Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(14):3112-3123. doi:10.7150/jca.30406 This issue Cite
Review
A long way to the battlefront: CAR T cell therapy against solid cancers
Zhicai Wang1,2 ![]() , Wei Chen2, Xing Zhang1, Zhiming Cai2,3,4
, Wei Chen2, Xing Zhang1, Zhiming Cai2,3,4 ![]() , Weiren Huang2,3,4
, Weiren Huang2,3,4 ![]()
1. Department of Medical Melanoma and Sarcoma, State Key Laboratory of Oncology in South China, Collaborative Innovation Center for Cancer Medicine, Sun Yat-sen University Cancer Center, Guangzhou, 510060, China
2. Key Laboratory of Medical Reprogramming Technology, Shenzhen Second People's Hospital, First Affiliated Hospital of Shenzhen University, Shenzhen, 518039, China
3. Department of Urology, Shenzhen Second People's Hospital, The First Affiliated Hospital of Shenzhen University, Carson International Cancer Center, Shenzhen University School of Medicine, Shenzhen 518039, China
4. Guangdong Key Laboratory of Systems Biology and Synthetic Biology for Urogenital Tumors, Shenzhen 518035, China
Received 2018-10-4; Accepted 2019-4-7; Published 2019-6-2
Abstract
Chimeric antigen receptors (CARs) are engineered synthetic receptors that redirect and reprogram T cells to tumor surface antigens for subsequent eradication. The unprecedented efficacy of CD19-CAR T cells against B-cell malignancies has inspired oncologists to extend these efforts for the treatment of solid tumors. However, limited success has been achieved so far, partially due to some of the formidable challenges, e.g. suppression of full activation, inhibition of T cell localization, lacking of ideal targets, inefficient trafficking and infiltration, immunosuppression of microenvironment, and the probability of off targets and associated side effects. Significant progresses have being made recently. Thus, an updated summary is urgently needed. Here in this review, we discuss the advantages and some of the key hurdles encountered by CAR T cell therapy in solid tumors as well as the strategies adopted to improve therapeutic outcomes of this approach. Continuing efforts to increase therapeutic potential and decrease the adverse effects of adaptive cell transfer are suggested as well.
Keywords: CAR T cell therapy, Solid cancer, Immunosuppression, Anti-tumor responses
Introduction
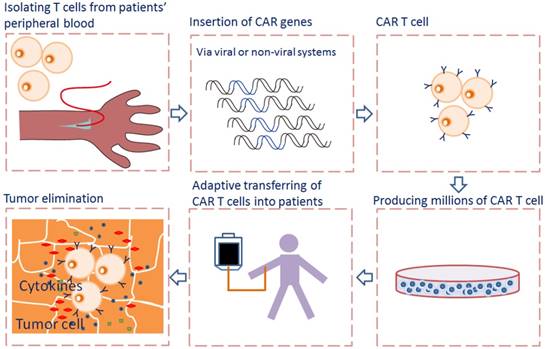
Adoptive cell transfer of genetically engineered T cells expressing synthetic chimeric antigen receptor (CAR), allows for collecting, redirecting and reprograming a patient's own T lymphocytes to treat his/her cancer. The rapidly emerging of CAR-T cell immunotherapy has shown encouraging results in treating advanced tumors by recognizing and binding to many different tumor-specific and tumor -associated antigens (TAA), particularly in treating hematological malignancies [1]. CARs allow T cells to recognize and attach to tumor surface antigens, while bypassing T cell specific activation checkpoints [2]. The CAR-transduced T cells are expanded ex vivo into hundreds of millions in a cell manufacturing facility and then adoptively transferred back to patient with the hope of selectively targeting and killing the antigen-expressing tumor cells [3] (Fig. 1).
Flowcharts of CAR T cell therapy. T cells are collected from peripheral blood of patients and then selected and activated. The CAR genes were transferred into T cells via viral or non-viral systems and expressed. The CAR T cells are expanded in vitro into hundreds of millions in a cell manufacturing facility and then adoptively transferred back to patient. When CAR recognizes the antigen on tumor cells, the intracellular signaling domains within the CAR produce a series of cytokines, resulting in the activation of CAR T cells.
The direct impact and potential mechanisms of CAR T cell therapy on cancer cells are mainly determined by the basic CAR structure. A typical CAR is composed of an extracellular antigen-specific immunoglobulin single-chain variable fragment (scFv) fused via a transmembrane domain to intracellular costimulatory signaling molecules. The binding of scFv to tumor antigens will trigger T cell receptor and costimulatory signaling, resulting in activation of T cells and subsequent killing of target cells. According to the intracellular signaling domains, there are four generations of CARs that have been used in clinical studies. The first generation receptors only use CD3ζ chain derived from the TCR as intracellular signaling domain to stimulate T cell activation. The second and third generation CARs consist of one or two co-stimulatory domains respectively, combining activation and costimulatory signaling domains like 4-1BB, CD28, or OX40, etc. to achieve robust expansion and persistence of CAR T cells in vivo [4,5]. The fourth generation CAR T cells contain additional genes encoding cytokines such as IL-12 and IL-15 [6].
The essential parameters for achieving high clinical efficacy in vivo depend on stable expression of the CAR, antigen specific recognition, activation, and subsequent expansion, engraftment, cytotoxicity and persistence of the effector cells [7]. To avoid fratricide events, the targeted antigen should be expressed at high level by all cancer cells in a large number of patients, and should not be expressed, nor rested or activated by some vital normal tissues [8]. By following these criteria, around 90% complete remissions have been observed in heavily pre-treated patients with relapsed or refractory B -cell malignancies [9], leading to the development and use of two CD-19-targeted therapeutic drugs approved by the US Food and Drug Administration (FDA), one is tisagenlecleucel (Kymriah) for children and adolescents, the other is axicabtagene ciloleucel (Yescarta) for adults with acute lymphoblastic leukemia (ALL) [10]. Despite these unprecedented clinical impacts, more potent and antigen specific CARs are needed to treat solid tumors [8]. This issue is due, at least in part, to the limited number of target antigens, the less efficient infiltration, accumulation and survival of transduced CAR-T cells to the tumor and the occurrence of immune escape [11].
Moreover, there are some unique challenges posed by solid tumors that limit full and persistent function of infused CAR T cells, including the oxidative, nutritional depleted, acidic, and hypoxic microenvironment, the suppressive soluble factors, cytokines, and immune cells as well as T cell intrinsic negative regulatory molecules [8]. Some extra caution is required with regard to side effects endured by patients due to cytokine release syndrome (CRS), neurotoxicity, or on-target off-tumor toxicity [12]. Given of these multiple hurdles, a growing numbers of preclinical studies and clinical trials are being initiated by targeting antigens expressed on tumor surfaces, such as human epidermal growth factor receptor 2 (HER2), carcinoembryonic antigen (CEA), epidermal growth factor receptor (EGFR), mesothelin (MSLN), and diganglioside GD2 [13]. Herein, we offer our prospects on some of the key immunosuppressive barriers and other negative elements within solid tumors, especially focusing on innovative strategies to overcome immunosuppression, reduce toxicities, and prevent antigen escape. Advances in the specific antigen selection, genetic engineering, side effects prevention are poised to broaden T cell based therapies and foster new applications.
Delivery, trafficking and infiltration of CAR T cells
Delivery of CAR T cells
Solid malignances impose considerable obstacles to effective CAR T cell immunotherapy, such as insufficient delivery of T cells to the tumor sites and inefficient functional persistence. One strategy to overcome this problem is direct administration of T cells to the lesions sites, thus avoiding their homing from the blood stream. In contrast to systematic intravenous application, local delivery of CAR T cells via intra-pleural or intra-cerebral injection exhibited enhanced immune responses [14,15] and improved safety profile with limited systemic absorption [16]. Intra-tumoral administration of ErbB retargeted CAR T cells in patients with head and neck cancer is safe without obvious dose limiting toxicity or systemic absorption [17]. Intrapleurally delivery of mesothelin-targeted CAR T cells bypasses trafficking barriers and redistributes to other tumor sites with superior efficacy than systemically infused T cells, thus allowing robust CAR T cell expansion and long-term complete remission [14]. Similarly, multiple infusion of CAR T cells directed to interleukin-13 receptor α2 (IL13Rα2) in one glioblastoma patient by intraventricular and intracavity administration induced a complete remission [18]. In a small phase I study, one out of 6 patients with liver metastases showed stable disease after percutaneous hepatic artery administration of CEA-CAR T cells [19]. In contrast to intravenous administration, intratumoral injection of AFP-CAR T cells induced a much more profound, rapid, and lasting effecter response, therefor might be a more attractive and safer clinical path [20]. Additionally, several clinical trials are underway for evaluating the merits of local delivery of CAR T cells in solid tumors (NCT02498912, NCT02414269, NCT01818323). However, further investigation are still needed to overcome the difficulties when injecting CAR T cells regionally, such as technical challenges and trafficking through the blood to other tumor locations [21].
Trafficking and infiltration of CAR-T cells
After a tumor antigen specific CAR was generated and infused into a patient, the first of many obstacles encountered is whether these CAR T cells can successfully target, accumulate and infiltrate into the tumor to efficiently exert antitumor effects. During these processes, the CAR T cells need to adhere to endothelial cells and initiate chemokine-chemokine receptor interactions to facilitate their extravasation into antigen-rich regions [22]. However, chemokines produced by most solid tumors do not favor T cell infiltration. The mismatch occurs quite frequently, with tumors secreting little chemokine or the CAR T cell has very small amount of chemokine receptors, resulting in little migration to the tumor site [23]. Therefore, better solutions are needed. One way to solve this problem is to profile the chemokine signature of a tumor or genetically engineer the better-matched chemokine receptor of the CAR T cell. Indeed, CAR T cells genetically modified to co-express CXCR2/CCR2b (receptor for CXCL1) showed enhanced targeting towards mesothelioma tumor cells expressing CXCL1 [24,25]. Similarly, increasing the expression level of CXCR3 by genetic inhibition of protein kinase A activity in CAR T cells improved trafficking and tumor control [26]. This effect has also been observed in CD30-CAR T cells bearing CCR4 (receptor for CCL17) to target Hodgkin's lymphoma [27]. Some other initiatives has also been taken to enhance CAR T cell targeting and trafficking by using oncolytic viruses armed with chemotactic chemokines. These viruses have the ability to specifically infect and lyse tumor cells. For examples, the use of oncolytic adenoviral vector co-expressing CCL5 and GD2-CAR T cells to target neuroblastoma has shown improved CAR T cell infiltration and better tumor control [28]. As an alternative strategy, the tumor chemokine secretion can be modulated by directly injection of oncolytic adenoviruses expressing RANTES and IL-15 into neuroblastoma, with enhanced CAR T cell infiltration and more efficient tumor control [28].
Targeting and disrupting the tumor vasculature provide an alternative solution to not only restrict blood flow and nutrient supply to the tumor, but also enhancing T cell infiltration. Therefore, CAR T cells engineered to express degrading enzymes such as heparanase, which degrades heparan sulfate proteoglycans, the main components of extra cellular matrix, improved tumor T cell penetration capacity and anti-tumor effects [29]. CAR T cells targeting VEGFR2, which was expressed on angiogenic endothelial cells and myeloid suppressor cells, resulted in increased tumor infiltration and anti-tumor effects [30]. Likewise, CAR T cells incorporating ligands for αvβ3, an integrin expressed on tumor vascular endothelium, improved migration [31]. Additionally, blocking the endothelin B receptor has been shown to enhance T cell infiltration into tumor lesions [32]. To circumvent the physical barriers that potentially impede CAR T cell infiltration, localized delivery of CAR T cells to the tumor surface has also been described [33].
Activation, proliferation and survival of CAR T cells
Activation of CAR-T cells
CAR T cell activity could be either regulated by tumor antigens or some intrinsic factors within T cells. The physiological activation of CAR T cells requires the density of tumor associated antigen to be above a certain threshold. Generally, a relative higher density is necessary to induce cytokine production and cell proliferation [34]. CAR T cell activation is also affected by the expression of CARs on the cell surface [35]. Lower CAR expression leads to subactivation of the CAR T cells, whereas overexpression can induce tumor antigen independent activation [36].
Inhibitory receptors including programed death-1 (PD-1) and cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) play vital roles in attenuating or terminating T cell responses. Some of them can be upregulated during T cell activation to inhibit immune responses [37]. Ligation of PD-1 or CTLA-4 during T cell priming blocks activation, cytokine production, and proliferation [38], whereas blockade of them restores T cell function [39]. Although IL-2 can support CAR T cells in vivo, it may also activate and induce proliferation of regulatory T cells (Tregs) [40]. Thus, the use of alternative cytokines instead of IL-2, such as IL-7 and IL-21 was explored, and enhanced CAR activity has been observed [41].
Proliferation of CAR-T cells
The varied in vivo expansion and persistence of CAR T cells is mainly attributed to the varied CAR transgene structure, co-stimulatory factors, gene integration methods, starting T cell population, culture techniques and final product composition. CAR T cells used in the initial studies contain only CD3ζ signal, thus may result in insufficient in vivo expansion. Therefore, costimulatory signals incorporating CD28, 4-1BB, and/or OX40 in series with CD3ζ are developed to produce the second- (one stimulatory domain) and third- (two stimulatory domains) generation CARs [42]. These genetically enhanced CARs significantly improved the clinical results, yielding robust in vivo cell expansion and near uniform efficacy [43].
For in vitro CAR T cell stimulation and expansion, anti-CD3/CD28 dynabeads and plate-bounded anti-CD3 antibody are most commonly used [44]. Furthermore, addition of homeostatic cytokines to T cell cultures can rescue the poor in vitro growth and inadequate expanding of T cells [45]. Incorporation of homeostatic cytokines including interleukin 7 (IL-7) and IL-15 have achieved a very high rate of manufacturing success in clinical trial [46]. For in vivo expansion and accumulation, CAR T cells have been created to constitutively secrete IL-12, a pro-inflammatory cytokine better known for increasing the proliferation of T cells [47]. Intraperitoneally injection of mice bearing MUC16 (CA125) positive ovarian cancer with MUC16 targeted CAR expressing IL-12 has shown increased antitumor response with a higher rate of CAR T cell accumulation in peritoneum and enhanced recruitment of endogenous T cells [47].
To achieve efficient transduction and expansion of patient-derived T cells, a chimeric cytokine receptor named 4αβ has been co-expressed with CAR T cells and tested in preclinical studies. This receptor incorporating the IL-4 receptor-α ectodomain and the shared β chain by IL-1/IL-15 delivers a potent mitogenic signal into the cell, promoting the selective enrichment and expansion of CAR T cells [48]. After IL-4 mediated cell expansion, the engineered CAR T cells produce a broader range of cytokines and show an increased anti-tumor activity against multiple cancers including head and neck squamous cell carcinoma (HNSCC), breast cancer and ovarian cancer [49].
Survival of CAR-T cells
Strategies to improve CAR T cell immunotherapy have mainly focused on sustained clinical remission in patients [50]. To have durable in vivo activity, significant in vivo expansion and long term persistence of CAR T cells are required. Factors including the CAR design, starting T cell population, ex vivo culture, T cell exhaustion, or host immunogenicity can influence the persistence of CAR T cells [51,52]. By initiating T cell cultures from prescribed subsets of T cells, such as central memory CD8+ T cells, but not commonly used peripheral blood mono-nuclear cells may facilitate the engraftment fitness and long term in vivo viability of the product [53,54].
It has been demonstrated that CD28 co-stimulation promotes T cell proliferation and persistence, glucose metabolism, and a potent effector response, while 4-1BB based CAR appears to stimulate lipid oxidation and enhance long term T cell persistence [55]. Incorporation of the ICOS and 4-1BB intracellular domains into a third-generation CAR augmented the effector function and in vivo persistence, with ICOS placed proximal to the cell membrane and linked to the ICOS transmembrane domain as the prerequisites [56,57]. It has also been shown that the incorporation of cytokine-encoding genes such as IL-7 into the constructs of CAR transgene enhances the proliferation and survival of T cells [41,58].
The murine-based scFv domain may elicit host immune-mediated rejection of CAR, shortening persistence of CAR T cells in vivo. Thus, the humanized or fully human CAR constructs are preferred in recent studies. Additionally, site specific integration of CAR within the T cell genome may enhance therapeutic efficacy, as shown in preclinical study that insertion of CAR into TCR locus sustained CAR transgene expression and successfully inhibited T cell exhaustion [35]. Other potential strategies for enhancing therapeutic efficacy including CAR modified T memory stem cells exhibiting improved engraftment fitness and long-lasting immunity [59], and Epstein Barr Virus (EBV) transformed lymphoblastoid cells enable post-CAR T cell vaccination, with enhanced persistence [44]. Another ongoing project in order to enhance CAR T cell persistence is using autologous T cells expressing a truncated form of CD19 protein to continuously stimulate CD19 CAR T cells in vivo [60].
The interactions of CAR T cells with other cells and molecules in the TME (tumor microenvironment)
Unlike the hematologic malignancies with circulating target cells, CAR T cells in solid tumor must successfully traffic to tumor sites and infiltrate the stromal elements in spite of chemokine/chemokine receptor mismatches, antigen loss or heterogeneity. Even after effective migration and infiltration, CAR T cells must surmount many more challenges characterized by physical and chemical barriers in the TME: high tissue pressure, extracellular matrix and abnormal vasculature, immunosuppressive molecules and immune regulators, as well as T cell intrinsic inhibitory mechanisms and potential immunogenicity compromise CAR T cell anti-tumor functions [61] (Fig. 2).
Tumor microenvironment (TME) in solid tumor imposes immunosuppression to CAR T cells. Strategies including using immune checkpoint blockade inhibitors or antibodies, activating cytokines and novel CARs were adopted to circumvent the TME suppression effects.
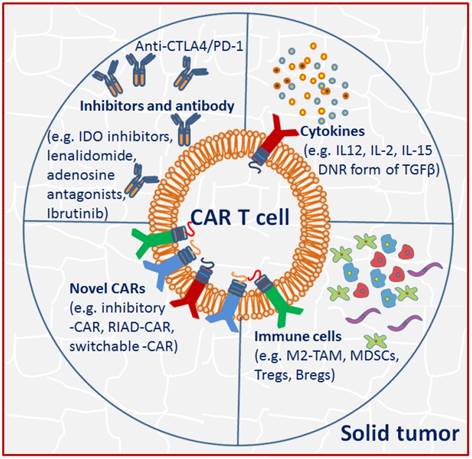
The interactions of CAR T cells with inhibitors and antibody
Notably, the inhibitory effect is tightly associated with the TME, as removal of it restores CAR T cell effectiveness. Thus, appropriate manipulation of the TME offers new opportunities for improving clinical therapeutic outcomes [13]. Pre-conditioning chemotherapy by using cyclophosphamide alone or coupled with fludarabine facilitates infused T cell engraftment and decreases suppressive immune cells in TME. Some small molecules including IDO inhibitors, lenalidomide, adenosine antagonists and ibrutinib are also used to interfere with immunosuppressive cells and pathways [62]. Multiple approaches by using blocking antibodies, dominant-negative receptors and targeted gene disruption have been developed to modulate checkpoint blockade, which may facilitate sustainable function and persistence of engineered T cells [63]. In fact, upon tumor-antigen encounter, multiple inhibitory immune receptors including CTLA-4, and/or PD-1 are upregulated on T cells [64], therefore allowing tumor progression. The use of PD-1 blockade by continuously secreting anti-PD-1 antibody [65] or directly application of monoclonal antibodies [66] attenuated the inhibitory signaling and enhanced anti-tumor response of CAR T cells in xenograft mouse model or in patients with melanoma and renal caner [67]. Alternatively, using a multiplex genome editing tools (CRISPR/Cas9), universal CAR T cells resistant to PD-1 inhibition has been generated, resulting in enhanced in vivo antitumor response [68].
The interactions of CAR T cells with cytokines and immune cells
Another obstacle to overcome in the inhibitory ascitic TME is the presence of immunosuppressive cytokines, which may inactivate adoptively transferred CAR T cell antitumor activity. One of the possible solutions is the development of armored CARs capable of constitutive secretion of activating cytokine like IL-12, which appears to be more effective in preclinical studies [69]. Specifically, constitutive IL-12 signaling repolarizes the TME, enhances T cell cytotoxicity, and promotes resistance against Treg immunosuppression [70]. Secretion of IL-12 by engineered CAR T cells limited tumor antigen escape events by destruction of antigen negative cancer cells [71]. Moreover, production of other activating cytokines such as IL-2 and IL-15 has been shown to improve CAR T cell functionality as well [72]. MUC1-CAR T cells incorporating chimeric IL-4 receptors mimicked IL-2 signaling and resulted in enhanced CAR efficacy [48]. TGFβ is another important inhibitory cytokine. In addition to promote tumor metastasis formation, it has direct negative effect on T cell effector functions [73]. To counteract TGFβ effects, a dominant negative TGFβ receptor has been developed and consequently displayed resistance to TGFβ suppression and augmented efficacy in animal models [74].
The solid tumor microenvironment is comprised of multiple immune suppressor cells including M2 tumor associated macrophages (M2-TAM), myeloid-derived suppressor cells (MDSCs), Tregs and B cells (Bregs), which likely blunt the efficacy of CAR T cells [75]. Among them, M2-TAM and MDSC are well-known producers of TGFβ, PGE2, reactive oxygen/nitrogen species [76], and MDSC may also recruit Tregs. It has been demonstrated that infusion of CEA-CAR T cells in combination with MDSC depletion enhanced anti-tumor efficacy [77].
Overcoming the TME immunosuppression by novel CAR designs
An alternative approach to overcome immunosuppression is to generate novel CARs incorporating dominant negative receptors (DNRs) that can override the inhibitory signals in the TME. DNRs often maintain the extracellular domain of a membrane receptor but have the mutant or nullified intracellular chain, thus competing with the endogenous receptor and disrupting the downstream signaling [78]. The use of DNR form of TGFβ and PD-1 have conferred the transduced EBV cells and CAR T cells with resistance to immune suppression, respectively [79,80].
CAR containing switchable receptors is another approach to circumvent immunosuppression. The switchable receptor has the extracellular region of an antibody specific for an immunosuppressive molecule, and an intracellular activation domain, with the aim of redirecting the inhibitory effects to an activating one [81], as shown by CAR T cells bearing PD-1-CD28 switch receptor that exhibiting enhanced infiltration and anti-tumor activity [81]. It has also been demonstrated that inhibition of Protein Kinase A with Ezrin using a regulatory subunit 1 anchoring disruptor (RIAD-CAR) resulted in enhanced resistance to immunosuppressive adenosine in the TME and anti-tumor response [26].
These strategies abate some of the resistant effects, but are not sufficient to remove all [82]. Additional steps including therapeutic combinations and design of more potent CARs are needed to confer resistance towards inhibition. Previous studies have shown that serial infusion of armed T cells or incorporation of cytokine receptors can alleviate immune suppression and enhance CAR T cell functionality [83,84]. Additionally, reducing the expression of diacylglycerol kinase alleviated intracellular negative feedback signaling and significantly increased efficacy [85]. FAP-CARs targeting tumor fibroblasts [86] and CAR T cells secreting matrix degrading enzymes [29] augmented antitumor function in animal models.
The direct impact and potential mechanisms of CAR T cell therapy on normal cells
One major challenge hindering the success of CAR T cell therapy against solid tumor is the relative low expression of target antigen on variety of potentially important normal tissues, leading to on-target, off-tumor toxicity. Even traces or transient expression of a target antigen in vital normal tissues may lead to undesired adverse events, as seen with EGFR, MART-1, MAGE-A3, CEA, or ERBB2 targeted T cell therapies [87,88]. Feasible strategies have been developed to enable CAR T cells to discriminate tumors overexpressing the target from normal tissues expressing the same antigen at relative low physiologic levels by tuning scFv affinity [89]. Decreasing the scFv affinity but maintaining robust antitumor efficacy of the EGFR-targeted CAR T cells can eliminate or strongly inhibit the reactivity against normal tissues [90].
Alternative strategies focusing on novel CAR design are necessary to avoid reactivity against normal tissues. Some CARs are designed to use suicide genes, such as EGFR mutation [91], inducible caspase 9 (iCas9) gene [92], and the herpes simplex virus thymidine kinase (HSV-TK) gene [93], which can be activated and mediate rapid T cell elimination after treatment of a prodrug or antibody in case of unwanted adverse events. Some other CARs are designed based on the concept of combinatorial antigen recognition [94], inhibitory receptors [95], split-signalling receptors [96], and sequentially acting receptors [97].
Exhaustion and senescence of T cells
Exhaustion and senescence of T cells are similar but not entirely the same [98]. Exhaustion is progressively loss of activated T cell function due to persistent antigen stimulation, whereas senescence is cell cycle arrest caused by aging [99]. In comparison to exhaustion, senescent T cells express different sets of markers and secrete high levels of pro-inflammatory cytokines [100].
Exhaustion
T cells exposed to persistent antigen are often associated with the deterioration of T cell function that inefficient to control tumors, known as 'exhaustion' [101]. Exhausted T cells lose cytotoxicity, reduce T cell proliferation and stimulatory cytokine production, alter chromatin structure and expression of key transcription factors [39,102]. The endogenous TCR can also impose negative effects on the persistence of CAR T cells when CAR with distinct TCR specificity is introduced into T cells [103]. Incorporation of CAR into the endogenous TCRα gene locus prevented exhaustion and improved the functionality of CAR T cells [35].
It has been demonstrated that the inhibitory receptor, PD-1- mediated CAR T cell exhaustion can be reversed by application of PD-1 antibody or co-transduction of PD-1 dominant negative receptor [79]. In addition to inhibitory receptors, co-stimulatory receptors, such as 4-1BBζ and CD28ζ are involved in T cell exhaustion as well [104]. Further studies indicated that antigen dependent CD28ζ based CAR T cells improve proliferation and persistence, whereas 4-1BBζ based CAR T cells induce early exhaustion, thereby limiting the effector functions [105]. However, early exhaustion caused by the antigen independent clustering of CAR scFv triggered persistent tonic signaling is augmented by CD28 co-stimulation, but reduced by 4-1BB [37], highlighting the importance of optimized CAR design.
Senescence
T cell senescence is different from T cell exhaustion. It has been recognized as a key player of immunosuppression in cancer patients and the aging population [106]. Although telomere length shortens with cell replication due to progressively lose telomerase activity in normal adult somatic cells, it can be maintained in human T lymphocytes because of reactivation of telomerase activity when encountered with persistent antigen stimulation [107]. However, with the differentiation of T cells, telomerase activity is reduced, resulting in telomere erosion and replicative senescence [108]. Thus, preventing or reversing replicative and early T cell senescence is required to increase lifespan and clinical outcome of cancer patients.
Importantly, senescent effector T cells induced by both naturally occurring Treg cell initiated DNA damage [109] and tumor-derived γδ Treg cells [110] have potent suppressive activities, which is cooperatively regulated by ERK1/2, P38 signaling and STAT1, STAT3 signaling [109]. Hopefully, the adverse effect of T cell senescence can be reversed via inhibition of DNA damage response and/or STATA signaling.
Markers for exhaustion and senescence
Exhausted T cells lose the capacity to produce IL-2, tumor necrosis factor (TNF) and interferon-γ, lose cytotoxicity and stop proliferation [39]. The increased expression of inhibitory receptors, including PD-1, CTLA-4, CD160, lymphocyte activation gene 3 (LAG-3), T cell immunoglobulin mucin 3 (TIM-3) has been identified as exhaustion associated markers [98]. Therefore, blocking the interaction of these and other T cell negative check-point receptor pathways using targeted reagents may serve as potential strategies for the reversal of T cell exhaustion.
T cells in replicative senescence have phenotypic changes, including down-regulation of co-stimulatory molecules such as CD27 and CD28 while expressing killer cell lectin-like receptor subfamily G (KLRG-1), CD57, the T cell immunoreceptor with Ig and tyrosine-based inhibitory motif (ITIM) domains (TIGIT), senescence-associated-β-galactosidase (SA-β-Gal), cell cycle-regulating proteins such as P15, P16 and P21 [110-112]. Molecules regulating the G1/S phase transition, including p16/Cdk6 and p21/WAF bounds are upregulated, while Cdk2 and cyclinD3 are down-regulated, forcing T cells entering into a replicative senescence [112].
Targets and associated heterogeneity
Promising targets
The first step for CAR T cell therapy is targeting an ideal TAA, which should be highly expressed on the surface of tumor mass but not or lowly expressed on normal tissues. Unfortunately, unlike the consistent expression of CD19 antigen on leukemia tumors, identification of specific antigens on solid tumor is rare. A growing number of tumor antigens are being evaluated for CAR T cell therapy (Table 1), including diganglioside GD2 [113] and HER2 [114].
Surface molecules as potential targets for CAR T cell therapies
| Malignancy | Target Antigen | Clinical evaluation | References |
|---|---|---|---|
| NSCLC | HER2/EGFR/MUC1/CEA | EGFR | [115,116] |
| Pancreatic cancer | PSCA/MUC1/HER2/EGFR | HER2 | [117,118] |
| Ovarian cancer | MUC16/PSMA/5T4/NKG2DL/HER2 | HER2 | [119,120,121,122] |
| GBM | EGFRvIII/HER2/IL13Rα2/EphA2 | EGFRvIII/HER2/IL13Rα | [124,129,131,132] |
In order to treat non-small cell lung cancer (NSCLC) by using CAR T cell therapy, several tumor antigens including MSLN, HER2, EGFR, MUC1, and CEA are tested [115] (Table 1). For example, EGFR-targeted CAR T cells for NSCLC in a Phase I clinical trial have been evaluated, with 2 out of 11 partial response and 5 stable disease for a period of 2-8 months [116]. For pancreatic cancer, CAR T cells targeting MSLN, prostate stem cell antigen (PSCA), MUC1, HER2 and EGFR are under evaluation in clinical trials [117]. In a Phase I study, HER2-positive biliary tract cancers and pancreatic patients are infiltrated with HER2-targeted CAR T cells, with 1 out of 11 patients obtained a partial response and 5 have stable disease [118]. In ovarian carcinoma, tumor antigens including MUC16 [119], prostate-specific membrane antigen (PSMA) [120] and 5T4 [121] have been tested for CAR T cell therapy. Aside from using scFv for CAR construction, different receptors, such as NKG2D, whose ligands NKG2DL are highly expressed in ovarian cancer but absent in normal tissues, have been incorporated into CARs. Preclinical results showed that NKG2D-targeted CAR T cells are efficient in recognizing and eradicating NKG2DL-expressing ovarian cancer cells [122].
Glioblastoma multiforme (GBM) is the most common and aggressive primary malignant brain cancer with grave prognosis [123]. Dysregulation of the transmembrane tyrosine kinase receptor EGFR can lead to various malignancies including GBM [124]. The deletion of 267 amino acids in extracellular domain of EGFR results in EGFRvIII variant, which is commonly and specifically expressed on GBM cancer cells [124]. CAR T cells targeting EGFRvIII can effectively cross the blood-brain barrier [125] and amount potent antitumor effects against EGFRvIII expressing glioblastoma stem cell lines without having any side effect on co-cultured normal cells [126]. A Phase I clinical trial using EGFRvIII-CAR T cell therapy to treat GBM patients demonstrated expansion of CAR T cells in peripheral blood and decrease in tumor burden [127]. Another attractive target for CAR T cell therapy in GBM patients is HER2, which is a transmembrane glycoprotein and belongs to EGFR family [128]. In GBM animal models, HER2-CAR T cells exhibited enhanced antitumor efficacy [129]. In a clinical study, 17 patients with HER2-positive GBM were infused with HER2-CAR T cells, out of which, one patient have partial response and 7 achieved stable disease [130]. Interleukin-13 receptor chain 2 (IL13Rα2), which is commonly overexpressed on GBM but absent on normal brain cells, is another important target for CAR T cell therapy in treating GBM patients [131]. Initial promising results have been obtained in a Phase I clinical trial for evaluation of CAR T cells targeting IL13Rα in recurrent GBM patients by intracranial delivery, with 2 out of 3 patients have complete remission [18]. Later on, the same group adopted another delivery strategy, intraventricular infusions of multiple times of genetically engineered T cells directed to the IL13Rα2, achieved complete regression in one metastatic GBM patient [18]. CAR T cells directed to alternative antigens against GBM including ephrin type A receptor 2 (EphA2), which is also highly expressed in GBM but not in normal brain tissue [132].
Heterogeneity
Antigens for solid tumor exhibit tissue and genetic heterogeneity with regards to varying levels of expression intensity and distribution of antigen positive cells, posing a major challenge for CAR T cell therapy [133]. It has published that, MSLN is overexpressed in NSCLC in comparison to normal tissue, and exhibits varying levels among tumor cells [134]. The intratumoral heterogeneity of other antigen targets, including HER2, MUC1, prostate stem cell antigen (PSCA) and epithelial cell adhesion molecule (EpCAM), has also been reported [135]. Tumor cells expressing high levels of specific antigen were preferentially eliminated, whereas those expressing low levels of antigen survived [136]. However, evidences begin to reveal that the immune-mediated anti-tumor effects and the ensuing inflammation induced by CAR T cell responses may initiate new immune responses and subsequent killing of any cancer cells [137]. Novel CAR designs, such as dual TAA-targeted CARs, tandem CARs, switchable CARs, inhibitory CARs, and the rational design of 'AND', 'OR', and 'NOT' gated approaches are being investigated to mitigate tumor antigen heterogeneity [138,139].
Minimizing variability between patient products
The interpatient variability of starting immune cell populations might result in unsuccessful CAR T cell production in vitro and unpredictable T cell expansion and tumor killing activity in vivo [140]. Strategies including control of CD4/CD8 ratio and CAR expression, generation of universal CAR T cells have been used to minimize variability among patient products. Enhanced efficacy has been demonstrated by culturing CD4 and CD8 cells separately and combining them in a defined ratio at the time of infusion [54]. Furthermore, uniform levels of CAR expression has been achieved by using in-process selection of the transduced cells [46].
The use of autologous T cells for CAR-based therapies requires individualized cell production, which makes interpatient variability very problematic with the risk of host immune rejection or GVHD. To circumvent these obstacles, genome-editing tools are harnessed for eliminating the endogenous HLA and/or TCRs on the surface of CAR T cells [68] or NK cells [7]. Combining CAR expression with deletion of TCRα and CD52 in a clinical setting [141] or down regulate expression of CD3/TCRαβ in preclinical studies [142] appears feasible and effective in CAR T cell production.
Most of the current CARs adopt a rigid design composing a fixed scFv and intracellular signaling domains, greatly limiting the controllability of CAR T cells [143]. To afford greater flexibility in antigen recognition, split CAR using a universal receptor as the signaling motif connected with a dissociable antigen recognition motif has been developed, allowing a great many antigens to be targeted without re-engineering the T cells [144,145]. In order to further expand the CAR T cell capability, a split, universal, and programmable (SUPRA) CAR system was developed [146]. It is a single, feature-rich and integrated system incorporating multiple “upgrades”, such as the ability to switch targets without re-engineering the immune cells via a split CAR configuration, fine-tune T cell activation strength via multiple mechanisms, sense and logically respond to multiple antigens for enhancing specificity.
Summary and future perspectives
The great potential of CAR T cell therapy has been validated in relapsed and refractory B-All malignancies, yet the clinical responses currently in solid tumors have been sporadic [13]. Here, we updated the major challenges and difficulties encountered by CAR T cells targeting against solid tumors, and discussed strategies to overcome these obstacles as well. Solid tumors constitute a challenge due to multiple reasons: lack of antigen specificity, poor trafficking and expansion, hostile immunosuppressive microenvironment [61], tumor escape and relapse [147], toxicity of CARs, as well as time-consuming and expensive manufacturing of CAR T cells. Given the limitations of current CAR T cell therapy, sophisticated strategies are under investigation to overcome these challenges, such as targeting multiple tumor antigens to lessen antigen escape, harnessing immunocheckpoint inhibitors to increase effecter response, and regional infusion of CAR T cells for direct effect. In particular, innovative CAR design and logic-gated approaches allow for exploiting the full therapeutic potential of CAR T cells and enabling treatment of a broader range of cancer patients in the near future. Apart from autologous T cell as the major source used at present, allogeneic cells including stem cell-derived, 'off the shelf' T cells may play an important role in the future.
Compared with conventional CAR T cells, CRISPR/Cas9- edited CAR T cells showed an enhanced potency, delayed differentiation and exhaustion, highlighting genome-editing technology as a promising approach to make additional changes and improvements to the infused CAR T cells. In addition to genome-editing technology, other therapeutic measures including chemotherapies, radiation, immune checkpoint blockade antibodies and cytokine treatment might be combined with CAR T cell therapy to broaden the application and enhance the efficacy for individual cancer elimination. Despite progress in managing severe cytokine release and neurotoxicity, the mechanisms behand are poorly understood, further improvement of the CAR T cell therapy for treating solid cancers is therefore deserves more attention.
Acknowledgements
Current research is funded by China Postdoctoral Science Foundation (2018M633220), the National Key Basic Research Program of China (973 Program) (2014CB745201), National Natural Science Foundation of China (81772737), National Science Foundation Projects of Guangdong Province (2017B030301015), the Shenzhen Municipal Government of China (JCYJ20170413161749433, JSGG20160301161836370), the Sanming Project of Shenzhen Health and Family Planning Commission (SZSM201412018, SZSM201512037), and the High Level University's Medical Discipline Construction (2016031638).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gill S, Maus MV, Porter DL. Chimeric antigen receptor T cell therapy: 25years in the making. Blood reviews. 2016;30(3):157-167
2. Gacerez AT, Sentman CL. T-bet promotes potent antitumor activity of CD4+ CAR T cells. Cancer gene therapy. 2018;25(5-6):117-128
3. Jensen MC, Riddell SR. Design and implementation of adoptive therapy with chimeric antigen receptor-modified T cells. Immunological reviews. 2014;257(1):127-144
4. Hay KA, Turtle CJ. Chimeric antigen receptor (CAR) T cells: lessons learned from targeting of CD19 in B-cell malignancies. Drugs. 2017;77(3):237-245
5. Savoldo B, Ramos CA, Liu E. et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. The journal of clinical investigation. 2011;121(5):1822-1826
6. Maus MV, Grupp SA, Porter DL. et al. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123(17):2625-2635
7. Krackhardt AM, Anliker B, Hildebrandt M. et al. Clinical translation and regulatory aspects of CAR/TCR-based adoptive cell therapies-the German Cancer Consortium approach. Cancer immunology immunotherapy. 2018;67(4):513-523
8. Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nature medicine. 2016;22(1):26-36
9. Kochenderfer JN, Dudley ME, Kassim SH. et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. Journal of clinical oncology. 2015;33(6):540-549
10. Grupp SA, Kalos M, Barrett D. et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. The New England journal of medicine. 2013;368(16):1509-1518
11. Kakarla S, Gottschalk S. CAR T cells for solid tumors: armed and ready to go? Cancer journal. 2014;20(2):151-155
12. Lamers CH, Klaver Y, Gratama JW. et al. Treatment of metastatic renal cell carcinoma (mRCC) with CAIX CAR-engineered T-cells-a completed study overview. Biochemical society transactions. 2016;44(3):951-959
13. Yong CSM, Dardalhon V, Devaud C. et al. CAR T-cell therapy of solid tumors. Immunology and cell biology. 2017;95(4):356-363
14. Adusumilli PS, Cherkassky L, Villena-Vargas J. et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Science translational medicine. 2014;6(261):261ra151
15. Choi BD, Suryadevara CM, Gedeon PC. et al. Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. Journal of clinical neuroscience. 2014;21(1):189-190
16. Parente-Pereira AC, Burnet J, Ellison D. et al. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID beige mice. Journal of clinical immunology. 2011;31(4):710-718
17. Van Schalkwyk MC, Papa SE, Jeannon JP. et al. Design of a phase I clinical trial to evaluate intratumoral delivery of ErbB-targeted chimeric antigen receptor T-cells in locally advanced or recurrent head and neck cancer. Human gene therapy clinical development. 2013;24(3):134-142
18. Brown CE, Alizadeh D, Starr R. et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. The New England journal of medicine. 2016;375(26):2561-2569
19. Katz SC, Burga RA, Mccormack E. et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clinical cancer research. 2015;21(14):3149-3159
20. Song MJ. Hepatic artery infusion chemotherapy for advanced hepatocellular carcinoma. World journal of gastroenterology. 2015;21(13):3843-3849
21. Newick K, Moon E, Albelda SM. Chimeric antigen receptor T-cell therapy for solid tumors. Molecular therapy oncolytics. 2016;3:16006
22. Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends in immunology. 2003;24(6):327-334
23. Harlin H, Meng Y, Peterson AC. et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer research. 2009;69(7):3077-3085
24. Craddock JA, Lu A, Bear A. et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. Journal of immunotherapy. 2010;33(8):780-788
25. Moon EK, Carpenito C, Sun J. et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clinical cancer research. 2011;17(14):4719-4730
26. Newick K, O'brien S, Sun J. et al. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase A localization. Cancer immunology research. 2016;4(6):541-551
27. Di Stasi A, De Angelis B, Rooney CM. et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392-6402
28. Nishio N, Diaconu I, Liu H. et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer research. 2014;74(18):5195-5205
29. Caruana I, Savoldo B, Hoyos V. et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nature medicine. 2015;21(5):524-529
30. Chinnasamy D, Yu Z, Theoret MR. et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. The journal of clinical investigation. 2010;120(11):3953-3968
31. Legler DF, Johnson-Leger C, Wiedle G. et al. The alpha v beta 3 integrin as a tumor homing ligand for lymphocytes. European journal of immunology. 2004;34(6):1608-1616
32. Kandalaft LE, Facciabene A, Buckanovich RJ. et al. Endothelin B receptor, a new target in cancer immune therapy. Clinical cancer research. 2009;15(14):4521-4528
33. Smith TT, Moffett HF, Stephan SB. et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. The journal of clinical investigation. 2017;127(6):2176-2191
34. Watanabe K, Terakura S, Martens AC. et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 zeta chimeric antigen receptor-modified effector CD8+ T cells. Journal of immunology. 2015;194(3):911-920
35. Eyquem J, Mansilla-Soto J, Giavridis T. et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113-117
36. Frigault MJ, Lee J, Basil MC. et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer immunology research. 2015;3(4):356-367
37. Long AH, Haso WM, Shern JF. et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature medicine. 2015;21(6):581-590
38. Liu H, Rhodes M, Wiest DL. et al. On the dynamics of TCR:CD3 complex cell surface expression and downmodulation. Immunity. 2000;13(5):665-675
39. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nature reviews Immunology. 2015;15(8):486-499
40. Kofler DM, Chmielewski M, Rappl G. et al. CD28 costimulation Impairs the efficacy of a redirected t-cell antitumor attack in the presence of regulatory T cells which can be overcome by preventing Lck activation. Molecular therapy. 2011;19(4):760-767
41. Markley JC, Sadelain M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood. 2010;115(17):3508-3519
42. Chang L, Chang WC, Mcnamara G. et al. Transgene-enforced co-stimulation of CD4+ T cells leads to enhanced and sustained anti-tumor effector functioning. Cytotherapy. 2007;9(8):771-784
43. Hudecek M, Sommermeyer D, Kosasih PL. et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer immunology research. 2015;3(2):125-135
44. Rossig C, Pule M. Vaccination to improve the persistence of CD19CAR gene-modified T cells in relapsed pediatric acute lymphoblastic leukemia. leukemia. 2017;31(5):1087-1095
45. Singh N, Perazzelli J, Grupp SA. et al. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Science translational medicine. 2016;8(320):320ra323
46. Gardner RA, Finney O, Annesley C. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322-3331
47. Chekmasova AA, Sandadi S, Spriggs DR. et al. Effect of modulation of the hostile tumor microenvironment through adoptive transfer of IL-12 expressing MUC 16 targeted T cells on ovarian tumors in vivo. Journal of clinical oncology. 2012;30(15):2586
48. Wilkie S, Burbridge SE, Chiapero-Stanke L. et al. Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. The journal of biological chemistry. 2010;285(33):25538-25544
49. Davies DM, Foster J, Van Der Stegen SJ. et al. Flexible targeting of ErbB dimers that drive tumorigenesis by using genetically engineered T cells. Molecular medicine. 2012;18:565-576
50. Maude S L, Frey N, Shaw PA. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. The New England journal of medicine. 2014;371(16):1507-1517
51. Jensen MC, Popplewell L, Cooper LJ. et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biology of blood and marrow transplantation. 2010;16(9):1245-1256
52. Van Der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nature reviews drug discovery. 2015;14(7):499-509
53. Berger C, Jensen MC, Lansdorp PM. et al. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. The journal of clinical investigation. 2008;118(1):294-305
54. Sommermeyer D, Hudecek M, Kosasih PL. et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30(2):492-500
55. Kawalekar OU, O'connor RS, Fraietta JA. et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380-390
56. Guedan S, Chen X, Madar A. et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 2014;124(7):1070-1080
57. Guedan S, Posey AD, Shaw C. et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI insight. 2018;3(1):pii 96976
58. Bradley LM, Haynes L, Swain SL. IL-7: maintaining T-cell memory and achieving homeostasis. Trends in immunology. 2005;26(3):172-176
59. Sabatino M, Hu J, Sommariva M. et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128(4):519-528
60. Annesley CE, Summers C, Ceppi F. et al. The evolution and future of CAR T cells for B-Cell acute lymphoblastic leukemia. Clinical pharmacology and therapeutics. 2018;103(4):591-598
61. Shiao SL, Ganesan AP, Rugo HS. et al. Immune microenvironments in solid tumors: new targets for therapy. Genes and development. 2011;25(24):2559-2572
62. Ruella M, Kenderian SS, Shestova O. et al. Kinase inhibitor ibrutinib to prevent cytokine-release syndrome after anti-CD19 chimeric antigen receptor T cells for B-cell neoplasms. Leukemia. 2017;31(1):246-248
63. Menger L, Sledzinska A, Bergerhoff K. et al. TALEN-mediated inactivation of PD-1 in tumor-reactive lymphocytes promotes intratumoral T-cell persistence and rejection of established tumors. Cancer research. 2016;76(8):2087-2093
64. Ruella M, Kalos M. Adoptive immunotherapy for cancer. Immunological reviews. 2014;257(1):14-38
65. Li S, Siriwon N, Zhang X. et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clinical cancer research. 2017;23(22):6982-6992
66. John LB, Devaud C, Duong CP. et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clinical cancer research. 2013;19(20):5636-5646
67. Topalian SL, Hodi FS, Brahmer JR. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine. 2012;366(26):2443-2454
68. Ren J, Liu X, Fang C. et al. Multiplex genome editing to generate universal CAR T cells resistant to PD-1 inhibition. Clinical cancer research. 2017;23(9):2255-2266
69. Yeku OO, Purdon TJ, Koneru M. et al. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Scientific reports. 2017;7(1):10541
70. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert opinion on biological therapy. 2015;15(8):1145-1154
71. Chmielewski M, Kopecky C, Hombach AA. et al. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer research. 2011;71(17):5697-5706
72. Wang W, Ma Y, Li J. et al. Specificity redirection by CAR with human VEGFR-1 affinity endows T lymphocytes with tumor-killing ability and anti-angiogenic potency. Gene therapy. 2013;20(10):970-978
73. Massague J. TGFbeta in cancer. Cell. 2008;134(2):215-230
74. Bollard CM, Rossig C, Calonge MJ. et al. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99(9):3179-3187
75. Schmid MC, Avraamides CJ, Dippold HC. et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer cell. 2011;19(6):715-727
76. Mussai F, Egan S, Hunter S. et al. Neuroblastoma arginase activity creates an immunosuppressive microenvironment that impairs autologous and engineered immunity. Cancer research. 2015;75(15):3043-3053
77. Burga RA, Thorn M, Point GR. et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer immunology immunotherapy. 2015;64(7):817-829
78. Sheppard D. Dominant negative mutants: tools for the study of protein function in vitro and in vivo. American journal of respiratory cell and molecular biology. 1994;11(1):1-6
79. Cherkassky L, Morello A, Villena-Vargas J. et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. The journal of clinical investigation. 2016;126(8):3130-3144
80. Foster AE, Dotti G, Lu A. et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-beta receptor. Journal of immunotherapy. 2008;31(5):500-505
81. Liu X, Ranganathan R, Jiang S. et al. A chimeric switch-receptor targeting PD-1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer research. 2016;76(6):1578-1590
82. Wrzesinski C, Paulos CM, Kaiser A. et al. Increased intensity lymphodepletion enhances tumor treatment efficacy of adoptively transferred tumor-specific T cells. Journal of immunotherapy. 2010;33(1):1-7
83. Mohammed S, Sukumaran S, Bajgain P. et al. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Molecular therapy. 2017;25(1):249-258
84. Stromnes IM, Schmitt TM, Hulbert A. et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma. Cancer cell. 2015;28(5):638-652
85. Riese MJ, Wang LC, Moon EK. et al. Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer research. 2013;73(12):3566-3577
86. Wang LC, Lo A, Scholler J. et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer immunology research. 2014;2(2):154-166
87. Linette GP, Stadtmauer EA, Maus MV. et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122(6):863-871
88. Casucci M, Hawkins RE, Dotti G. et al. Overcoming the toxicity hurdles of genetically targeted T cells. Cancer immunology immunotherapy. 2015;64(1):123-130
89. Drent E, Themeli M, Poels R. et al. A rational strategy for reducing on-target off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Molecular therapy. 2017;25(8):1946-1958
90. Caruso HG, Hurton LV, Najjar A. et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer research. 2015;75(17):3505-3518
91. Wang X, Chang WC, Wong CW. et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255-1263
92. Di Stasi A, Tey SK, Dotti G. et al. Inducible apoptosis as a safety switch for adoptive cell therapy. The New England journal of medicine. 2011;365(18):1673-1683
93. Cooper LJ, Ausubel L, Gutierrez M. et al. Manufacturing of gene-modified cytotoxic T lymphocytes for autologous cellular therapy for lymphoma. Cytotherapy. 2006;8(2):105-117
94. Kloss CC, Condomines M, Cartellieri M. et al. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nature biotechnology. 2013;31(1):71-75
95. Fedorov VD, Sadelain M, Kloss CC. Novel approaches to enhance the specificity and safety of engineered T cells. Cancer journal. 2014;20(2):160-165
96. Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Current opinion in immunology. 2015;33:9-15
97. Roybal KT, Rupp LJ, Morsut L. et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell. 2016;164(4):770-779
98. Wherry EJ. T cell exhaustion. Nature immunology. 2011;12(6):492-499
99. Crespo J, Sun H, Welling TH. et al. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Current opinion in immunology. 2013;25(2):214-221
100. Coppe JP, Desprez PY, Krtolica A. et al. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual review of pathology. 2010;5:99-118
101. Davoodzadeh GM, Kardar GA, Saeedi Y. et al. Exhaustion of T lymphocytes in the tumor microenvironment: significance and effective mechanisms. Cellular immunology. 2017;322:1-14
102. Amezquita RA, Kaech SM. Immunology: the chronicles of T-cell exhaustion. Nature. 2017;543(7644):190-191
103. Yang Y, Kohler ME, Chien CD. TCR engagement negatively affects CD8 but not CD4 CAR T cell expansion and leukemic clearance. Science translational medicine. 2017;9(417):eaag1209
104. Odorizzi PM, Wherry EJ. Inhibitory receptors on lymphocytes: insights from infections. Journal of immunology. 2012;188(7):2957-2965
105. Zhang C, Liu J, Zhong JF. et al. Engineering CAR-T cells. Biomarker research. 2017;5:22
106. Kasakovski D, Xu L, Li Y. T cell senescence and CAR-T cell exhaustion in hematological malignancies. Journal of hematology and oncology. 2018;11(1):91
107. Hodes RJ, Hathcock KS, Weng NP. Telomeres in T and B cells. Nature reviews immunology. 2002;2(9):699-706
108. Plunkett FJ, Franzese O, Finney HM. et al. The loss of telomerase activity in highly differentiated CD8+ CD28-CD27- T cells is associated with decreased Akt (Ser473) phosphorylation. Journal of immunology. 2007;178(12):7710-7719
109. Liu X, Mo W, Ye J. et al. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nature communications. 2018;9(1):249
110. Ye J, Ma C, Hsueh EC. et al. Tumor-derived gammadelta regulatory T cells suppress innate and adaptive immunity through the induction of immunosenescence. Journal of immunology. 2013;190(5):2403-2414
111. Takasugi M. Emerging roles of extracellular vesicles in cellular senescence and aging. Aging cell. 2018;17(2):e12734
112. Xu W, Larbi A. Markers of T cell senescence in humans. International journal molecular sciences. 2017;18(8):1-13
113. Louis CU, Savoldo B, Dotti G. et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118(23):6050-6056
114. Ahmed N, Brawley VS, Hegde M. et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. Journal of clinical oncology. 2015;33(15):1688-1696
115. Zeltsman M, Dozier J, Mcgee E. et al. CAR T-cell therapy for lung cancer and malignant pleural mesothelioma. Translational research. 2017;187:1-10
116. Feng K, Guo Y, Dai H. et al. Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Science China life sciences. 2016;59(5):468-479
117. Deselm CJ, Tano ZE, Varghese AM. et al. CAR T-cell therapy for pancreatic cancer. Journal of surgical oncology. 2017;116(1):63-74
118. Feng K, Liu Y, Guo Y. et al. Phase I study of chimeric antigen receptor modified T cells in treating HER2-positive advanced biliary tract cancers and pancreatic cancers. Protein and cell. 2018;9(10):838-847
119. Chekmasova AA, Rao TD, Nikhamin Y. et al. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clinical cancer research. 2010;16(14):3594-3606
120. Santoro SP, Kim S, Motz GT. et al. T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer immunology research. 2015;3(1):68-84
121. Owens GL, Sheard VE, Kalaitsidou M. et al. Preclinical assessment of CAR T-cell therapy targeting the tumor antigen 5T4 in ovarian cancer. Journal of immunotherapy. 2018;41(3):130-140
122. Song DG, Ye Q, Santoro S. et al. Chimeric NKG2D CAR-expressing T cell-mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Human gene therapy. 2013;24(3):295-305
123. Iacob G, Dinca EB. Current data and strategy in glioblastoma multiforme. Journal of medicine and life. 2009;2(4):386-393
124. Villa GR, Mischel PS. Old player, new partner: EGFRvIII and cytokine receptor signaling in glioblastoma. Nature neuroscience. 2016;19(6):765-767
125. Miao H, Choi BD, Suryadevara CM. et al. EGFRvIII-specific chimeric antigen receptor T cells migrate to and kill tumor deposits infiltrating the brain parenchyma in an invasive xenograft model of glioblastoma. PloS one. 2014;9(4):e94281
126. Morgan RA, Johnson LA, Davis JL. et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Human gene therapy. 2012;23(10):1043-1053
127. O'rourke DM, Nasrallah MP. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Science transnational medicine. 2017;9:399
128. Cho HS, Mason K, Ramyar KX. et al. Structure of the extracellular region of HER2 alone and in complex with the herceptin Fab. Nature. 2003;421(6924):756-760
129. Hegde M, Mukherjee M, Grada Z. et al. Tandem CAR T cells targeting HER2 and IL13R alpha2 mitigate tumor antigen escape. The journal of clinical investigation. 2016;126(8):3036-3052
130. Ahmed N, Brawley V, Hegde M. et al. HER2-specific chimeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA oncology. 2017;3(8):1094-1101
131. Brown CE, Warden CD, Starr R. et al. Glioma IL13R alpha2 is associated with mesenchymal signature gene expression and poor patient prognosis. PloS one. 2013;8(10):e77769
132. Yi Z, Prinzing BL, Cao F. et al. Optimizing EphA2-CAR T cells for the adoptive immunotherapy of glioma. Molecular therapy Methods and clinical development. 2018;9:70-80
133. Mcgranahan N, Furness AJ, Rosenthal R. et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463-1469
134. Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer discovery. 2016;6(2):133-146
135. Ali A, Brown V, Denley S. et al. Expression of KOC, S100P, mesothelin and MUC1 in pancreatico-biliary adenocarcinomas: development and utility of a potential diagnostic immunohistochemistry panel. BMC clinical pathology. 2014;14:35
136. Song DG, Ye Q, Poussin M. et al. Effective adoptive immunotherapy of triple-negative breast cancer by folate receptor-alpha redirected CAR T cells is influenced by surface antigen expression level. Journal of hematology and oncology. 2016;9(1):56
137. Slaney CY, Von Scheidt B, Davenport AJ. et al. Dual-specific chimeric antigen receptor T cells and an indirect vaccine eradicate a variety of large solid tumors in an immunocompetent, self-antigen setting. Clinical cancer research. 2017;23(10):2478-2490
138. Chen N, Li X, Chintala NK. et al. Driving CARs on the uneven road of antigen heterogeneity in solid tumors. Current opinion in immunology. 2018;51:103-110
139. Ebert LM, Yu W, Gargett T. et al. Logic-gated approaches to extend the utility of chimeric antigen receptor T-cell technology. Biochemical society transactions. 2018;46(2):391-401
140. Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545(7655):423-431
141. Qasim W, Zhan H, Samarasinghe S. et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Science translational medicine. 2017;9:374
142. Kamiya T, Wong D, Png YT. et al. A novel method to generate T-cell receptor-deficient chimeric antigen receptor T cells. Blood advances. 2018;2(5):517-528
143. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127(26):3321-3330
144. Cartellieri M, Feldmann A, Koristka S. et al. Switching CAR T cells on and off: a novel modular platform for retargeting of T cells to AML blasts. Blood cancer journal. 2016;6(8):e458
145. Rodgers DT, Mazagova M, Hampton EN. et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proceedings of the national academy of sciences of the United States of America. 2016;113(4):E459-468
146. Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173(6):1426-1438
147. Wang QS, Wang Y, Lv HY. et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Molecular therapy. 2015;23(1):184-191
Author contact
![]() Corresponding authors: wangzhc6sysu.edu.cn (Zhicai Wang); caizhiming2000com (Zhiming Cai); pony8980com (Weiren Huang).
Corresponding authors: wangzhc6sysu.edu.cn (Zhicai Wang); caizhiming2000com (Zhiming Cai); pony8980com (Weiren Huang).