-569 as the research target were as follows: Firstly, our bioinformatic analysis showed that the generic transcriptional pathway was the top enriched pathway associated with the lymph node metastasis of HNSCC. Most of the deregulated transcriptional factors were ZNFs family members. Secondly, currently the role of ZNF-569 in carcinogenesis of HNSCC or even cancer is poorly known. Our results demonstrated that our interested target gene ZNF569 was hypermethylated in HNSCC specimens especially those with lymph node metastasis. In addition, ZNF569 was significantly downregulated in HNSCC cell lines and tissues. Moreover, ectopic expression of ZNF569 suppressed the proliferation, migration and invasion capacity of HNSCC cells. Furthermore, reduced ZNF569 level was associated with unfavorable overall survival of HNSCC. Taken together, loss of ZNF569 promoted the tumorigenesis of HNSCC and ZNF569 might play a tumor suppressive role in HNSCC.
Epigenetic modification of the DNA is extremely important for transforming the normal cells to cancer cells. Loss of tumor suppressor genes by abnormal CpG island methylation is a common epigenetic event for tumorigenesis [12-13]. Many MDMs we identified are very novel or even first time reported to be associated with the tumorigenesis of HNSCC. For instance, many members from HOX family including HOXB2 has been demonstrated to be hypermethylated in oral cancer cell lines [14]. COX7A1 was significantly reduced in lung cancer tissues and cell lines. Overexpression of COX7A1 suppressed the proliferation and induced apoptosis of cancer cells, indicating that COX7A1 played a tumor suppressive role in lung cancer [15]. Our pathway analysis of hypermethylated genes showed that generic transcription pathway was upregulated. Abnormal activation of transcriptional factors plays an essential role in the initiation and progression of cancer [16]. Interestingly, most of the hypermethylated transcriptional factors belong to the ZNF family. Several members of ZNF family we identified has been shown to be closely associated with carcinogenesis of HNSCC. Our results showed that ZNF582 was highly methylated in tumor specimens with lymph node metastasis compared to those without lymph node metastasis. Consistent with our findings, the methylation level of ZNF582 was remarkably higher in moderate/severe dysplasia/oral cancer than mild dysplasia/normal control [17].
In addition, hypermethylated ZNF582 in the oral scrapings collected from adjacent normal oral mucosal sites of cancer tissues was positively correlated with aggressive progression and poor prognosis of OSCC [18]. Similarly, ZNF382 was hypermethylated in many cancer types such as nasopharyngeal cancer, esophageal cancer and gastric cancer. Ectopic expression of ZNF-382 inhibited the malignant behaviors of cancer cells by suppressing many important oncogenic pathways [19-20]. We were not able to perform pathway analysis of the hypomethylated genes as only two statistically significant hypomethylated genes were identified.
To the best of our knowledge, this was the first time we showed that ZNF569 acted as a tumor suppressor in HNSCC or even human cancer. ZNF569 is a transcription factor characterized by an N-terminal Kruppel-associated box domain and tandem C-terminal C2H2-type zinc fingers. It is highly conserved across different species and widely distributed in human tissues [21]. Barrett's oesophagus is a premalignant condition that predisposes to the progression of oesophageal adenocarcinoma. ZNF569 was found to be highly methylated in Barrett's oesophagus, indicating that downregulation of ZNF569 might be associated with initiation of Barrett's oesophagus [22]. A recent study reported that ZNF569 was highly methylated in the tissue and plasma samples from patients with gastric adenocarcinoma (GAC). In addition, a panel of three plasma methylated DNA markers (ELMO1, ZNF569, C13orf18) was able to detect most of GAC at high specificity [23]. These data suggested that ZNF569 methylation might be a common phenomenon in human cancer. Stark et al showed that ZNF569 could regulate the cellular response to paclitaxel. Knockdown of ZNF569 in lymphoblastoid cell lines led to apoptosis induction and cell survival promotion in response to paclitaxel, suggesting that ZNF569 levels might be closely with the chemosensitivity of cancer cells [24]. Activation of mitogen activated kinases (MAPKs) plays a crucial role in the initiation and development to HNSCC [25]. ZNF569 seems to be a suppressor of MAPK signaling pathway, which might partially explain its tumor inhibition effects in HNSCC [21]. Further studies are warranted to explore the molecular mechanisms accounting for the tumor suppressive role of ZNF569. In addition, monitoring the levels of methylated ZNF569 in the biofluids such as serum, plasma and saliva might have promising potential for early detection, prognosis prediction and therapeutic efficacy evaluation for patients with HNSCC.
In conclusion, we have successfully identified many novel MDMs that might be highly correlated with the lymph node metastasis of HNSCC. The novel MDM ZNF569 is downregulated in HNSCC tissues and cell lines, and overexpression of ZNF569 suppresses the oncogenic activities of HNSCC cells. Therefore, ZNF569 might play a tumor suppressive role in tumorigenesis of HNSCC.
Acknowledgements
The study was supported by Natural Science Foundation of Guangdong Province (Grant No: 2016A030310176) the National Natural Science Foundation of China (Grant No: 81801454).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhao X, Sun S, Zeng X, Cui L. Expression profiles analysis identifies a novel three-mRNA signature to predict overall survival in oral squamous cell carcinoma. Am J Cancer Res. 2018;8:450-61
2. Argiris A, Karamouzis MV, Raben D, Ferris RL. Head and neck cancer. Lancet. 2008;371:1695-709
3. Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212-36
4. Forastiere A, Koch W, Trotti A, Sidransky D. Head and neck cancer. N Engl J Med. 2001;345:1890-900
5. Moore LD, Le T, Fan G.DNA methylation, its basic function. Neuropsycho-pharmacology. 2013; 38:23-38.
6. Robertson KD.DNA methylation and human disease. Nat Rev Genet. 2005; 6:597-610.
7. Liang G, Weisenberger DJ. DNA methylation aberrancies as a guide for surveillance and treatment of human cancers. Epigenetics. 2017;12:416-32
8. Maruya S, Issa JP, Weber RS, Rosenthal DI, Haviland JC, Lotan R. et al. Differential methylation status of tumor-associated genes in head and neck squamous carcinoma: incidence and potential implications. Clin Cancer Res. 2004;10:3825-30
9. El-Naggar AK, Lai S, Clayman G, Lee JK, Luna MA, Goepfert H. et al. Methylation, a major mechanism of p16/CDKN2 gene inactivation in head and neck squamous carcinoma. Am J Pathol. 1997;151:1767-74
10. Yang CM, Wang TH, Chen HC, Li SC, Lee MC, Liou HH. et al. Aberrant DNA hypermethylation-silenced SOX21-AS1 gene expression and its clinical importance in oral cancer. Clin Epigenetics. 2016;8:129
11. Lee JS. Exploring cancer genomic data from the cancer genome atlas project. BMB Rep. 2016;49:607-11
12. Wajed SA, Laird PW, DeMeester TR. DNA methylation: an alternative pathway to cancer. Ann Surg. 2001;234:10-20
13. Klutstein M, Nejman D, Greenfield R, Cedar H. DNA methylation in cancer and aging. Cancer Res. 2016;76:3446-50
14. Xavier FC, Destro MF, Duarte CM, Nunes FD. Epigenetic repression of HOXB cluster in oral cancer cell lines. Arch Oral Biol. 2014;59:783-9
15. Mishra N, Timilsina U, Ghimire D, Dubey RC, Gaur R. Downregulation of cytochrome c oxidase subunit 7A1 expression is important in enhancing cell proliferation in adenocarcinoma cells. Biochem Biophys Res Commun. 2017;482:713-9
16. Bhagwat AS, Vakoc CR. Targeting transcription factors in cancer. Trends Cancer. 2015;1:53-65
17. Cheng SJ, Chang CF, Ko HH, Liu YC, Peng HH, Wang HJ. et al. Hypermethylated ZNF582 and PAX1 genes in oral scrapings collected from cancer-adjacent normal oral mucosal sites are associated with aggressive progression and poor prognosis of oral cancer. Oral Oncol. 2017;75:169-77
18. Cheng SJ, Chang CF, Ko HH, Lee JJ, Chen HM, Wang HJ. et al. Hypermethylated ZNF582 and PAX1 genes in mouth rinse samples as biomarkers for oral dysplasia and oral cancer detection. Head Neck. 2018;40:355-68
19. Cheng Y, Geng H, Cheng SH, Liang P, Bai Y, Li J. et al. KRAB zinc finger protein ZNF382 is a proapoptotic tumor suppressor that represses multiple oncogenes and is commonly silenced in multiple carcinomas. Cancer Res. 2010;70:6516-26
20. Liu Z, Zhang J, Gao Y, Pei L, Zhou J. et al. Large-scale characterization of DNA methylation changes in human gastric carcinomas with and without metastasis. Clin Cancer Res. 2014;20:4598-612
21. Huang X, Yuan W, Huang W, Bai Y, Deng Y, Zhu C. et al. ZNF569, a novel KRAB-containing zinc finger protein, suppresses MAPK signaling pathway. Biochem Biophys Res Commun. 2006;346:621-8
22. Chettouh H, Mowforth O, Galeano-Dalmau N, Bezawada N, Ross-Innes C, MacRae S. et al. Methylation panel is a diagnostic biomarker for Barrett's oesophagus in endoscopic biopsies and non-endoscopic cytology specimens. Gut. 2018;67:1942-9
23. Anderson BW, Suh YS, Choi B, Lee HJ, Yab TC, Taylor WR. et al. Detection of gastric cancer with novel methylated DNA markers: discovery, tissue validation, and pilot testing in plasma. Clin Cancer Res. 2018;24:5724-34
24. Stark AL, Hause RJ Jr, Gorsic LK, Antao NN, Wong SS, Chung SH. et al. Protein quantitative trait loci identify novel candidates modulating cellular response to chemotherapy. PLoS Genet. 2014;10:e1004192
25. Leelahavanichkul K, Amornphimoltham P, Molinolo AA, Basile JR, Koontongkaew S, Gutkind JS. A role for p38 MAPK in head and neck cancer cell growth and tumor-induced angiogenesis and lymphangiogenesis. Mol Oncol. 2014;8:105-18
Author contact
![]() Corresponding author: Dr. Xiangzhen Liu, First Affiliated Hospital, Sun Yat-sen University, Guangzhou, China. Email: lxzh3sysu.edu.cn, Phone: +8620-82379472
Corresponding author: Dr. Xiangzhen Liu, First Affiliated Hospital, Sun Yat-sen University, Guangzhou, China. Email: lxzh3sysu.edu.cn, Phone: +8620-82379472