Journal of Cancer
ISSN: 1837-9664
3.2
Impact Factor
ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(11):2415-2424. doi:10.7150/jca.32071 This issue Cite
Research Paper
Cordycepin Induces Apoptosis and G2/M Phase Arrest through the ERK Pathways in Esophageal Cancer Cells
Jia-Cheng Xu1*, Xue-Ping Zhou2*, Xu-An Wang2, Mei-Dong Xu1, Tao Chen1, Tian-Yin Chen1, Ping-Hong Zhou1 ![]() , Yi-Qun Zhang1
, Yi-Qun Zhang1 ![]()
1. Endoscopy Center and Endoscopy Research Institute, Zhongshan Hospital, Fudan University, No. 180 FengLin Road, Shanghai 200032, China.
2. Department of General Surgery, School of Medicine, Shanghai Jiao Tong University, No. 1665 Kongjiang Road, Shanghai 200092, China.
* These authors contributed equally to this work.
Received 2018-12-7; Accepted 2019-4-10; Published 2019-5-26
Citation:
Xu JC, Zhou XP, Wang XA, Xu MD, Chen T, Chen TY, Zhou PH, Zhang YQ. Cordycepin Induces Apoptosis and G2/M Phase Arrest through the ERK Pathways in Esophageal Cancer Cells. J Cancer 2019; 10(11):2415-2424. doi:10.7150/jca.32071. https://www.jcancer.org/v10p2415.htm
Other stylesAbstract
Esophageal cancer is one of the most aggressive and lethal gastrointestinal tract malignancies, with a poor overall five-year survival rate. Cordycepin, a major compound of Cordyceps sinensis, has been shown to have anticancer potential. This study focuses on the anticancer properties of cordycepin that target esophageal cancer and reveals molecular aspects underlying these effects. In our CCK-8 assays and colony formation assays, cordycepin significantly suppressed esophageal cancer cell proliferation. Moreover, cordycepin induced chromatin condensation in esophageal cancer cells and significantly increased the number of apoptotic cells through activation of caspase cascades, apoptotic signaling, and the regulation of Bcl-2 family members. Cell cycle assays showed that cordycepin altered cyclin-dependent kinase1 and cyclinB1 expression, which resulted in a G2/M phase blockade. Mechanistically, ERK pathway inactivation was involved in the anti-tumor functions of cordycepin. The same results were also observed in vivo. Taken together, these findings reveal that cordycepin induces pro-apoptosis and anti-proliferation mechanisms in cancer cells, and may represent a novel therapeutic agent.
Keywords: cordycepin, esophageal cancer, apoptosis, cell cycle, ERK
Introduction
Esophageal cancer is one of the most aggressive and highly lethal malignant tumors in China [1]. Associated morbidity and mortality has jumped to the fourth highest of all forms of malignant tumors. Esophageal squamous cell carcinoma (ESCC) accounts for more than 90% of cases of esophageal cancer worldwide [2]. Although the incidence of ESCC has decreased over the past two decades, it remains one of the most significant contributors to cancer death [2]. The overall 5-year survival rate from the time diagnosis is only 15%. The most comprehensive and primary curative surgical treatment of esophageal cancer is esophagostomy. Unfortunately, at diagnosis most patients already have advanced local disease with regional lymph node metastasis, which leads to tumor lesion recurrence or treatment failure [3, 4]. Chemotherapy is an important component of combination therapy to improve outcomes for patients with non-resectable tumors [5]. Therefore, the development of non-toxic and effective chemotherapy agents against esophageal cancer is urgently needed.
Recent studies show that there is growing interest in the use of traditional Chinese medicine as a complementary or alternative therapy. Cordycepin (3′-Deoxyadenosine) is a natural, plant-derived compound extracted from the traditional Chinese medicine herbs Cordyceps sinensis and Cordyceps militaris [6]. Cordycepin is a cytotoxic nucleoside with a structure similar to that of adenosine, but which lacks the 3′-hydroxyl group of the ribose moiety that is involved in DNA/RNA polymerase function [7, 8]. The putative pharmacological activities of this drug include anti-inflammatory, antioxidant, anti-aging, and anti-tumor actions, among others [9-12]. Moreover, cordycepin has been demonstrated to induce cancer cell death in multiple types of cancer, including colon cancer cells, renal cancer cells, breast cancer cells and gallbladder cancer cells, by regulating specific molecules pathways [13-16]. However, cordycepin has not been investigated in esophageal cancer cells until now. In this study, our results show that cordycepin inhibits the proliferation of esophageal cancer cells by inducing apoptosis and G2/M phase arrest. We identified that the functional mechanism was the ERK pathway, a finding that sheds light on why cordycepin may be a singular therapeutic intervention for esophageal cancer.
Materials and Methods
Drugs and reagents
Cordycepin was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Sample purity was at least 98%. Cordycepin was dissolved in 100% dimethyl sulfoxide (DMSO) and stored at -20°C. The final DMSO concentration did not exceed 0.4% (>0.1%). For negative controls, isovolumetric DMSO was added to the culture medium. Propidium iodide (PI), Annexin V-FITC, and Hoechst 33342 were purchased from Invitrogen (Carlsbad, CA, USA). All antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA).
Cell cultures
The ECA109 and TE-1 cells were purchased from the cell bank of Shanghai Institutes for Biological Sciences (Shanghai, China). HEK-293T cells were obtained from the American Type Culture Collection (ATCC). ECA109 and 293T cells were cultured in Dulbecco`s Modified Eagle`s Medium (Gibco); TE-1 cells were maintained in 1640 medium (Gibco). All media were supplemented with 10% fetal bovine serum (Gibco, NY, USA), 100 µg/ml penicillin/ streptomycin. Both cell types were cultured at 37 °C and 5% CO2.
Cell Proliferation Assay
Cell proliferation was assessed using a cell counting Kit-8 assay (Dojindo, Kumamoto, Japan). Approximately 1×103 of ECA109, 293T and TE-1 cells were seeded in a 96-well plate and cultured for 24h. The cells were subsequently treated with cordycepin at sequential concentrations (0, 20, 40, 60, 80, 100μg/mL). After 24h, 48h, and 72h of treatment, the 450 nm absorbance of the solution was measured using a microtiter plate reader (Bio-Rad, CA, USA). IC50 was measured by Graphpad Prism 7.
Colony formation assay
ECA109 and TE-1 cells were plated during exponential growth phases into 6-well plates (500 cells/well). After adherence, cells were exposed to cordycepin for 14 days at concentrations of 0, 40, 60, and 80 μg/ml. Then, colonies were fixed with 4% paraformaldehyde for 30 min and stained with 0.1% crystal violet solution (Sigma, USA) for 30 min. The plates were dried and the colonies (>50 cells/colony) were counted under microscopy.
Cell apoptosis assay
For the cell apoptosis assays, ECA109 and TE-1 cells were plated in 6-well plates and treated with various doses of cordycepin (0, 40, 60 and 80 μg/mL). After 48h of treatment, adherent cells and floating cells were collected. The cells were then washed with PBS twice and suspended with 100 µl of 1 x binding buffer containing 5 µl of AnnexinV-FITC and 5 µl of PI staining solution in accordance with the manufacturer′s instruction. After incubation for 30 min at 37 °C, the samples were measured using flow cytometry (BD Biosciences, USA).
Hoechst 33342 staining
ESCC Cells were treated with 0, 40, 60 and 80 μg/mL cordycepin for 48h, then washed twice with PBS and subsequently fixed in methanol:acetic acid (3:1) for 20 min. The cells were again washed twice with PBS and stained with 5 µg/ml Hoechst 33342 for 10 min at room temperature. Next, a fluorescence microscope (Lecia) was used to analyze the morphological changes.
Cell cycle analysis
The effect of cordycepin on cell cycle progression was analyzed by flow cytometry. ECA109 and TE-1 cells were harvested after 48h treatment with cordycepin and washed with PBS. The cell pellets were then fixed in 70% ethanol overnight at 4 °C. After incubating in darkness for 30 minutes at 37°C the cells were treated with RNase and propidium iodide (PI). Finally, samples were analyzed by flow cytometry and the percentage of G0/G1, S, and G2/M phases cells were measured by BD Bioscience.
Western blot analysis
Following the previously described treatments, the cells were harvested and lysed in RIPA buffer (CellSignaling, Danvers, MA, USA). Cell lysate proteins were separated using 10% sodium dodecyl sulfate polyacrylamide gel and transferred to polyvinylidene difluoride membranes (Millipore, USA). Membranes were then blocked for 1h at room temperature and incubated with primary antibodies against cleaved-caspase9, cleaved-caspase3, cleaved-|PARP, PARP, ERK, MEK, P-ERK, P-MEK, Bcl-2, BAX, GAPDH overnight at 4°C. Next, the membranes were incubated with suitable HRP-conjugated secondary antibodies for 1h at 37 °C. Proteins were scanned using a Gel Doc 2000 (Berkeley, California, USA).
In vivo efficacy of cordycepin
Four- to six-week-old male nude mice (20 ±2 g) were obtained from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). The animals were housed under specific pathogen-free conditions. The animal experiments were approved by the National Institutional Animal Care and Use Committee of Shanghai Jiao Tong University, and were carried out in strict accordance with the institutional guidelines of Shanghai Jiao Tong University. ECA109 cells were resuspened in PBS and injected into the right axilla of nude mice (2x106 /mice). After 1 week, these mice were randomly divided into three groups (control, 5mg/kg, 10mg/kg), with five animals in each respective group. Control animals received an intraperitoneal injection of 10% DMSO and 90% PBS every two days. The other two groups were received cordycepin (5 and 10 mg/kg) every two days. On Day 25, mice were sacrificed and primary tumors were removed and weighed for further Western blot and immunohistochemical staining analysis.
Figure 1
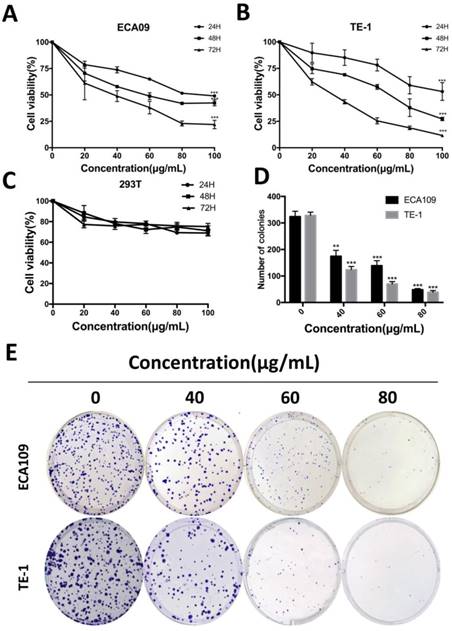
Cordycepin suppresses ESCC cell proliferation. (A,B) ECA109 and TE-1 cells were treated with cordycepin (0, 20,40, 60, 80 and 100μg/ml) for 24, 48, and 72 h. Cell viability was assessed using a CCK-8 assay. (C) 293T cell viability was evaluated by CCK-8 analysis after treatment with cordycepin. (D,E) ECA109 and TE-1 cells were exposed to cordycepin and were allowed to form colonies for 14 days. Colony numbers were counted and recorded. Results are presented as mean ± SD (n= 3 independent experiments). *p<0.05, **p<0.01, and ***p<0.001.
HE staining and IHC in xenograft tumor
After the mice were sacrificed, tumors were removed and formalin-fixed, embedded in paraffin and cut into sections. Paraffin-embedded sections of the mouse livers were stained for histopathological examination according to standard protocols. Immunohistochemical staining was performed using a standard immunoperoxidase staining procedure. Expression of Ki67, cleaved-caspase 3, P-MEK, P-ERK were analyzed.
Statistical analysis
Analyses were performed with SPSS software, version 22.0. All quantified data are expressed as the mean ± standard. Statistical significance was calculated using the Student′s t-test. A P value of P<0.05 (*p<0.05, **p<0.01) was regarded as statistically significant. All experiments were performed at least three times.
Results
Cordycepin suppresses the proliferation of ESCC cells
To determine the function of cordycepin on cancer cells proliferation, we performed cell counting Kit-8 assays on ECA109 and TE-1 human esophagus squamous cancer cells. After treatment with sequential doses of cordycepin (0, 20, 40, 80, 160, 320, 640 μg/mL for both two cell lines) for 0, 24, 48 and 72h, the growth curve of cell viability decreased significantly in a dose-dependent manner with increased duration of treatment with cordycepin (Figure 1A,B). As normal esophageal cells were not incubated, human kidney epithelial cells (293T) were treated with cordycepin to evaluate whether normal cells underwent inhibitation following treatment [17, 18](Figure 1C). CCK-8 assays found no significant difference between cordycepin-treated 293T cells and the control group. The 50% growth-inhibitory concentrations (IC50) after treatment with cordycepin for 48h were 64.8 μg/mL in ECA109 cells and 60.6μg/mL in TE-1 cells. A flat plate colony formation assay was also performed to investigate the independent antiproliferation effects of cordycepin. The colony count decreased significantly after incubation with 40, 60, and 80μg/mL concentrations of cordycepin compared to the control group, indicating dose- dependent inhibition (Figure 1D,E). These findings support the hypothesis that cordycepin has anticancer effects on human ESCC in vitro.
Cordycepin induces apoptosis in ECA109 and TE-1 cells
To determine whether cordycepin effects apoptosis in esophagus squamous cancer cells in response to the inhibition of cell proliferation, we performed apoptosis assays on ECA109 and TE-1 cells treated with various concentration of cordycepin for 48h. To calculate the apoptosis outcomes, Annexin V-FITC/PI dual staining and Hoechst 33345 staining assays were used to assess cell death (Figure 2). Compared with untreated group, cells treated with cordycepin show typical apoptotic morphology that included chromatin condensation and apoptotic body formation (Figure 2D,E). Moreover, the fraction of early apoptotic cells strikingly increased in a dose-dependent manner in cordycepin pre-treated ESCC cells (Figure 2A-C). Additionally, despite the fact that the fractions of early apoptotic cells increased in both lines, the percentages of late apoptosis cells strikingly increased in ECA109. We hypothesize that this was because Annexin V positivity and PI positivity can also stain senescent and dead cells and may have influenced the results. Collectively, cordycepin demonstrated properties that inhibited the viability of esophagus cancer cells by inducing apoptosis.
Cordycepin arrests cells in G2/M stage and regulates expression of G2/M phase cell cycle-related proteins
To assess whether the suppressive effects of cordycepin on ESCC cells proliferation was due to arrest of cell cycle progression, we analyzed of the ESCC cell cycles. Flow cytometry showed significant accumulation of cells in the G2/M stage after exposure to cordycepin for 48h with the previously described concentrations, and induced cell cycle arrest in the G2/M phase (Figure 3A-C). Western Blot analyses were then performed to investigate the expression of cell cycle-related proteins (Figure 3D). The results indicated that there was a substantial decrease in the expression of the G2/M-phase-related proteins CDK1 and cyclin B1, both of which are key molecular regulators of G2/M progression [19]. Together, these results indicate that cordycepin induces arrest of the G2/M phase, a finding that accounts for its antiproliferative function by disturbing the level of cell cycle related proteins.
Cordycepin regulates the expression of caspase and Bcl-2 families
We examined the expression of apoptosis-related proteins, including caspase and Bcl-2 family proteins, in ECA109 and TE-1 cells after treatment with cordycepin for 48h by Western Blot analysis. As shown in Figure 4A-C, exposure to cordycepin significantly increased levels of cleaved-caspase3, cleaved-caspase9, cleaved-PARP, and Bax proteins, and also downregulated Bcl-2 and the Bax/Bcl-2 ratio. This indicates that cordycepin activates intrinsic apoptosis pathways [20]. To further investigate these results, we used a caspase inhibitor (Z-VAD-FMK) to evaluated cell viability after treatment with cordycepin. Gain of Z-VAD-FMK abrogated the pro-apoptosis and anti-proliferation effect after cordycepin treatment (Figure D). Collectively, these results indicate that cordycepin alters the levels of caspase and Bcl-2 family proteins in response to the apoptosis in ECA109 and TE-1 cells.
Cordycepin suppresses ESCC progression by regulating the ERK pathway
Multiple signal transduction pathways take part in the proposed cordycepin-induced anticancer effects [14-16]. To elucidate the molecular mechanisms by which cordycepin modulates the antitumor properties of ESCC cells, we measured the expression of cell cycle- and cell apoptosis related signaling molecules in ESCC cells. MAPK-mediated apoptotic induction and anti-proliferation plays a central role in many cordycepin depend tumor therapies. Therefore, we focused our study on this signaling pathway. Western blot analyses showed that the levels of the phosphorylated forms of MEK, ERK were significantly suppressed in both the cordycepin treated and control group in ECA109 and TE-1 cell lines. However, there was no obvious change in total expression of MEK,ERK (Figure 4E). To determine whether targeting MAPK/ERK attenuates the effects of cordycepin, we treat ECA109 and TE-1 cells with Sch772984, an ERK inhibitor. As anticipated, after treatment with cordycepin with Sch772984, we found that the ERK inhibitor significantly suppressed cancer cell viability (Figure 4F). These data suggest that inactivation of ERK pathway may be attributed to the suppressive effects of cordycepin on esophageal cancer.
Cordycepin is efficacious against ECA109 cell xenograft tumor growth
As cordycepin exhibits anti-cancer proliferation effect in vitro, we hypothesized that cordycepin could also inhibit tumorigenesis of ESCC cells in vivo. Xenograft tumors were grown in nude mice inoculated with ECA109 cells in log-phase. We injected mice with cordycepin (5mg/kg and 10mg/kg) or vehicle (DMSO+PBS) every two days. Both tumor volumes and weights dramatically decreased in a dose-dependent manner in the cordycepin-treated group compared with the vehicle group without obvious appearance differation (Figure 5A-C). Moreover, HE staining of the mice livers found no evident effect on organ structure and indicated that cordycepin exposure did not cause systemic toxicity (Figure 5D).
Figure 2
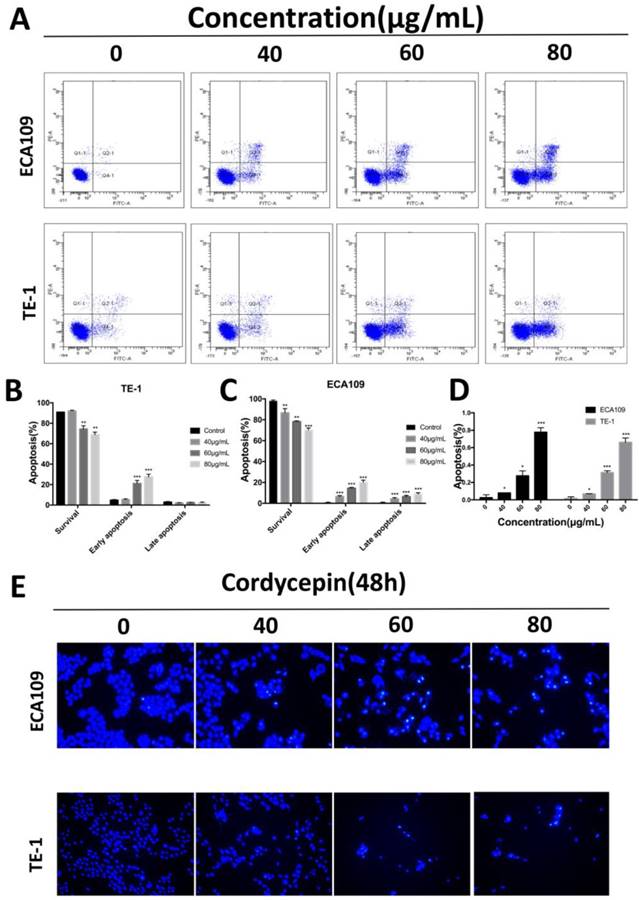
Cordycepin induces apoptosis in ECA109 and TE-1 cells. (A) ECA109 and TE-1 cells were treated with cordycepin (0,40, 60, 80μg/ml) for 48h. Apoptosis was assessed by flow cytometry with annexin V-FITC/propidium iodide (PI) staining. The x-axis represents annexinV-FITC, and the y-axis represents propidium iodide (PI) staining. (B,C) The Q3 quadrant (annexin V/PI), Q4 quadrant (annexin V+/PI), and Q2 quadrant (Annexin V+/PI+) indicate the percentages of normal cells, early apoptotic and late apoptotic cells, respectively. (D,E) Nuclear morphological changes associated with apoptosis were evaluated by Hoechst 33342 staining. Results are presented as mean ± SD (n = 3 independent experiments). *p<0.05, **p<0.01, and ***p<0.001.
Encourage by these results, we performed Western blot and IHC to analyze the expression of proteins in xenograft tumors. Based on the Western blot analysis in ECA109 cells, cleaved-caspase3, cleaved-caspase9, cleaved PARP, and Bax all significantly increased. Moreover, the downregulation of P-MEK, P-ERK and Bcl-2 was consistent with the results obtained in vitro (Figure 5F). IHC analyses revealed that the expression levels of cleved-caspase3 were higher in the control group than in the cordycepin group, and that expression of ERK, MEK and Ki67 was significantly downregulated in xenograft tumors after cordycepin treatment (Figure 5D). Together, these data demonstrates that cordycepin suppresses tumor growth in vivo.
Figure 3
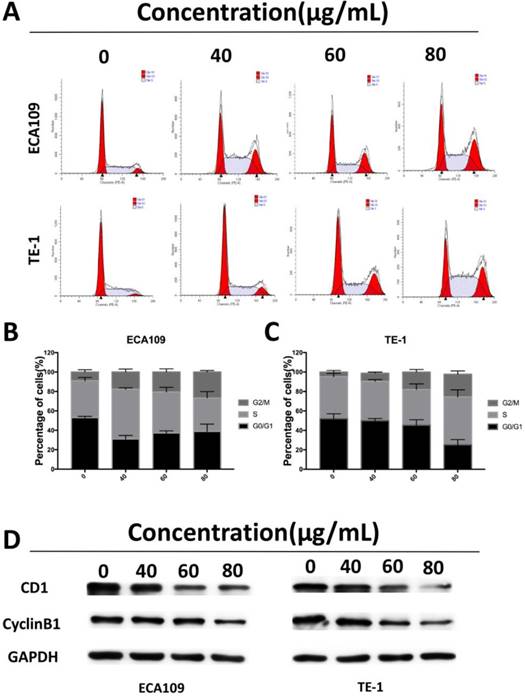
Cordycepin induces cell cycle arrest at G2/M phase and regulates the expression of cell cycle-related proteins. (A) ECA109 and TE-1 cells were treated with cordycepin (0,40, 60, 80μg/ml) for 48h.The cell cycle phases of treated cells were evaluated by flow cytometry. (B,C) The percentage of cells in the G1, S , and G2/M phases of the cell cycle are shown. (D) The protein levels of cell cycle regulators, such as CDK1 and cyclinB1, were examined by western blotting analysis and GAPDH was used as a loading control.
Discussion
Esophageal cancer is characterized as one of the most malignant and fatal cancers. Recent studies have shown that a traditional Chinese medicine herb, cordycepin, exhibits anticancer potential. Studies indicate that cordycepin has many intracellular targets, such as PLC/PKC PI3K/AKT GSK-3β and MAPK [13-16, 21-24]. However, there have been no studies evaluating the anti-cancer functions of cordycepin in esophageal cancer. In the present study, we investigated the antitumor growth effects of cordycepin in ESCC cell using CCK-8 and colony formation assays. The results show that cordycepin treatment dramatically inhibited the viability of ECA109 and TE-1 cell lines in a time- and dose-dependent manner. Supporting this, cordycepin significantly decreased tumor weight and volumes in ECA109 cell xenografted nude mice compared with control groups. Furthermore, based on the HE staining of livers and previously mentioned assays in 293T cells, we demonstrated that cordycepin exerted nontoxic anticancer efficacy in esophageal cancer cells.
Apoptosis is an important target in many chemotherapy-induced tumor cell deaths [25, 26]. Apoptotic cells were characterized by specific morphological changes such as shrinkage of the nucleus, DNA fragmentation, chromatin aggregation, and the formation of apoptotic bodies [26]. To further ascertain the antitumor function, Annexin V-FITC/PI, Hoechst 33342 staining analysis and flow cytometry assays were performed. The percentages of cells in apoptotic stages significantly increased following cordycepin treatment. Furthermore, ECA109 and TE-1 cells showed clear chromatin condensation and fragmentation compared with non-treatment cells as the dosages of cordycepin increased.
The apoptosis signaling cascade occurs through two major pathways: the death receptor pathway, known as extrinsic pathway, and the mitochondria-apoptosome- mediated intrinsic pathway. Both of these pathways converge to activate caspases3 and the cleavage of specific substrates [27]. Bcl-2 family proteins, which include pro-and anti-apoptotic members, play a central role in the regulation of the intrinsic pathway[28]. In response to apoptotic stimuli induced by chemotherapy, cytochrome c is released from the mitochondria into cytosol because of mitochondrial outer membrane permeabilization (MOMP) [29]. Released cytochrome c interacts with Apaf-1 and triggers the activation of upstream caspase, caspase-9. Caspase9 then activates downstream caspase-3, which activates subsequent caspases in a cascade-like model that leads to apoptosis. Caspase-3 is a crucial executioner that it is capable of cleaving specific substrates, such as PARP [30]. Bcl-2 proteins act as gatekeepers for cytochrome c [31]. These effects are more dependent on the ratio between Bcl-2 and anti-apoptosis protein Bax than on Bcl-2 quantity alone [32] (Figure 4B,C). In this study, we found that the intrinsic pathway was involved in cordycepin-induced cell death. We observed that cordycepin pre-treatment upregulated BAX and suppressed Bcl-2 protein expression. Furthermore, cordycepin significantly promoted the levels of caspase-3, caspase-9, and PARP cleavage both in vitro and in vivo. Further supporting this, the inhibition of apoptosis with pan-caspase inhibitor Z-VAD-FMK effectively blocked cordycepin-induced cell death.
Figure 4
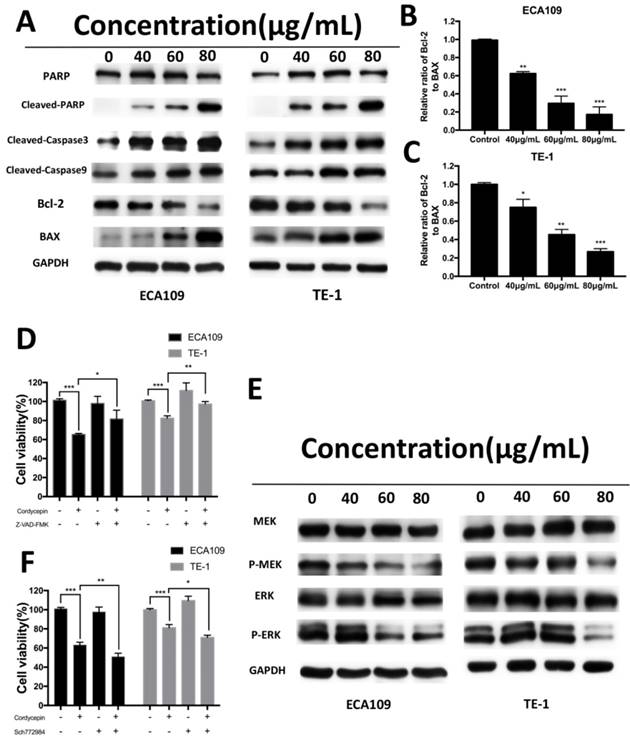
Cordycepin alters the expression of apoptosis-related proteins and MEK/ERK signaling proteins. (A) ECA109 and TE-1 cells were treated with cordycepin (0,40, 60, 80μg/ml) for 48h. Apoptosis-related proteins, PARP,cleaved PARP, cleaved caspase-3, cleaved caspase-9, Bax and Bcl-2 were analysed by Western blot analysis. GAPDH was used as a loading control. (B,C) The Bcl-2/Bax ratio was evaluated by the band density compared with the control. (D) After pretreatment with 10mM Z‑VAD‑FMK for 30min, ECA109 and TE-1 cells were incubated with 60 μg/ml cordycepin for 24h, and cellular viability was determined. (E) Western blotting analysis of MAPK/ERK signalling-related proteins in ECA109 and TE-1 cell lines. GAPDH was used as a loading control. (F) ECA109 and TE-1 cells treated with or without the ERK inhibitor SCH772984 were analysed by CCK-8 assay. Results are presented as mean ± SD (n = 3 independent experiments). *p<0.05, **p<0.01, and ***p<0.001.
Figure 5
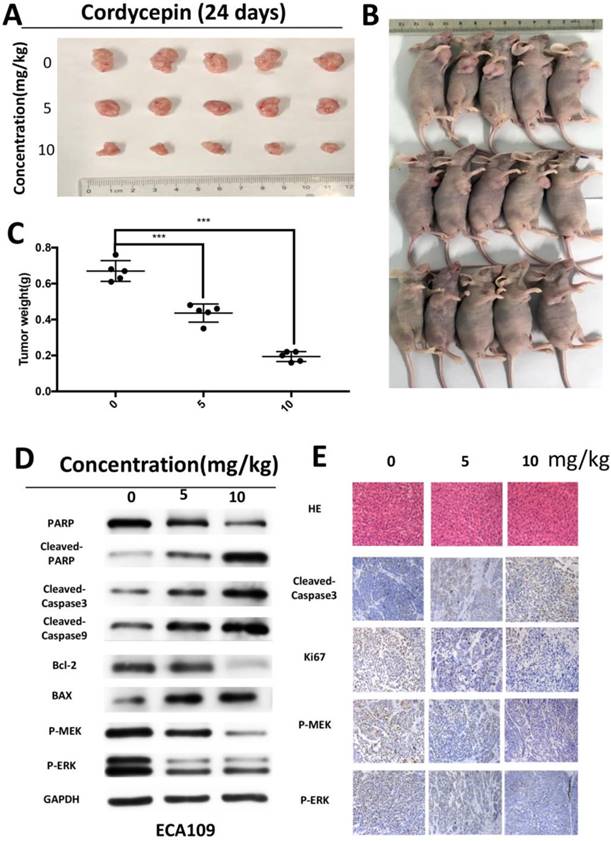
Cordycepin suppressed tumor growth in vivo. (A-C) Mice were treated with different concentrations (0, 5 mg/kg and 10 mg/kg) of cordycepin for 24 days. Images of 5 representative mice from each group are shown. Tumor sizes and tumor weights were measured. (D) PARP,cleaved PARP, cleaved-caspase3, cleaved-caspase9, Bax and Bcl-2, P-MEK, P-ERK expression levels in xenograft tumors were analyzed using western blot analysis. (E) The expression of cleaved caspase-3, Ki67, P-MEK and P-ERK were evaluated by IHC and HE staning in xenograft tumor. Results are presented as mean ± SD (n = 3 independent experiments). *p<0.05, **p<0.01, and ***p<0.001.
The extracellular signal-regulated kinase (ERK) has a critical role in the MAPK pathway by phosphorylating multiple target proteins that control cell proliferation [33]. The results of Western blot also demonstrated that cordycepin significantly inhibited P-MEK and P-ERK expression. Previous studies have also confirmed that the MAPK-mediated pathway alters the cell cycle distribution caused by G2/M stage arrest and promotes cell apoptosis [34, 35]. ERK can also phosphorylate two phosphorylation sites on the cytoplasmic retention sequence (CRS) of cyclin B1 , which is necessary for nuclear localization of cyclin B1 and mitosis progression [36]. The expression of cell cycle proteins were also analyzed. The results indicated that inactivation of the MEK/ERK pathway leads to enhanced levels of cyclinB1 and CDK-1, which contributed to G2/M stage arrest and cell death. Furthermore, Western blot indicated that cordycepin decreased PI3K and p-AKT levels in ECA109 and TE-1 cell lines, which suggests that cordycepin plays an essential role in mediating the activation of both constitutive MEK/ERK and PI3K/ Akt signaling.
Thus, the effects of cordycepin on proliferation, apoptosis, and cell cycle arrest in esophageal cancer cells can be explained by inactivation of the MEK/ERK pathway, at least in part. In addition, one mechanism of cancer cell resistance to chemotherapeutic agents is overproduction of Bcl-2 proteins [37]. We hypothesize that cordycepin synergizes with cisplatin and 5-FU against esophageal cancer and that the underlying mechanism for this is could be cordycepin-induced ERK inactivation. However, animal studies and possible clinical trials are needed to confirm this hypothesis.
Abbreviations
MAPK: mitogen-activated protein kinase; ERK: extracellular regulated protein kinases; P-ERK: phosphorylated extracellular regulated protein kinases; ESCC: esophageal squamous cell carcinoma; DMSO: dimethyl sulfoxide; PBS: Phosphate Buffered Saline; FITC: fluorescein isothiocyanate; CCK-8: Cell Counting Kit-8; PI: propidium iodide; IHC: immunohistochemical streptavidin-peroxidase staining; HE: hematoxylin and eosin; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; PVDF: polyvinylidene difluoride; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; PARP: poly ADP-ribose polymerase; Z-VAD-FMK: Pan-caspase inhibitor; PKC: protein kinase C; PI3K: phosphatidylinositol 3-kinase; AKT: protein kinase B; p-AKT: phosphorylated protein kinase B; GSK-3β: Glycogen synthase kinase 3 beta; MOMP: mitochondrial outer membrane permeabilization; 5-FU: 5-fluorouracil.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No. 81370588 and 81570595), the Training Program Foundation for the Talents of Zhongshan Hospital (2015ZSYXGG), the Shanghai Engineering and Research Center of Diagnostic and Therapeutic Endoscopy (No. 16DZ2280900). The authors thank Dr Xiang-lin Hu (Zhongshan Hospital, Fudan University, Shanghai, China) for his very warm and professional suggestions on the manuscript revision.
Author Contributions
Yi-Qun Zhang and Ping-Hong Zhou did the conception and design. Jia-Cheng Xu, Xue-Ping Zhou, Xu-An Wang, Mei-Dong Xu, Tao Chen, Tian-Yin Chen designed and conducted the study. Jia-Cheng Xu, Xue-Ping Zhou analyzed the data and wrote the manuscript. Xu-An Wang, Mei-Dong Xu reviewed the manuscript and gave conceptual advice. All authors approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA: a cancer journal for clinicians. 2016;66:115-32
2. Rustgi AK, El-Serag HB. Esophageal carcinoma. The New England journal of medicine. 2014;371:2499-509
3. Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438:946-53
4. Hsu WH, Hsu PK, Hsieh CC, Huang CS, Wu YC. The metastatic lymph node number and ratio are independent prognostic factors in esophageal cancer. Journal of gastrointestinal surgery: official journal of the Society for Surgery of the Alimentary Tract. 2009;13:1913-20
5. Cassileth BR. Alternative and Complementary Cancer Treatments. The oncologist. 1996;1:173-9
6. Cunningham KG, Manson W, Spring FS, Hutchinson SA. Cordycepin, a metabolic product isolated from cultures of Cordyceps militaris (Linn.) Link. Nature. 1950;166:949
7. Kuchta RD. Nucleotide Analogues as Probes for DNA and RNA Polymerases. Current protocols in chemical biology. 2010;2:111-24
8. Holbein S, Wengi A, Decourty L, Freimoser FM, Jacquier A, Dichtl B. Cordycepin interferes with 3' end formation in yeast independently of its potential to terminate RNA chain elongation. RNA (New York, NY). 2009;15:837-49
9. Zhang DW, Wang ZL, Qi W, Lei W, Zhao GY. Cordycepin (3'-deoxyadenosine) down-regulates the proinflammatory cytokines in inflammation-induced osteoporosis model. Inflammation. 2014;37:1044-9
10. Lee EJ, Kim WJ, Moon SK. Cordycepin suppresses TNF-alpha-induced invasion, migration and matrix metalloproteinase-9 expression in human bladder cancer cells. Phytotherapy research: PTR. 2010;24:1755-61
11. Nakamura K, Shinozuka K, Yoshikawa N. Anticancer and antimetastatic effects of cordycepin, an active component of Cordyceps sinensis. Journal of pharmacological sciences. 2015;127:53-6
12. Chaicharoenaudomrung N, Jaroonwitchawan T, Noisa P. Cordycepin induces apoptotic cell death of human brain cancer through the modulation of autophagy. Toxicology in vitro: an international journal published in association with BIBRA. 2018;46:113-21
13. Wang XA, Xiang SS, Li HF, Wu XS, Li ML, Shu YJ. et al. Cordycepin induces S phase arrest and apoptosis in human gallbladder cancer cells. Molecules. 2014;19:11350-65
14. Lee SJ, Moon GS, Jung KH, Kim WJ, Moon SK. c-Jun N-terminal kinase 1 is required for cordycepin-mediated induction of G2/M cell-cycle arrest via p21WAF1 expression in human colon cancer cells. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2010;48:277-83
15. Wang Z, Wu X, Liang YN, Wang L, Song ZX, Liu JL. et al. Cordycepin Induces Apoptosis and Inhibits Proliferation of Human Lung Cancer Cell Line H1975 via Inhibiting the Phosphorylation of EGFR. Molecules. 2016:21
16. Yamamoto K, Shichiri H, Uda A, Yamashita K, Nishioka T, Kume M. et al. Apoptotic Effects of the Extracts of Cordyceps militaris via Erk Phosphorylation in a Renal Cell Carcinoma Cell Line. 2015; 29: 707-13.
17. Li M, Zhang Z, Li X, Ye J, Wu X, Tan Z. et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nature genetics. 2014;46:872-6
18. Song X, Wang Z, Liang H, Zhang W, Ye Y, Li H. et al. Dioscin Induces Gallbladder Cancer Apoptosis by Inhibiting ROS-Mediated PI3K/AKT Signalling. International journal of biological sciences. 2017;13:782-93
19. Williams GH, Stoeber K. The cell cycle and cancer. The Journal of pathology. 2012;226:352-64
20. Spencer SL, Sorger PK. Measuring and modeling apoptosis in single cells. Cell. 2011;144:926-39
21. Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug discovery today. 2012;17:359-66
22. Lee SJ, Kim SK, Choi WS, Kim WJ, Moon SK. Cordycepin causes p21WAF1-mediated G2/M cell-cycle arrest by regulating c-Jun N-terminal kinase activation in human bladder cancer cells. Archives of biochemistry and biophysics. 2009;490:103-9
23. Lee HH, Jeong JW, Lee JH, Kim GY, Cheong J, Jeong YK. et al. Cordycepin increases sensitivity of Hep3B human hepatocellular carcinoma cells to TRAIL-mediated apoptosis by inactivating the JNK signaling pathway. Oncology reports. 2013;30:1257-64
24. Wong YY, Moon A, Duffin R, Barthet-Barateig A, Meijer HA, Clemens MJ. et al. Cordycepin inhibits protein synthesis and cell adhesion through effects on signal transduction. The Journal of biological chemistry. 2010;285:2610-21
25. Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA: a cancer journal for clinicians. 2005;55:178-94
26. Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. The EMBO journal. 2011;30:3667-83
27. Hu W, Kavanagh JJ. Anticancer therapy targeting the apoptotic pathway. The Lancet Oncology. 2003;4:721-9
28. Kelly PN, Strasser A. The role of Bcl-2 and its pro-survival relatives in tumourigenesis and cancer therapy. Cell death and differentiation. 2011;18:1414-24
29. Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science (New York, NY). 2004;305:626-9
30. Thornberry NA, Lazebnik Y. Caspases: enemies within. Science (New York, NY). 1998;281:1312-6
31. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES. et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479-89
32. Reed JC. Bcl-2 family proteins: regulators of apoptosis and chemoresistance in hematologic malignancies. Seminars in hematology. 1997;34:9-19
33. Deschenes-Simard X, Kottakis F, Meloche S, Ferbeyre G. ERKs in cancer: friends or foes? Cancer research. 2014;74:412-9
34. Liao Y, Ling J, Zhang G, Liu F, Tao S, Han Z. et al. Cordycepin induces cell cycle arrest and apoptosis by inducing DNA damage and up-regulation of p53 in Leukemia cells. Cell cycle (Georgetown, Tex). 2015;14:761-71
35. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E. et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656-70
36. Chambard JC, Lefloch R, Pouyssegur J, Lenormand P. ERK implication in cell cycle regulation. Biochimica et biophysica acta. 2007;1773:1299-310
37. Reed JC, Miyashita T, Takayama S, Wang HG, Sato T, Krajewski S. et al. BCL-2 family proteins: regulators of cell death involved in the pathogenesis of cancer and resistance to therapy. Journal of cellular biochemistry. 1996;60:23-32
Author contact
![]() Corresponding authors: zhang.yiqunsh.cn; zhou.pinghongsh.cn; Tel.: +86-21-64041990.
Corresponding authors: zhang.yiqunsh.cn; zhou.pinghongsh.cn; Tel.: +86-21-64041990.
Citation styles
APA
Xu, J.C., Zhou, X.P., Wang, X.A., Xu, M.D., Chen, T., Chen, T.Y., Zhou, P.H., Zhang, Y.Q. (2019). Cordycepin Induces Apoptosis and G2/M Phase Arrest through the ERK Pathways in Esophageal Cancer Cells. Journal of Cancer, 10(11), 2415-2424. https://doi.org/10.7150/jca.32071.
ACS
Xu, J.C.; Zhou, X.P.; Wang, X.A.; Xu, M.D.; Chen, T.; Chen, T.Y.; Zhou, P.H.; Zhang, Y.Q. Cordycepin Induces Apoptosis and G2/M Phase Arrest through the ERK Pathways in Esophageal Cancer Cells. J. Cancer 2019, 10 (11), 2415-2424. DOI: 10.7150/jca.32071.
NLM
Xu JC, Zhou XP, Wang XA, Xu MD, Chen T, Chen TY, Zhou PH, Zhang YQ. Cordycepin Induces Apoptosis and G2/M Phase Arrest through the ERK Pathways in Esophageal Cancer Cells. J Cancer 2019; 10(11):2415-2424. doi:10.7150/jca.32071. https://www.jcancer.org/v10p2415.htm
CSE
Xu JC, Zhou XP, Wang XA, Xu MD, Chen T, Chen TY, Zhou PH, Zhang YQ. 2019. Cordycepin Induces Apoptosis and G2/M Phase Arrest through the ERK Pathways in Esophageal Cancer Cells. J Cancer. 10(11):2415-2424.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.