Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(16):3678-3690. doi:10.7150/jca.32803 This issue Cite
Research Paper
Identification of Chaetocin as a Potent non-ROS-mediated Anticancer Drug Candidate for Gastric Cancer
Xinwen Liao1,#, Yaqiong Fan1,#, Jihuan Hou1,#, Xiong Chen1, Xiaolin Xu1, Yifan Yang1, Jinying Shen1, Panying Mi1, Xiaohua Huang2, Wenqing Zhang1, Hanwei Cao1, Xiaoting Hong2, Tianhui Hu1, Yan-yan Zhan1, ![]()
1. Cancer Research Center, School of Medicine, Xiamen University, Xiamen 361102, Fujian, P.R. China
2. Department of Basic Medicine, School of Medicine, Xiamen University, Xiamen 361102, Fujian, P.R. China
#These authors contribute equally to this work.
Received 2019-1-4; Accepted 2019-5-14; Published 2019-6-9
Abstract
Chaetocin, a natural product extracted from Chaetomium species, possesses anticancer effects in several kinds of tumors. However, it remains unclear whether the potential indication for chaetocin could also include human gastric cancer. We found here that chaetocin induced caspase-dependent and -independent apoptosis in human gastric cancer cell lines, which greatly depended on BID-mediated AIF translocation. Despite not increasing the intercellular ROS levels in gastric cancer cells, chaetocin did cause a reduction in mitochondrial membrane potential probably through its regulation on the expression of Bcl-2 and BAX. Chaetocin could also induce autophagy in gastric cancer cells; blocking autophagy by chloroquine enhanced the cytotoxicity of chaetocin. Chaetocin was further found to suppress the growth of gastric cancer xenograft in nude mice. Therefore, our study provides first evidence that chaetocin has an anticancer efficacy against gastric cancer and the combined use of chaetocin with autophagy inhibitors may enhance the therapeutic effect for gastric cancer. As chronic and exorbitant ROS levels instigate drug resistance, chaetocin, which eradicates gastric cancer cells without increasing ROS levels, may initiate a new line of non-ROS-mediated anti-tumor strategy.
Keywords: Chaetocin, gastric cancer, apoptosis, AIF, non-ROS-mediated, autophagy
1. Introduction
Gastric cancer is one of the most serious malignancies that threaten human health [1]. Surgical resection and chemoradiotherapy are the main treatments for gastric cancer [2]. Despite advances in gastric cancer diagnosis and treatment, the overall 5-year survival for gastric cancer remains extremely low due to the recurrence, metastasis and chemo-resistance [3]. Therefore, it is an urgent task to explore new therapeutic agents or new combination of agents to improve the treatment of gastric cancer.
Chaetocin, a natural metabolite isolated from Chaetomium fungi, belongs to the diketoepiperazines [4, 5]. Chaetocin was reported to be a nonspecific inhibitor of histone lysine methyltransferases such as SUV39H1, thereby affecting gene expression [6]. Recently, accumulating evidence reveals that chaetocin exhibits anticancer effects in several types of solid tumors including hepatocellular carcinoma, melanoma and non-small cell lung cancer [7-10]. It seems that chaetocin employs diverse mechanisms to induce cell death in a context-dependent manner. Chaetocin was found to induce apoptosis in human hepatoma cell lines HepG2, Hep3B and Huh7, which could be suppressed by the inhibition of caspase-3/7 activity with z-VAD-fmk [7]. Chaetocin manifests a proapoptotic activity on human melanoma cells through stimulating ROS generation and the downstream intrinsic mitochondrial pathway [8]. Chaetocin provokes endoplasmic reticulum stress by inhibiting SUV39H1 activity, leading to the death receptor 5-dependent apoptosis in human non-small cell lung cancer cells [9]. However, it remains unknown whether chaetocin could also be a potential therapeutic agent for gastric cancer.
The apoptosis-inducing factor (AIF), a mitochondrial oxidoreductase, is most famous for its critical role in caspase-independent programmed cell death (apoptosis) [11-13]. Upon cell death induction, AIF is released from mitochondria and translocated to the nucleus to mediate chromatin condensation and DNA fragmentation [11-13]. In addition to this pro-death function, AIF play an opposite pro-survival role in healthy cancer cells [14]. The enzymatic activity of AIF modulates energy metabolism to promote the growth and invasiveness of advanced prostate cancer cells [15]. AIF suppresses cancer metastasis by physically interacting with and inhibiting the oxidation of PTEN (phosphatase and tensin homolog on chromosome ten) [16]. Therefore, transforming AIF from an oncogenic protein into a lethal protein by inducing its cellular translocation was considered to be a promising anticancer strategy. For instance, the diarylheptanoid hirsutenone sensitizes chemo-resistant ovarian cancer cells to cisplatin at least partially through enhancing the translocation of AIF from mitochondria to the nucleus [17].
In the present study, we revealed that chaetocin induced apoptosis in gastric cancer cells mainly through BID/AIF-mediated caspase-independent pathway without stimulating ROS generation. Chaetocin-induced cell death could be enhanced by autophagy inhibitor for chaetocin also induced autophagy in gastric cancer cells. Our study thus for the first time provided evidence for the cytotoxicity of chaetocin on human gastric cancer and the combined use of chaetocin with autophagy inhibitors for the treatment of gastric cancer. More importantly, as chronic and abnormally high ROS levels instigate drug resistance [18], chaetocin may provide a new direction for the development of non-ROS-mediated cancer-targeting strategy to prevent or defer drug resistance.
2. Materials and Methods
2.1. Cell culture and drug treatment
Human embryonic kidney cell line 293T, human gastric cancer cell lines HGC-27 and NCI-N87, and human cervical carcinoma cell line HeLa were purchased from the Institute of Cell Biology, Shanghai, China. Human gastric cancer cell line AGS were kindly provided by Songlin Shi (Xiamen University, Xiamen, Fujian, China). All cell lines were identified by STR profiling and confirmed to be mycoplasma negative by the source. 293T and HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Gibco, Grand Island, NY, USA). HGC-27 and AGS cells were maintained in RPMI 1640 medium (Gibco, Grand Island, NY, USA). NCI-N87 cells were maintained in RPMI 1640 medium supplemented with 1% glutamax (Invitrogen, Carlsbad, CA, USA) and 1 mM sodium pyruvate (Invitrogen, Carlsbad, CA, USA). All media were supplemented with 10% fetal bovine serum (FBS, HyClone, Logan, UT, USA), 100 U/ml penicillin and 100 μg/ml streptomycin (Life Technologies, Carlsbad, CA, USA), and cultured at 37 °C in humidified incubator containing 5% CO2. Before drug treatment, the full medium was exchanged with 1640 medium that did not contain serum.
2.2. Antibodies and reagents
Rabbit anti-BAX (Cat. #ab32503), rabbit anti-AIF (Cat. #ab32516), mouse anti-P62 (Cat. #ab56416) and mouse anti-β-tubulin (Cat. #ab32503) antibodies were purchased from Abcam, Cambridge, MA, USA. Rabbit anti-Bcl-2 (Cat. #2870) and anti-cleaved PARP (Cat. #9541) were purchased from Cell Signaling Technology, Beverly, MA, USA. Rabbit anti-Lamin B (Cat. #sc-6216) antibodies was purchased from Santa Cruz Biotechnology, Santa Cruz, CA, USA. β-actin antibody was purchased from Sigma-Aldrich, St. Louis, MO, USA. Rabbit anti-Caspase-3 (Cat. #19677-1-AP) and mouse anti-Caspase-9 (Cat. #66169-1-Ig) antibodies were purchased from Proteintech Group, Inc., Wuhan, China. Rabbit anti-Caspase-8 (Cat. #CY5038) antibody was purchased from Abways Technology, Wynne, AR, USA. Rabbit anti-LC3B (NB100-2220) antibody was purchased from Novus Biological, Inc., Littleton, CO, USA. Chaetocin (Cat. #C9294), BI-6C9 (Cat. #B0186), acridine orange (AO, Cat. #A6014), propidium iodide (PI, Cat. # A601112), N-acetyl-L-cysteine (NAC, Cat. #A7250) and chloroquine (CQ, Cat. #C6628) were purchased from Sigma-Aldrich, St. Louis, MO, USA. Z-VAD-FMK (Cat. #T6013) was purchased from Target Molecule Corp., Boston, MA, USA. Alexa Fluor 488 labelled goat anti-rabbit IgG secondary antibody (Cat. #A27034) was purchased from Thermo Fisher Scientific, San Jose, CA, USA. Necrostatin-1 (Cat. #480065) was purchased from Merck Millipore, Billerica, MA, USA.
2.3. Cell viability assay
Cell viability was measured by MTT (3-[4,5-dimethylthiazol-2-yl]-2,3-diphenyltetrazodium bromide) assay. Chaetocin was dissolved in DMSO to a final concentration of 1 mM and then stored at -20˚C. For viability assays, cells were seeded at a density of 1.5×104 cells (for AGS and HGC-27) or 2.5×104 cells (for NCI-N87) per well in 96-well plates and cultured overnight, and then treated with different concentrations of chaetocin as indicated or DMSO (vehicle control) in serum-free medium for 24 h. After treatment, 20 μL of MTT solution was added into each well at a final concentration of 5 mg/mL and incubated for 4 h at 37˚C. The medium was then replaced with 150 μL of DMSO, and the plates were incubated at room temperature for 15 min to dissolve the formed violet formazan crystals within the metabolically viable cells. The number of viable cells was evaluated by measuring the absorbance at an OD of 590 nm using a microplate reader. IC50 was calculated as the concentration (μM) of chaetocin causing a 50% inhibition of cell viability.
2.4. Quantification of cell apoptosis by Annexin V-FITC/PI assay
Cells were plated in 12-well plates and, after adhesion, incubated with indicated concentrations of chaetocin for 24 h. Cells were then collected, and subjected to Annexin V-FITC/PI assay with an Annexin V-FITC/PI Apoptosis Detection Kit (Cat. # 40302ES60, YEASEN, Shanghai, China). Briefly, cells were resuspended in 100 μL binding buffer, and stained with 10 μL annexin V-FITC and 5 μL PI in the dark at room temperature for 15 min after the addition of 400 μL binding buffer. The FITC/PI fluorescence intensity was assayed by flow cytometry.
2.5. Analysis of cell death by PI staining
Cells were plated in 12-well plates and then was incubated with chaetocin for 24 h. After that, cells were collected and resuspended in PBS, and then stained with PI (50 μg/mL, Sigma-Aldrich, St. Louis, MO, USA) for 10 min in the dark. The dyed cells were washed twice in PBS. The PI fluorescence was assayed by flow cytometry.
2.6. Hoechst staining and observation of typical apoptotic nuclei
Cells were seeded in 12-well culture plate and incubated at 37˚C for 20 h and treated with indicated concentrations of chaetocin for 24 h. Then, the cells were washed twice with phosphate-buffered saline (PBS) and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. Fixed cells were washed with PBS, and stained with 4 µg/ml Hoechst 33342 (Sigma-Aldrich, St. Louis, MO, USA) for 30 min at room temperature in the dark. The stained cells were washed twice with PBS, and then observed and photographed by a fluorescent microscope.
2.7. Western blotting
Cells were lysed in cold ELB buffer (50 mM Tris, pH 7.6, 140 mM NaCl, 100 mM NaF, 0.5% NP-40, 2 mM NaVO3, 5 mM β-Glycerol phosphate and 1 mM PMSF), and then centrifuged at 14,000 g, 4°C for 30 min. The supernatant was collected and protein concentration was determined by BCA assay kit (Bio-Rad, Hercules, CA, USA). Cell lysate was mixed with equal volume of 2 × loading buffer and boiled at 95˚C for 10 min, and then subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membrane. After blocked with 5% non-fat dry milk in TBS-T buffer (20 mM Tris, pH 7.5, 100 mM NaCl, and 0.1% Tween-20) for 1 h at room temperature, the membranes were incubated with primary antibody overnight at 4°C, washed 3 times with TBS-T (5 min each time), incubated with secondary antibody for 1 h at room temperature, and then washed 3 times with TBS-T (10 min each time). The immune complexes were visualized by an ECL kit (Millipore, Bedford, MD, USA).
2.8. Lentivirus-mediated knockdown of AIF protein or stably expression of GFP-LC3
Lentiviruses were generated by transfecting 293T cells with the lentiviral vector (pLKO or pLV) and packaging plasmids (pMDLg-pRRE, pRSV-REV and pCMV-VSV-G) using polyethylenimine (PEI, Cat. #23966, Polysciences, Warrington, PA). Viral supernatants were collected 48 h after transfection, centrifuged at 3,000 g for 15 min, and filtered through 0.45 μm filters (Millipore, Billerica, MA, USA). Freshly plated AGS and HGC-27 cells were infected with the packaged lentivirus and selected by puromycin (2 μg/mL).
The shRNA sequences targeting human AIF was 5′-GGGTTAAGGTGATGCCCAATG-3′ (for shAIF-1) and 5′-GGTAAATGCAGAGCTACAAGC-3′ (for shAIF-2). The shRNA control (scramble) sequence was 5′-GTAACACGTCTATACGCCCA-3′. Oligonucleotides (Invitrogen, Guangzhou, China) were annealed and inserted into the pLKO vector. pEGFP-LC3 (kindly provided by Sheng-cai Lin, Xiamen University, China) was subcloned into pLV-puro plasmid using ClonExpress II One Step Cloning Kit (Vazyme Biotech, Nanjing, China).
2.9. Total RNA extraction and real-time PCR
Total RNA was extracted using the RNAiso Plus reagent as described by the manufacturer (Takara, Dalian, China). cDNA was reverse-transcribed from 1 μg of total RNA using PrimescriptTM RT reagent kit (TaKaRa, Dalian, China). Real-time PCR was carried out using the SYBR Green I fluorescent dye (SYBR® Premix Ex TaqTM II, TaKaRa, Dalian, China) and the StepOnePlusTM real- time PCR system (Applied Biosystems, Australia). GAPDH was used as an internal control. The following are the primer sequences:
AIFM1-F: 5'-TCAACAGTAGGAGCTGGTGC-3'.
AIFM1-R: 5'-TGCCTCCACCAATTAGCAGG-3'.
GAPDH-F: 5'-GGAAGGTGAAGGTCGGAGTCA-3'.
GAPDH-R: 5'-GTCATTGATGGCAACAATATCCACT-3'.
2.10. Immunofluorescence
Cells plated on glass coverslips were fixed by 4% paraformaldehyde solution for 10 min at room temperature and then permeabilized with PBS-T (phosphate-buffered saline (PBS) with 0.3% Triton X-100) for 10 min on ice, and then blocked with PBS-T containing 5% BSA for 1 h. Cells were incubated with the primary antibodies recognizing AIF (1:500) overnight at 4 °C and then incubated with Alexa Fluor 488-conjugated secondary antibodies (1:500) for 2 h at room temperature. The nuclei of the cells were stained with 4,6-diamidino-2-phenylindole (DAPI). Cells were visualized and photographed with a FV1000 confocal laser-scanning microscope.
2.11. Isolation of nuclear and cytoplasmic fractions
Cells were collected and resuspended in 200 µL Buffer A (10 mM HEPES, PH 7.9, 10 mM KCL, 0.1 mM EDTA, 0.1 mM EGTA, 0.15% NP-40, 2 mM NaVO3, 5 mM β-Glycerol phosphate and 1 mM PMSF) and put on the ice for 15 min. The homogenates were centrifuged at 12,000 g, 4°C for 1 min. The supernatant was collected to obtain the cytosolic fraction. For the extraction of the nuclear proteins, the pellet was washed five times with buffer A and centrifuged at 10,000 g, 4°C for 1 min after each washing. The supernatant was discarded and the pellet was resuspended in 100 µL Buffer B (20 mM HEPES, pH7.9, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.5% NP-40, 2 mM NaVO3, 5 mM β-Glycerol phosphate and 1 mM PMSF) and lyzed in an ultrasonic field. The lysate was centrifuged at 12,000 g, 4°C for 30 min. The supernatant was collected as the nuclear fraction.
2.12. Intracellular reactive oxygen species (ROS) measurement
Intracellular ROS were measured by flow cytometry using ROS assay kit (Cat. #S0033, Beyotime, Beijing, China) with 2,7-dichlorofluorescein diacetate (DCFH-DA) as a fluorescent probe. Briefly, 2×105 cells were seeded into 12-well plates. After adherence, cells were treated with DMSO or the indicated concentrations of chaetocin for 24 h. Thereafter, cells were resuspended in 500 μL of serum-free RPMI 1640 medium containing DCFH-DA (10 μM) and incubated at 37 °C for 20 min in the dark. The labeled cells were washed with PBS for three times, and evaluated immediately by flow cytometry.
2.13. Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was assessed by flow cytometry using mitochondrial membrane potential assay kit with JC-1 (Cat. #C2006, Beyotime, Beijing, China). Cells were collected and stained with freshly prepared JC-1 solution (10 μg/mL) at 37 °C for 20 min in the dark, and then washed twice in PBS, according to the manufacturer's protocol. The prepared cells were evaluated by flow cytometry.
2.14. Acridine orange staining to detect autophagy
Cells were seeded in 6-well culture plate and incubated at 37˚C for 20 h. After that, cells were treated with chaetocin for 24 h, and then stained with 1 µg/mL acridine orange (AO, Sigma-Aldrich, St. Louis, MO, USA) for 15 min. Cells were examined under a fluorescent microscope, with the acidic vesicular organelles (AVOs) appearing as orange/red fluorescent cytoplasmic vesicles and the nuclei displaying bright green fluorescence.
2.15. Tumor xenograft
Female BALB/C nude mice (5-week-old, 15-17 g) were obtained from Laboratory Animal Center of Xiamen University, and housed in the barrier facility of the Laboratory Animal Center, Xiamen University, as approved by the Animal Ethics Committee of Xiamen University. 8 × 106 HGC-27 cells were resuspended in 100 μL RPMI-1640 medium and injected subcutaneously into the right armpit of each mice. One week later, tumor size was measured using vernier caliper and tumor volume was calculated with the following formula: TV = L (Length) × W2 (Width) /2. Paired mice with equal tumor volume were divided into the chaetocin-treated group and the vehicle-treated group (n = 6 for each group), and then injected intraperitoneally (i.p.) daily with chaetocin (0.5 mg/kg) and the vehicle (DMSO), respectively. After 10 days of drug treatment, mice were sacrificed and the tumor were weighed.
2.16. Statistical analysis
Results were presented as means ± SEM. Statistical significance was determined by one-way ANOVA or Student's t-test using GraphPad Prism 6 (Graphpad Software). Significance was recognized as *p < 0.05, **p < 0.01 and ***p < 0.001 vs control.
3. Results
3.1. Chaetocin induced both caspase-dependent and -independent apoptosis in human gastric cancer cells
To investigate the cytotoxic effect of chaetocin on human gastric cancer, three human gastric cancer cell lines, including AGS, HGC-27 and NCI-N87, were treated with different doses of chaetocin for 24 h and their survival rate was estimated by MTT assay. Results showed that chaetocin induced cell death in all these cell lines in a dose-dependent manner, and the IC50 values of chaetocin were 120 nM, 400 nM and 820 nM for AGS, HGC-27 and NCI-N87 cell lines, respectively (Fig. 1A). When these cell lines were treated with chaetocin at the concentration of IC50 for different durations including 12, 24, 36 and 48 hours, time-dependent cell mortalities were observed (Fig. 1B).
Chaetocin induced cell death in human gastric cancer cells. (A-B) Dose- and time-dependent cell death was induced by chaetocin in gastric cancer cells. Three human gastric cancer cell lines (AGS, HGC-27 and NCI-N87) were treated with different concentrations of chaetocin for 24 h (A), or with chaetocin at the concentration of IC50 (in figure A) for different time periods (0, 12, 24, 36, 48 h) (B), and then subjected to MTT assay for the determination of cell viability. (C) Chaetocin-induced cell apoptosis was examined by Annexin V-FITC/PI staining and flow cytometry. (D) Chaetocin-induced apoptosis was detected by morphological observation. AGS and HGC-27 cells were treated with chaetocin at the concentration of IC50 for 24 h in figures C-D. Typical apoptotic nuclei were indicated by white arrows. (E) Cleavage of apoptosis-related proteins was analyzed by western blotting. AGS and HGC-27 cells were treated with different concentrations of chaetocin for 24 h. (F) Effects of Z-VAD-FMK and necrostatin-1 on chaetocin-induced cell death. AGS and HGC-27 cells were pretreated with Z-VAD-FMK (30 μM, 2 h) or necrostatin-1 (20 μM, 2 h) before chaetocin treatment (at the concentration of IC50, 24 h), and the cell viability was analyzed by MTT assay. All data are presented as means ± SEM. The basal level of cell viability was normalized to 1. ***P < 0.001, ns nonsignificant.
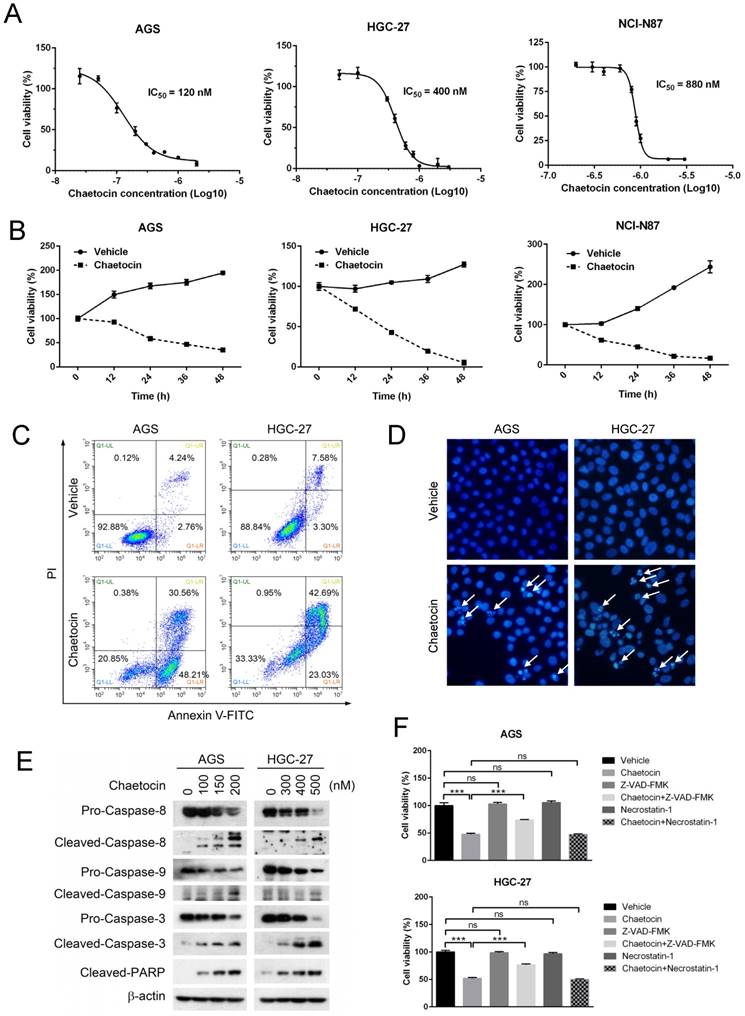
To determine the mode of cell death induced by chaetocin, two most sensitive cell lines, AGS and HGC-27 cells (Fig. 1A), were treated with chaetocin at the concentration of IC50 for 24 h and then subjected to Annexin V-FITC/PI assay and Hoechst 33258 staining. Early and late stages of apoptosis, as well as typical apoptotic nuclei—condensed or fragmented, were observed in chaetocin-treated cells (Fig. 1C & 1D), indicating that chaetocin elicited apoptosis in gastric cancer cells. The induction of apoptosis by chaetocin was further verified by the increase of apoptotic markers, including the cleavage of caspase-3, -8, -9 and poly ADP ribose polymerase (PARP), upon chaetocin treatment (Fig. 1E). However, pan-caspase inhibitor Z-VAD-FMK partly suppressed but not eliminated the cell death induced by chaetocin (Fig. 1F). We also found that chaetocin didn't induce necroptosis, as necroptosis inhibitor necrostatin-1 had no influence on chaetocin-induced cell death in AGS and HGC-27 cells (Fig. 1F). The above results suggested that chaetocin triggered apoptosis through both caspase-dependent and -independent pathway in gastric cancer cells.
3.2. AIF is critical for chaetocin to induce cell death in gastric cancer cells
AIF was reported to be a central factor in caspase-independent programmed cell death (apoptosis) [11-13]. To investigate whether AIF was required for chaetocin to induce cell death in gastric cancer cells, we stably knocked down endogenous AIF expression in AGS and HGC-27 cells using a lentivirus vector-based shRNA technique, with two different AIF-shRNA (shAIF-1 and shAIF-2). As shown in Fig. 2A-B, both protein and mRNA levels of AIF were dramatically impaired in AGS and HGC-27 cells expressing shAIF-1 or shAIF-2 when compared to their respective ctrl-shRNA (shCtrl)-expressing cells.
AIF was essential for chaetocin to induce cell death in gastric cancer cells. (A-B) AIF knockdown efficiency in AGS and HGC-27 cells was examined by western blotting (A) and real-time PCR (B). (C-D) Knocking down AIF attenuated cytotoxicity of chaetocin in AGS and HGC-27 cells. Cell sublines, including shCtrl, shAIF-1 and shAIF-2, were treated with chaetocin at the concentration of IC50 for 24 h, and then subjected to MTT assay (C), or PI staining and flow cytometry (D). Data were presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ns nonsignificant.
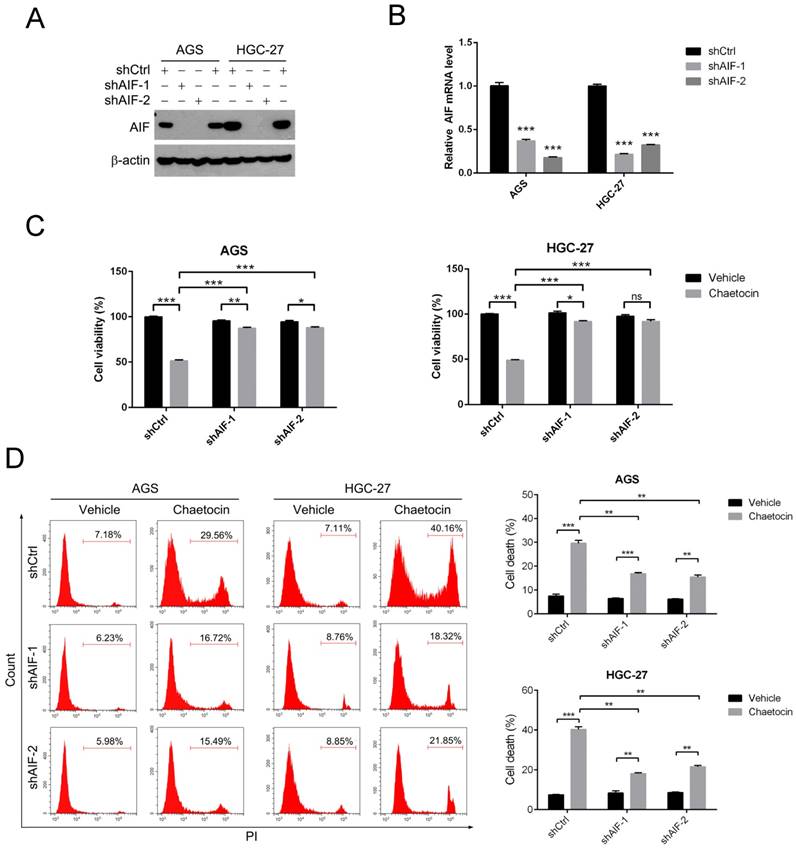
We then employed MTT assay to examine the cell viability under chaetocin treatment in these AGS and HGC-27 cell sublines. Knocking down AIF with either of these two shRNA significantly promoted cell viability in the present of chaetocin in both AGS and HGC-27 cells (Fig. 2C). Similar results were observed using propidium iodide (PI) staining and flow cytometry in AGS and HGC-27 cells (Fig. 2D). All these results indicated that AIF was required for chaetocin to induce cell death, therefore suggesting that caspase-independent events play more important role than the caspase-dependent cascades in the process of cell death induced by chaetocin.
3.3. BID-mediated AIF translocation is involved in chaetocin-induced apoptosis in gastric cancer cells
AIF translocation from the mitochondria in the cytoplasm to the nucleus is an essential step to provoke caspase-independent apoptosis [11-13], we therefore examine whether chaetocin could alter the subcellular location of AIF. As demonstrated by the results from immunostaining and isolation of nuclear and cytoplasmic fractions assays, AIF predominantly located in the cytoplasm of untreated AGS and HGC-27 cells; however, after chaetocin treatment, a great part of AIF appeared in the nucleus (Fig. 3A-B), indicating that chaetocin induced the translocation of AIF from cytoplasm to the nucleus in gastric cancer cells.
BID mediated the cell death induced by chaetocin via promoting AIF translocation. (A-B) Chaetocin induced the translocation of AIF from cytoplasm to the nucleus in gastric cancer cells as indicated by immunostaining (C) and isolation of nuclear and cytoplasmic fractions (D) assays. AGS and HGC-27 cells were treated with chaetocin at the concentration of IC50 for 24 h. Cytoplasmic and nuclear fractions were prepared for western blotting analysis using anti-AIF, anti-β-tubulin (cytosolic marker), and anti-Lamin B (nuclear marker) antibodies. C: cytoplasm; N: nuclei. (C-D) BID inhibitor BI-6C9 suppressed AIF translocation and cell death induced by chaetocin in AGS and HGC-27 cells. Cells were pretreated with BI-6C9 (10 μM) for 2 h, and then treated with chaetocin at the concentration of IC50 for 24 h, followed by immunostaining (C) and PI staining and flow cytometry (D) analyses. Data were presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
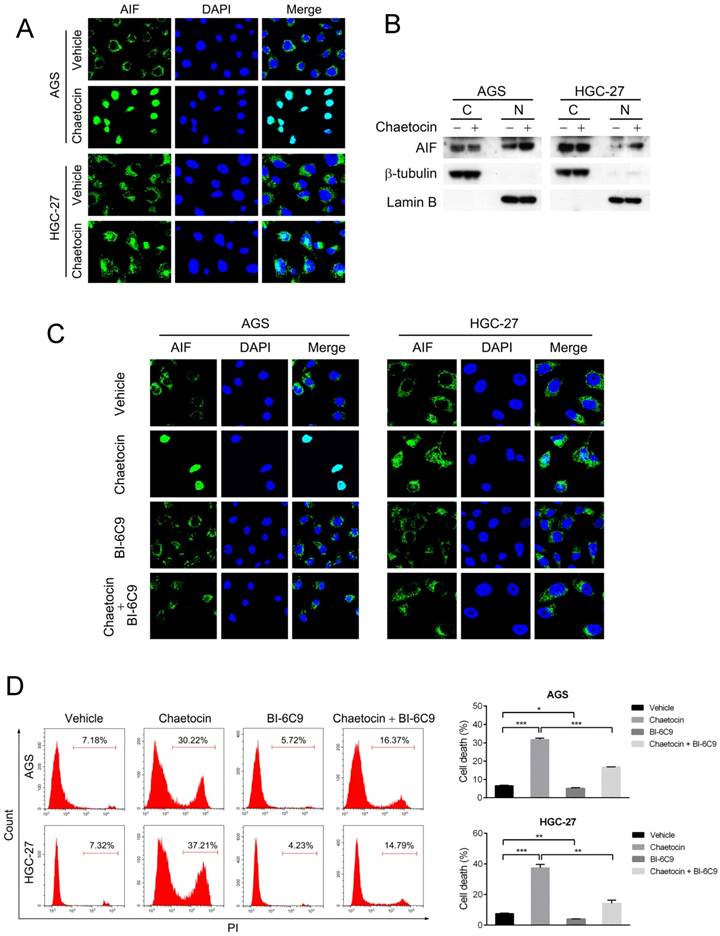
We further explored how AIF subcellular location was regulated when subjected to chaetocin. It was previously reported that BID promoted mitochondrial release of AIF to the nucleus to mediate the caspase-independent cell death triggered by N-methyl-N'-nitro-N'-nitrosoguanidine (MNNG) and matrine [19, 20]. The specific BID inhibitor BI-6C9 was therefore utilized to test whether BID regulated AIF translocation and cell death upon chaetocin treatment. Results from immunostaining showed that BI-6C9 could prevent AIF from translocating to the nucleus under chaetocin treatment in both AGS and HGC-27 cells (Fig. 3C). Moreover, pretreatment with BI-6C9 effectively attenuated chaetocin-induced cell death in these two cell lines (Fig. 3D). Therefore, BID-mediated AIF translocation to the nucleus was critical for chaetocin to induce cell death in gastric cancer cells.
3.4. Chaetocin decreased mitochondrial membrane potential without increasing intercellular ROS levels
Increased reactive oxygen species (ROS) production, followed by opened mitochondrial permeability transition pore (MPTP) and the subsequent decreased mitochondrial transmembrane potential, is a universal mechanism utilized by antitumor chemicals to induce cell death in multiple types of cancer [20-23]. To study whether ROS participated in chaetocin-induced cell death in gastric cancer cells, we analyzed chaetocin's effect on cellular ROS levels. Surprisingly, chaetocin treatment at different doses for 24 h did not increased but slightly decreased the intracellular ROS level of AGS and HGC-27 cell lines (Fig. 4A). Neither did chaetocin treatment at the concentration of IC50 for shorter duration, including 2, 4, 6, 8, 10 and 12 hours, elevate the intracellular ROS levels of AGS and HGC-27 cells (Fig. 4B). However, as a contrast, 24 h of chaetocin treatment did dose-dependently increased the ROS generation in cervical carcinoma HeLa cells (Fig. 4A), with the IC50 value of chaetocin to be 285 nM (Fig. 4C). Moreover, chaetocin-induced cell death was not influenced by the pretreatment with the ROS scavenger N-acetyl-L-cysteine (NAC) (Fig. 4D). These results suggested that ROS production was not involved in chaetocin-induced cell death in gastric cancer cells.
Chaetocin reduced mitochondrial membrane potential without increasing intercellular ROS levels. (A-B) Effect of chaetocin on the intercellular ROS levels of AGS, HGC-27 and HeLa cells. Cells were treated with different doses of chaetocin as indicated for 24 h (A) or with chaetocin at the at the concentration of IC50 for different time periods (0, 2, 4, 6, 8, 10, 12 h) (B). ROS levels were then detected by flow cytometry. (C) IC50 of chaetocin on cell viability of HeLa cells. Cells were treated with different concentrations of chaetocin for 24 h, and the cell viability was determined by MTT assay. (D) Effect of ROS scavenger NAC on chaetocin-induced cell death. AGS and HGC-27 cells were pretreated with NAC (10 μM) for 2 h before chaetocin treatment at the concentration of IC50 for 24 h, and then subjected to MTT assay. (E) Chaetocin decreased the mitochondrial membrane potential of AGS and HGC-27 cells. Cells were collected after chaetocin treatment at the concentration of IC50 for 24 h and the mitochondrial membrane potential was measured by JC-1 staining and flow cytometry. (F) Expression levels of BAX and Bcl-2 proteins after chaetocin treatment (24 h) were determined by western blotting. Data were presented as the mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ns nonsignificant.
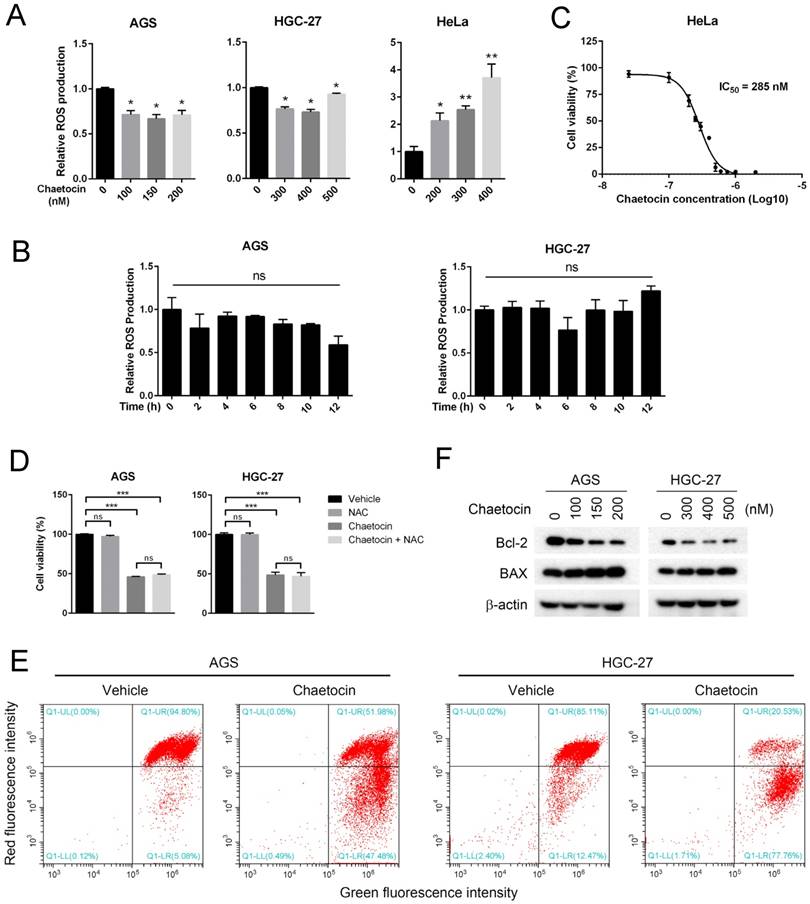
We further examined the effect of chaetocin on the mitochondrial membrane potential of gastric cancer cells by JC-1 staining, which labels high-membrane-potential mitochondria orange and low-membrane-potential mitochondria green [24]. An obviously reduction in the mitochondrial membrane potential was observed after chaetocin treatment in both AGS and HGC-27 cells (Fig. 4E). We then analyzed how chaetocin advanced mitochondrial depolarization in gastric cancer cells. It was reported that BCL-2 family proteins including BAX and Bcl-2, localized in the mitochondrial membrane, play important roles in the regulation of mitochondrial membrane permeabilization and potential in the apoptosis cascade [25, 26]. We then test whether chaetocin modulated the expression of these proteins using western blotting. As shown in Fig. 4F, chaetocin increased the expression of pro-apoptotic protein BAX while decreased the expression of anti-apoptotic protein Bcl-2. Together, the above results suggested that chaetocin decreased mitochondrial membrane potential through regulating the expression of Bcl-2 and BAX but not the generation of ROS.
3.5. The anticancer effect of chaetocin is enhanced by inhibition of autophagy
Cancer therapeutic agents, such as cisplatin and 5-FU, are recently found to trigger autophagy which supports the survival of cancer cells under therapeutic stress and leads to low curative efficacy or chemotherapy resistance [27, 28]. We therefore examined whether chaetocin induced autophagy in gastric cancer cells. Treatment of AGS and HGC-27 cells with chaetocin for 24 h resulted in a dose-dependent upregulation on the expression of microtubule-associated protein 1 light chain 3B (LC3B)-II (Fig. 5A), a marker for the presence of completed autophagosomes [29]. Induction of autophagic flux was further proven by the dose-dependent reduction of p62 protein levels, an LC3-interacting protein degraded in the autophagolysosomes [30, 31], in chaetocin-treated AGS and HGC-27 cells (Fig. 5A). Additionally, we assessed the production of acidic vesicular organelles (AVOs)/autophagolysosomes with acridine orange staining, in which AVOs/autophagosomes fluoresce red while cytoplasm and nuclei fluoresce green [32]. As showed in Fig. 5B, red vehicles appeared only in chaetocin-treated AGS and HGC-27 cells. We also test whether chaetocin was able to induce LC3 puncta, a widely-used indicator of autophagosome formation. AGS and HGC-27 cell sublines stably expressing GFP-LC3 were constructed and used. We observed very few GFP-LC3 puncta following vehicle treatment, while the GFP-LC3 punctate dots greatly increased under chaetocin treatment (Fig. 5C). These results provided strong evidence that chaetocin could also induce autophagy in gastric cancer cells.
Suppression of autophagy enhanced cytotoxicity of chaetocin in gastric cancer cells. (A) Effect of chaetocin on the expression of autophagy markers was analyzed by western blotting. Cells were treated with different concentrations of chaetocin for 24 h. (B-C) Chaetocin-induced autophagy was examined by acridine orange staining (B) and GFP-LC3 puncta (C) after treatment at the concentration of IC50 for 24 h. (D-E) Chloroquine (CQ) strengthened chaetocin's effects on cell death and the cleavage of caspase-3 and PARP. AGS and HGC-27 cells were pretreated with CQ (10 μM) for 2 h before chaetocin treatment at the concentration of IC50 for 24 h, and then subjected to western blotting (D) or MTT assay (E). Data were presented as the mean ± SEM. *P < 0.05, ***P < 0.001, ns nonsignificant.
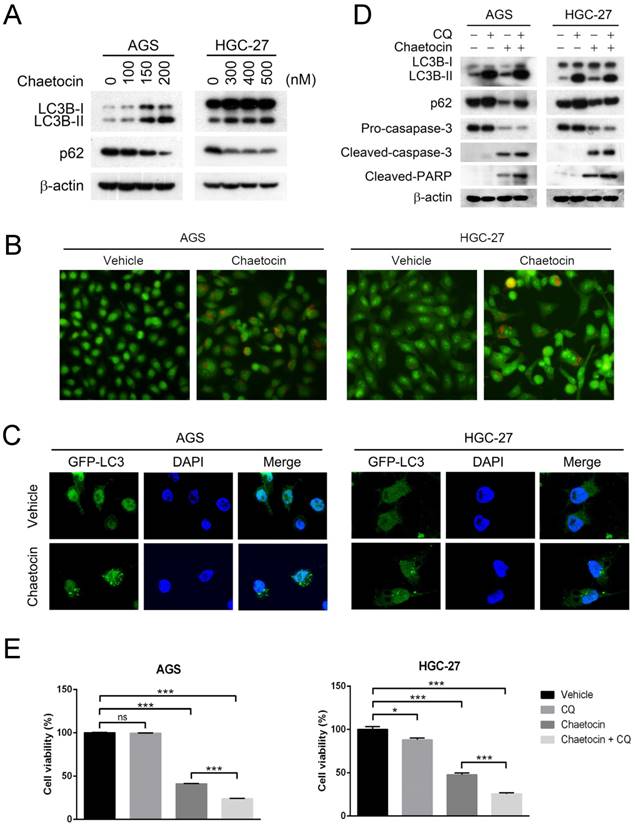
To determine whether suppression of autophagy improved chaetocin sensitivity in gastric cancer, we examined the effect of chloroquine (CQ), a late-stage inhibitor of autophagy, on chaetocin-induced cell death. As expected, the protein levels of LC3B-II and p62 were dramatically increased by CQ treatment due to its inhibition on autophagic flux (Fig. 5D). Moreover, CQ significantly enhanced chaetocin's effects on cell death and the cleavage of caspase-3 and PARP in both AGS and HGC-27 cells (Fig. 5D-E). Therefore, chaetocin-induced cell death could be strengthened by blocking autophagy in gastric cancer cells.
3.6. Chaetocin suppressed growth of HGC-27 xenografts in nude mice
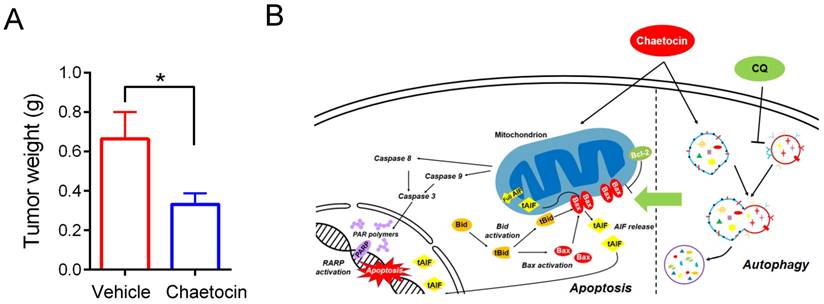
Tumor xenograft assays were further carried out to validate the anti-tumor effects of chaetocin in vivo. 8 × 106 HGC-27 cells were injected subcutaneously into each of the BALB/C nude mice. One week later, tumor size was measured and tumor volume was calculated. As the growth of HGC-27 xenografts varies remarkably in each nude mouse, paired mice with equal tumor volume were divided into the chaetocin-treated group and the vehicle-treated group (n = 6 for each group), and then injected intraperitoneally (i.p.) daily with chaetocin (0.5 mg/kg) or vehicle for 10 days. Mice were then sacrificed and the tumors were weighted. As showed in Fig. 6A, tumors from the chaetocin-treated group are significantly smaller than that from the vehicle-treated group (unpaired t-test, n = 6), confirming the tumor inhibitory role of chaetocin on gastric cancer xenograft in nude mice.
Chaetocin inhibited growth of HGC-27 xenografts in nude mice. (A) Weight of tumor samples from the nude mice treated with vehicle or chaetocin. HGC-27 cells were injected subcutaneously into the right armpit of nude mice. One week later, paired mice with equal tumor volume were separately arranged into the chaetocin group (n = 6) and the vehicle group (n = 6), and then injected intraperitoneally (i.p.) daily with chaetocin (0.5 mg/kg) or vehicle for 10 days. Data were presented as the mean ± SEM. *P < 0.05 vs control (unpaired t-test). (B) Proposed working model of chaetocin in human gastric cancer cells.
4. Discussion
Chaetocin, a fungal metabolite, was recently found to exhibit anti-tumor functions in several types of solid tumors [7-10]. However, it remains unknown whether chaetocin could also be a potential therapeutic agent for gastric cancer. In the present study, we found that chaetocin induced caspase-dependent and -independent apoptosis in human gastric cancer cell lines (Fig. 1). Caspase-independent apoptosis seemed to play a leading role as knocking down AIF greatly attenuated cell death induced by chaetocin (Fig. 2). BID-mediated AIF translocation to the nucleus was further found to be critical for chaetocin to induce cell death (Fig. 3). Although chaetocin did not raise ROS levels in gastric cancer cells, it did cause a decrease in mitochondrial membrane potential, which might be attributed to the changed expression of Bcl-2 and BAX controlling mitochondrial membrane permeability (Fig. 4). Chaetocin was further found to suppress the growth of xenograft tumors of gastric cancer HGC-27 cells in nude mice (Fig. 6A). This study demonstrated for the first time the cytotoxicity and the underlying mechanisms of chaetocin in gastric cancer cells (Fig. 6B), which were quite different from the mechanisms of chaetocin's action in other types of solid tumors [7-10]. Therefore, chaetocin may employs distinct mechanisms to induce cell death in different kinds of cancers.
Increased ROS production, followed by the opened MPTP and the decreased mitochondrial transmembrane potential, represents a major anti-cancer strategy [20-23]. However, accumulating reports revealed that chronic and abnormally high ROS levels may bring about some unwanted side-effects, such as drug resistance [33, 34]. Due to tumor heterogeneity, only some sub-populations of cancer cells benefit from ROS-based therapies, while the oxidative stress may cause genetic instability and mutations within other cancer cells or the adjacent normal cells, which eventually turn into drug-resistant cells [18, 35]. Therefore, antitumor agents that kill cells without elevating intercellular ROS levels stand a good chance to avoid this problem. Chaetocin, as demonstrated in the present study in the treatment of gastric cancer, seems to be the first antitumor agent fitting this description. Although some agents, such as plumbagin, were found to inhibit cell proliferation independent of ROS production, however, they instigated dramatic increase in ROS production [36]. Nevertheless, chaetocin could still increase ROS levels in cancer cells from other tissues, including cervical carcinoma HeLa cells (Fig. 4A & 4C) and melanoma cells [8]. Further investigation on the mechanism why chaetocin could not stimulate ROS production in gastric cancer may help to design and develop a new type of antitumor agents to avoid or postpone drug resistance.
Some cancer therapeutic agents were found to give rise to autophagy which leads to low curative efficacy or chemotherapy resistance, such as cisplatin and 5-FU [27, 28]. Inhibition of autophagy, in combination with anti-tumor agents, may be an effective therapeutic strategy for cancer treatment. As chaetocin was recently reported to initiate autophagy in hepatoma cells [7], we therefore investigated and found that chaetocin could also arouse autophagy in gastric cancer cells and chaetocin-induced cell death was enhanced by autophagy inhibitor (Fig. 5), providing evidence for the combined use of chaetocin with autophagy inhibitors to enhance the therapeutic effect for gastric cancer.
In conclusion, our study provided a promising therapeutic agent—chaetocin—for gastric cancer, which did not elevate the intercellular ROS production and thus potentially avoid or postpone drug resistance; additionally, the combined use of chaetocin with autophagy inhibitors could enhance the therapeutic effect for gastric cancer.
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (81572589 and 31770860) and the Natural Science Foundation of Fujian grant (2017J06020, 2017R1036-4 and 2018R1036-5).
Abbreviations
Reactive oxygen species (ROS); apoptosis-inducing factor (AIF); BH3 interacting-domain death agonist (BID); Bcl-2-associated X protein (BAX); Dimethyl sulfoxide (DMSO); 3-[4,5-dimethylthiazol-2-yl]-2,3-diphenyltetrazodium bromide (MTT); optical density (OD); propidium iodide (PI); half maximal inhibitory concentration (IC50).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferro A, Peleteiro B, Malvezzi M. et al. Worldwide trends in gastric cancer mortality (1980-2011), with predictions to 2015, and incidence by subtype. Eur J Cancer. 2014;50:1330-44
2. Macdonald JS, Smalley SR, Benedetti J. et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N Engl J Med. 2001;345:725-30
3. De Angelis R, Sant M, Coleman MP. et al. Cancer survival in Europe 1999-2007 by country and age: results of EUROCARE-5-a population-based study. Lancet Oncol. 2014;15:23-34
4. Sekita S, Yoshihira K, Natori S. et al. Mycotoxin production by Chaetomium spp. and related fungi. Can J Microbiol. 1981;27:766-72
5. Hauser D, Weber HP & Sigg HP. Isolation and configuration of Chaetocin. Helv Chim Acta. 1970;53:1061-73
6. Cherblanc F, Chapman-Rothe N, Brown R. et al. Current limitations and future opportunities for epigenetic therapies. Future Med Chem. 2012;4:425-46
7. Jung HJ, Seo I, Casciello F. et al. The anticancer effect of chaetocin is enhanced by inhibition of autophagy. Cell Death Dis. 2016;7:e2098
8. Han X, Han Y, Zheng Y. et al. Chaetocin induces apoptosis in human melanoma cells through the generation of reactive oxygen species and the intrinsic mitochondrial pathway, and exerts its anti-tumor activity in vivo. PLoS One. 2017;12:e0175950
9. Liu X, Guo S, Liu X. et al. Chaetocin induces endoplasmic reticulum stress response and leads to death receptor 5-dependent apoptosis in human non-small cell lung cancer cells. Apoptosis. 2015;20:1499-507
10. Isham CR, Tibodeau JD, Bossou AR. et al. The anticancer effects of chaetocin are independent of programmed cell death and hypoxia, and are associated with inhibition of endothelial cell proliferation. Br J Cancer. 2012;106:314-23
11. Susin SA, Lorenzo HK, Zamzami N. et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441-6
12. Bano D & Prehn JHM. Apoptosis-Inducing Factor (AIF) in Physiology and Disease: The Tale of a Repented Natural Born Killer. EBioMedicine. 2018;30:29-37
13. Hangen E, Blomgren K, Benit P. et al. Life with or without AIF. Trends Biochem Sci. 2010;35:278-87
14. Sevrioukova IF. Apoptosis-inducing factor: structure, function, and redox regulation. Antioxid Redox Signal. 2011;14:2545-79
15. Lewis EM, Wilkinson AS, Jackson JS. et al. The enzymatic activity of apoptosis-inducing factor supports energy metabolism benefiting the growth and invasiveness of advanced prostate cancer cells. The Journal of biological chemistry. 2012;287:43862-75
16. Shen SM, Guo M, Xiong Z. et al. AIF inhibits tumor metastasis by protecting PTEN from oxidation. EMBO Rep. 2015;16:1563-80
17. Farrand L, Kim JY, Byun S. et al. The diarylheptanoid hirsutenone sensitizes chemoresistant ovarian cancer cells to cisplatin via modulation of apoptosis-inducing factor and X-linked inhibitor of apoptosis. The Journal of biological chemistry. 2014;289:1723-31
18. Okon IS. & Zou MH. Mitochondrial ROS and cancer drug resistance: Implications for therapy. Pharmacol Res. 2015;100:170-4
19. Cabon L, Galan-Malo P, Bouharrour A. et al. BID regulates AIF-mediated caspase-independent necroptosis by promoting BAX activation. Cell Death Differ. 2012;19:245-56
20. Zhou H, Xu M, Gao Y. et al. Matrine induces caspase-independent program cell death in hepatocellular carcinoma through bid-mediated nuclear translocation of apoptosis inducing factor. Mol Cancer. 2014;13:59
21. Wang D, Qu X, Zhuang X. et al. Seed Oil of Brucea javanicaInduces Cell Cycle Arrest and Apoptosis via Reactive Oxygen Species-Mediated Mitochondrial Dysfunction in Human Lung Cancer Cells. Nutr Cancer. 2016;68:1394-1403
22. Xu B, Xu M, Tian Y. et al. Matrine induces RIP3-dependent necroptosis in cholangiocarcinoma cells. Cell Death Discov. 2017;3:16096
23. Dasari S & Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364-78
24. Smiley ST, Reers M, Mottola-Hartshorn C. et al. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci U S A. 1991;88:3671-5
25. Youle RJ & Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47-59
26. Breckenridge DG & Xue D. Regulation of mitochondrial membrane permeabilization by BCL-2 family proteins and caspases. Curr Opin Cell Biol. 2004;16:647-52
27. Harhaji-Trajkovic L, Vilimanovich U, Kravic-Stevovic T. et al. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J Cell Mol Med. 2009;13:3644-54
28. Zhang H, Tang J, Li C. et al. MiR-22 regulates 5-FU sensitivity by inhibiting autophagy and promoting apoptosis in colorectal cancer cells. Cancer Lett. 2015;356:781-90
29. Kondo Y, Kanzawa T, Sawaya R. et al. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726-34
30. Bjorkoy G, Lamark T & Johansen T. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy. 2006;2:138-9
31. Mizushima N, Yoshimori T & Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313-26
32. Paglin S, Hollister T, Delohery T. et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439-44
33. Chen G, Wang F, Trachootham D. et al. () Preferential killing of cancer cells with mitochondrial dysfunction by natural compounds. Mitochondrion. 2010;10:614-25
34. Okon IS, Coughlan KA, Zhang M. et al. Gefitinib-mediated reactive oxygen specie (ROS) instigates mitochondrial dysfunction and drug resistance in lung cancer cells. J Biol Chem. 2015;290:9101-10
35. Sabharwal SS & Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat Rev Cancer. 2014;14:709-21
36. Checker R, Sharma D, Sandur SK. et al. Plumbagin inhibits proliferative and inflammatory responses of T cells independent of ROS generation but by modulating intracellular thiols. J Cell Biochem. 2010;110:1082-93
Author contact
![]() Corresponding author: Y.-Y.Z. (Email: yyzhanedu.cn, Tel: +86-592-2188223)
Corresponding author: Y.-Y.Z. (Email: yyzhanedu.cn, Tel: +86-592-2188223)