Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(16):3735-3745. doi:10.7150/jca.32022 This issue Cite
Research Paper
BDH2 is downregulated in hepatocellular carcinoma and acts as a tumor suppressor regulating cell apoptosis and autophagy
Hao Liang1,2#, Zhiyong Xiong2#, Ruixi Li3, Kunpeng Hu2, Mingbo Cao1, Jiarui Yang1, Zhaozhong Zhong1, Changchang Jia4, Zhicheng Yao2 ![]() , Meihai Deng1
, Meihai Deng1 ![]()
1. Department of Hepatobiliary Surgery, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou, 510000, China
2. Department of General Surgery, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou, 510000, China
3. Department of Hepatobiliary Surgery, The Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, 518000, China
4. Department of Cell-gene Therapy Translational Medicine Research Center, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou, 510000, China
#These authors contributed equally to this work
Received 2018-12-5; Accepted 2019-5-5; Published 2019-6-9
Abstract
BDH2 is a short-chain dehydrogenase/reductase family member involved in several biological and pathological processes, including the utilization of cytosolic ketone bodies, immunocyte regulation and tumor progression. In this study, we first revealed that BDH2 was downregulated in HCC tissues by qRT-PCR and immunohistochemistry analysis and that low BHD2 expression was significantly associated with poor overall survival, poor tumor differentiation, increased tumor size, venous invasion and an advanced BCLC stage. Moreover, the results of a univariate analysis and multivariate analysis revealed that BDH2 may be regarded as an independent prognostic marker. As a member of a gene family involved in ketone metabolism, BDH2 upregulated the level of β-HB in liver cells as well as the level of H3 histone acetylation. Functional analysis showed that BDH2 expression inhibited tumor cell growth, proliferation and migration. The results of the mechanistic analysis revealed that BDH2 induced mitochondrial apoptosis and inhibited autophagy through the unfolded protein response. Therefore, BDH2 may be a new HCC prognostic marker and a useful treatment target.
Keywords: BDH2, hepatocellular carcinoma, tumor suppressor, apoptosis, autophagy
Introduction
Hepatocellular carcinoma (HCC) is the sixth most common malignancy worldwide and is the fourth most common cause of cancer-related death. Approximately 841,080 new cases were diagnosed as HCC, and 781,631 people die from HCC around the world annually [1]. Although there have been advances in the diagnosis and various ways to treat HCC clinically, the incidence of HCC has increased in recent years. Although surgical resection is the first choice for HCC patients [2], the recurrence of HCC is still found in a high proportion of cases in clinical practice [3, 4]. Moreover, most patients are often diagnosed at a quite advanced stage that can only be treated with palliative therapy; only 30-40% of patients who are diagnosed early may receive curative therapy, which accounts for the overall poor prognosis of HCC [5-8]. HCC is a tumor with high invasion and aggression and its molecular pathogenesis is complicated. Previous studies have proposed many molecular markers with high sensitivity and specificity, but none were justifiable for routine use in clinical practice until now [7, 9]. Therefore, novel molecular markers and pathways in the assessment of HCC carcinogenesis and prognosis are urgently needed.
BDH2 (3-hydroxybutyrate dehydrogenase 2) is a short-chain dehydrogenase/reductase family member whose original name was DHRS6 [10]. BDH2 is a novel cytosolic-type 2-hydroxybutyrate dehydrogenase that plays a physiological role in the utilization of cytosolic ketone bodies that can subsequently enter mitochondria and participate in the tricarboxylic acid cycle [10]. BDH2 can also catalyze the synthesis of 2,5-dihydroxybenzoic acid (2,5DHBA) during the biosynthesis of enterobactin, and 2,5DHBA was identified as an iron-binding molecule in mammalian cells [10, 11]. Additionally, the expression of BDH2 in macrophages was affected upon bacterial infection, endoplasmic reticulum stress and inflammation, suggesting that BDH2 may participate in iron-limiting innate immunity [12, 13]. Moreover, BDH2 may be a novel independent poor prognostic marker in acute myeloid leukemia through its influence on cell apoptosis [14], and BDH2 was regulated by long noncoding RNA TP73-AS1, which affects cell proliferation and apoptosis in esophageal squamous cell carcinoma [15]. Overall, BDH2 may be a multifunctional gene in mammalian cells.
Recently, Huang et al. reported that HCC might redirect the mechanism of ketolysis under nutritional deprivation stress, resulting in the use of ketone bodies as a fuel source for cancer progression through the upregulation of 3-oxoacid CoA-transferase 1 (a rate-limiting ketolytic enzyme) triggered by mTORC2-AKT-SP1 signaling, which was invalid in normal adult liver cells[16]. However, the biological function of BDH2, an enzyme in the utilization of cytosolic ketone bodies, in HCC is undefined. There are no relevant studies about BDH2 expression in HCC. In the current study, we first analyzed BDH2 expression in HCC tissues and its clinical relevance, association with overall patient survival and potential biological function in HCC cells. Our results suggest that BDH2 might be a novel prognostic biomarker and treatment target for HCC.
Materials and Methods
Patients and tissue samples
Our study previously collected HCC tissues with adjacent normal tissues from our hospital (the Third Affiliated Hospital of Sun Yat-sen University) with informed consent of all included patients. The study was conducted according to ethical and legal standards and received approval from the Research Ethics Committee of Sun Yat-sen University, Guangzhou, China. A total of 229 patient tissues were analyzed in this study. Among the tissue samples, 184 tissues had associated patient information and patient clinical characteristics; however, the other 45 tissues did not. Two independent retrospective HCC patient cohorts, called the training cohort and validation cohort, were used to investigate the role of BDH2 in this study. In the training cohort, 75 HCC patients whose tissue was embedded in paraffin were recruited. Simultaneously, in order to validate the findings in the training cohort, we recruited tissue microarray (TMA) containing another 109 HCC patients' tissues to build the validation cohort. The enrollment criteria for the patients in the present study were as follows: patients had histologically diagnosed as hepatocellular carcinoma; patients had no history of previous anticancer treatment before surgery; patients had no lymph node or distant metastasis; patients had no other diagnosed tumors; patients underwent adequate clinical follow-up time (≥6 months); and patients provided representative tissue samples for immunohistochemical analysis. The patient characteristics of age, gender, tumor differentiation, tumor size, nodal metastasis, venous invasion, Barcelona Clinic Liver Cancer (BCLC) stage and AFP level were collected through the patient medical records. The clinicopathological parameters of the included samples are briefly summarized in Table 1. The patients were followed up every 3 months for the first year after the surgery, then every 6 months thereafter in the next few years. Blood tests for AFP and liver function, abdominal ultrasonography and magnetic resonance imaging (MRI) were adopted to monitor the patient's condition. The median follow-up time was 36 months (range, 6-88 months).
Association between BDH2 expression and clinicopathological parameters in two cohorts of patients with HCC.
| Variable | Training cohort (n=75) | p value | Validation cohort (n=109) | p value | ||
|---|---|---|---|---|---|---|
| Low(n=44) | High(n=31) | Low(n=70) | High(n=39) | |||
| Age | 0.265 | 0.483 | ||||
| <60 | 27 | 15 | 39 | 19 | ||
| ≥60 | 17 | 16 | 31 | 20 | ||
| Gender | 0.907 | 0.533 | ||||
| Male | 35 | 25 | 42 | 21 | ||
| Female | 9 | 6 | 28 | 18 | ||
| Differentiation | 0.033 | 0.031 | ||||
| Well-moderate | 16 | 19 | 28 | 24 | ||
| Poor | 28 | 12 | 42 | 15 | ||
| Tumor size | <0.001 | 0.019 | ||||
| ≤5 cm | 14 | 24 | 25 | 23 | ||
| >5 cm | 30 | 7 | 45 | 16 | ||
| Nodal metastasis | 0.082 | 0.721 | ||||
| Positive | 18 | 19 | 33 | 17 | ||
| Negative | 26 | 12 | 37 | 22 | ||
| Venous invasion | 0.002 | 0.022 | ||||
| Positive | 30 | 10 | 48 | 18 | ||
| Negative | 14 | 21 | 22 | 21 | ||
| BCLC stage | 0.037 | 0.034 | ||||
| 0 | 6 | 0 | 7 | 5 | ||
| A | 12 | 17 | 18 | 20 | ||
| B | 21 | 11 | 34 | 11 | ||
| C | 5 | 3 | 11 | 3 | ||
| AFP level | 0.082 | 0.103 | ||||
| <400 | 18 | 19 | 23 | 19 | ||
| >400 | 26 | 12 | 47 | 20 | ||
Notes: P < 0.05 by χ2 test. P < 0.05 was considered statistically significant. Abbreviations: BCLC, Barcelona Clinic Liver Cancer; AFP, alpha-fetoprotein.
Cell lines and culture
In this study, BDH2 expression was detected in 5 HCC cell lines (HepG2, Hep3B, MHCC-97L, MHCC-97H) and 1 human hepatocyte cell line (L02) as a control. All cell lines were purchased from the Shanghai Cell Bank (Chinese Academy of Science) and were cultured in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA, USA). The cells were put in a humidified 5% CO2 incubator at 37°C. DMEM containing 25 mM glucose, 4 mM L-glutamine and 1 mM pyruvate (Gibco, 12800), and lithium acetoacetate (Sigma) was used to culture the L02, HepG2 and Hep3B cells to test the β-hydroxybutyrate (β-HB) and acetoacetate (AcAc) levels.
β-HB and AcAc measurements
Ketone body (acetoacetate and β-hydroxybutyrate) levels were measured by colorimetric assay kits (BioVision Inc., Milpitas, CA, and Cayman Chemicals, Ann Arbor, MI). All measurements followed the manufacturer's instructions.
Quantitative reverse transcription-PCR (qRT-PCR)
Total RNA from 45 pairs of HCC specimens and HCC cell lines was extracted by the TRIzol solution (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer's instructions. The concentrations of total RNA were determined by a NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Inc.), and each complementary DNA (cDNA) sequence was synthesized by 2 μg total RNA with a reverse transcriptase kit (Invitrogen, Carlsbad, CA, USA). The Power SYBR Green qPCR SuperMix-UDG (Invitrogen, Grand Island, NY, USA) was used for the qRT-PCR analyses on a Light Cycler 480 SYBR Green I Master (Roche, Indianapolis, IN, USA). GAPDH was used as an internal control. The BDH2 primers used for RT-qPCR were as follows: BDH2 forward, 5'-TCCAGCGTCAAAGGAGTTGT-3' and reverse, 5'-CTGATGCCCTGCTGGATGAA-3'. The relative expression of BDH2 was determined using the 2-ΔΔCt method.
Immunohistochemistry (IHC)
A total of 75 HCC tissues that were embedded in paraffin were cut into 4-µm thick sections and 109 microarray slides used to determine BDH2 expression by immunohistochemical (IHC) staining. The paraffin sections were first heated in a thermotank at 55°C for an hour and the TAM slides were heated for three hours. Then, the tissue sections were deparaffinized in dimethylbenzene and rehydrated in 100%, 95%, 75% ethanol solutions and distilled water. The slides were boiled in a pressure cooker filled with citrate‑hydrochloric acid (pH 6.0) for 25 minutes for antigen retrieval. Then, the tissue sections were submerged in 3% hydrogen peroxide at room temperature for 15-20 minutes to block endogenous peroxidase activity. After being washed with PBS, the slides were blocked with 5% sheep serum albumin (cat. No. ab7481; Abcam) for 20 minutes. Subsequently, the tissue sections were incubated with rabbit anti-BDH2 (cat. no. PA5-57501; dilution 1:400; Thermo Fisher Scientific) overnight at 4°C in a thermostatic chamber. The next day, the tissue sections were washed with PBS and then incubated with a secondary antibody for one hour at 37°C. Then, the tissue sections were stained with 3,3-diaminobenzidine (DAB) for 1 minute and counterstained with hematoxylin for 1 minute at room temperature. Finally, the tissue sections were dehydrated and mounted. The primary antibody was replaced by normal rabbit serum as a negative control.
Two independent pathologists without knowledge of the clinical status of the patients evaluated each tissue section individually. BDH2 expression was evaluated according to the extent of staining score based on the percentage of positive cells and the staining intensity score (0, no staining; 1, weak staining; 2, moderate staining; 3, strong staining). The final immunostaining score was calculated by the extent of staining score multiplied by the intensity score.
Construction and transfection of overexpressed-BDH2 cell lines
The BDH2 gene (NM_020139.3) was cloned into the pReceiver-M03 vector backbone for fusion with the GFP reporter gene, which was obtained from Genecopoeia (USA, EX-A4307-M03). The vector also contained the neomycin resistance gene. The HCC cell lines HepG2 and Hep3B were transfected with the BDH2 plasmid (EX-A4307-M03, Genecopoeia, USA) or control vector (EX-NEG-M03, Genecopoeia, USA) by using Lipofectamine 2000 (cat. no. 11668019, Thermo Fisher Scientific). Two clones of the stably transfected cell lines were used for further experiments.
Colony formation assay
One-thousand transfected cells were seeded in 6-well plates containing 10% FBS in DMEM for colony formation assays. Two weeks later, the cell colonies were immobilized with 70% ethanol for 15 min, and 0.1% crystal violet was used to stain the colonies for 10 min. The number of colonies was counted from three different experiments.
Cell viability assay
The cell viability assay was conducted with a CCK8 cell counting kit (Dojindo, Kumamoto, Japan). The cells were seeded in 96-well plates containing 10% FBS in DMEM at a density of 2500 cells/well. At the indicated time point, the cells of each well were incubated with 10 μL CCK8 reagent for 1 hour at 37°C, and the absorbance was measured at 450 nm.
Cell migration assay
Cell migration and invasion were evaluated with a transwell assay (Cambridge, MA, USA) according to the standard procedure. The treated cells were trypsinized and resuspended in serum-free DMEM. Then, 1 x 105 cells were seeded into the upper chamber of transwells containing serum-free DMEM. The lower transwell chambers were filled with 10% FBS in DMEM. The cells in the upper chamber were carefully removed by a cotton swab after the indicated hours of incubation. The other cells that migrated across the membrane and grew on the underside of membrane were fixed with 20% methanol and stained with 0.1% crystal violet (Bogoo, Shanghai, China). The stained cells from five different fields were photographed under an inverted microscope, and the cell numbers were calculated.
Cell apoptosis analysis
Cell apoptosis was detected with Annexin V-FITC/PI double staining assays according to the manufacturer's instructions (Apoptosis Detection kit; KeyGen, Nanjing, China). The percentage of apoptosis was analyzed by flow cytometry using a FACSCalibur machine (BD Biosciences, Baltimore, MD, USA).
Western blot
The proteins of the cells were extracted by using RIPA buffer (Pierce, Rockford, USA). The protein amounts were determined using a BCA Protein Assay Kit (Pierce). Equivalent amounts of protein (20 μg) were separated using 6-12% SDS-PAGE and transferred to PDVF membranes (Millipore, MA, USA). Afterwards, 5% nonfat milk was used to block the membranes for 1 hour and then incubated overnight at 4°C with the indicated primary antibodies. The next day, the membranes were washed with PBS and incubated with HRP-labeled secondary antibody (1:2000) at room temperature for 1 hour. The western blots were visualized and detected using chemiluminescence (ECL, Guangzhou, China).
Statistical analysis
The association between BDH2 expression and clinicopathological characteristics was analyzed by chi-square tests. The overall survival curve was analyzed by the Kaplan-Meier method and compared by the log-rank test. Univariate and multivariate Cox proportional hazards regression analyses were used to assess the prognostic variables in predicting overall survival. All experimental data were analyzed by t-tests (two-sided), and P < 0.05 was considered as a significant difference. All statistical analyses were completed with the SPSS 22.0 software package (SPSS, IBM, Chicago, IL, USA).
Results
BDH2 expression was downregulated in HCC and was associated with poor clinical outcome
We evaluated BDH2 expression in primary HCC and adjacent normal tissues by RT-qPCR in 45 HCC patient samples and IHC staining in 184 HCC patient samples. The BDH2 expression between tumor specimens and paired adjacent tissues from patients with HCC were obviously different in both RT-qPCR and IHC results (Fig 1A, Fig 1B, Fig 1C). The expression of BDH2 mRNA in HCC tissues was significantly lower than that in match noncancerous liver tissues. In IHC analysis, we also found that BDH2 expression was significantly lower in HCC tissues compared with that of noncancerous liver tissues in the same patient. Overall, the expression of BDH2 was downregulated in HCC tissues.
BDH2 expression is downregulated in HCC patients and is correlated with poor prognosis. A. The expression of BDH2 mRNA in HCC tissue and matched normal tissues (45 cases) was determined by qRT-PCR (HCC tissue vs. normal liver tissue, **P<0.01). GAPDH was used as an internal quantitative control. B. The immunohistochemical results of BDH2 expression in training cohort. (B1) and (B2) are representative immunohistochemical staining areas for low BDH2 expression in HCC tissues. (B3) and (B4) are representative immunohistochemical staining areas for high BDH2 expression in normal liver tissues. C. The immunohistochemical results of BDH2 expression in validation cohort. (C1) is representative immunohistochemical staining areas for low BDH2 expression in HCC tissues. (C2) is representative immunohistochemical staining areas for high BDH2 expression in normal liver tissues. D. The overall Kaplan-Meier plots according to BDH2 expression in patients with HCC, which revealed poorer survival in patients with low BDH2 expression than in patients with high BDH2 expression (n=75, P=0.001, log-rank test).
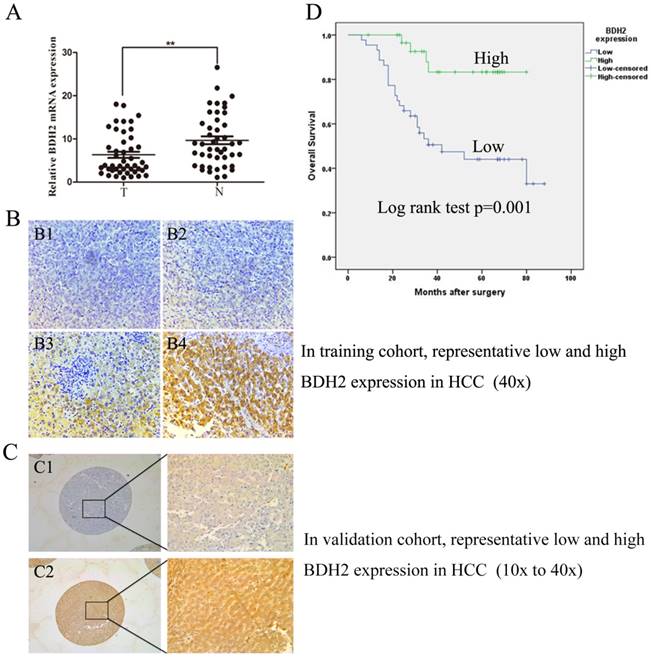
Subsequently, the clinicopathologic characteristics of the training cohort and the validation cohort of HCC biopsies were summarized in Table 1. In training cohort, low BHD2 expression was significantly related to poor tumor differentiation (P=0.033), tumor size (P<0.001), venous invasion (P=0.002) and BCLC stage (P=0.037). These findings were confirmed in the validation cohort of HCC patients (Table 1).
To further study whether reduced BDH2 staining in HCC patients correlates with a worse prognosis, Kaplan-Meier survival curves were constructed using overall cumulative survival to compare the patients with high BDH2 staining to those with low BDH2 staining. (n=75, follow-up time, 6-88 months). Our data revealed that the HCC patients with low BDH2 expression had shorter OS compared with HCC patients with high BDH2 expression (P<0.05, log-rank test) (Fig 1D).
Univariate Cox regression analysis showed that BDH2 expression (P=0.003), tumor differentiation (P=0.029), tumor size (P<0.001), venous invasion (P<0.001) and BCLC stage (P<0.001) were significantly related to OS in HCC patients (Table 2). Multivariate analysis was carried out for all variables with significance after the univariate analysis. The multivariate analysis indicated that BDH2 expression (HR: 3.358, P=0.044), tumor differentiation (HR: 2.340, P=0.049), venous invasion (HR: 4.727, P=0.017) and BCLC stage (HR: 4.061, P=0.013) were independent prognostic factors for OS (Table 2).
Cox regression analyses of prognostic factors for overall survival.
| Variable | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
| Hazard ratio (95% CI) | P value | Hazard ratio (95% CI) | P value | |
| Differentiation | 2.498(1.099-5.679) | 0.029 | 2.340(1.004-5.450) | 0.049 |
| Tumor size | 6.139(2.317-16.264) | 0.000 | 1.126(0.345-3.673) | 0.845 |
| Nodal metastasis | 0.752(0.351-1.610) | 0.463 | 1.052(0.441-2.512) | 0.908 |
| Venous invasion | 8.925(2.692-29.582) | 0.000 | 4.727(1.322-16.895) | 0.017 |
| BCLC stage | 5.833(2.201-15.462) | 0.000 | 4.061(1.351-12.211) | 0.013 |
| AFP level | 0.921(0.439-1.935) | 0.829 | 0.523(0.222-1.231) | 0.138 |
| BDH2 expression | 5.052(1.749-14.595) | 0.003 | 3.385(1.035-11.072) | 0.044 |
Note: P < 0.05 was considered statistically significant. Abbreviations: BCLC, Barcelona Clinic Liver Cancer; AFP, alpha-fetoprotein; CI, confidence interval.
BDH2 expression affects the ketone body concentration in the cell
Because BDH2 is one of the gene families involved in ketone metabolism, we performed the following experiments to verify whether BDH2 expression affected the ketone body level. We transfected a plasmid to construct BDH2-expressing cell lines (L02-BDH2, HepG2-BDH2, Hep3B-BDH2). Meanwhile, control vector plasmids were transfected to construct control cells (L02-Vector, HepG2-Vector, Hep3B-Vector). For serum starvation, the cells were washed twice with PBS and cultured in medium without FBS but with 10 mM acetoacetate for 48 h. Then, the β-HB assay kit and AcAc assay kit were used to measure the β-HB and AcAc levels. We found that BDH2 could catalyze AcAc to convert to β-HB in liver cells and hepatoma cells. The β-HB level in BDH2-expressing cells was higher than that in control cells (Fig 2A), but the AcAc level in BDH2-expressing cells was lower than that in control cells (Fig 2B). It is well-known that ketogenesis mainly occurs in the liver where acetyl-CoA is first converted to AcAc, which is further converted into β-HB by the catalytic activity of β-hydroxybutyrate dehydrogenase (BDH1). Both AcAc and β-HB are then transported to the bloodstream and reach extrahepatic tissues, such as muscle, kidney or brain [17]. For ketone body catabolism in extrahepatic tissues, β-HB is first oxidized back to AcAc, also by BDH1. Then, AcAc is converted into acetoacetyl-CoA (AcAc-CoA) by the catalysis of 3-oxoacid CoA-transferase 1 (OXCT1, also known as SCOT). Finally, AcAc-CoA is catalyzed into two acetyl-CoA complexes by acetyl-CoA acetyltransferase 1 (ACAT1) [16]. Previous studies have reported that BDH2 is also a hydroxybutyrate dehydrogenase that is adjacent to BDH1 and has a catalytic specificity that is 2-3-orders of magnitude lower toward β-HB, which may play a possible role in the “backup” or lower affinity system for utilizing hydroxybutyrate [10, 18, 19]. From the information above, we hypothesized a mechanism for BDH2 in ketone body metabolism that is described in Fig 2C.
BDH2 expression affected acetoacetate and β-hydroxybutyrate concentrations in L02 and liver cancer cells. A. The AcAc level was lower in BDH2-overexpressing cells than in control cells (*P<0.05). B. The β-HB level was higher in BDH2-overexpressing cells than in control cells (*P<0.05). C. The mechanism of BDH2 in ketone body metabolism. D. Determination of the expression of H3ac by western blot. H3ac was upregulated in BDH2-overexpressing cells. All the relative quantitative values are shown below the corresponding blots. All experiments were performed using vector-transfected cells as a control.
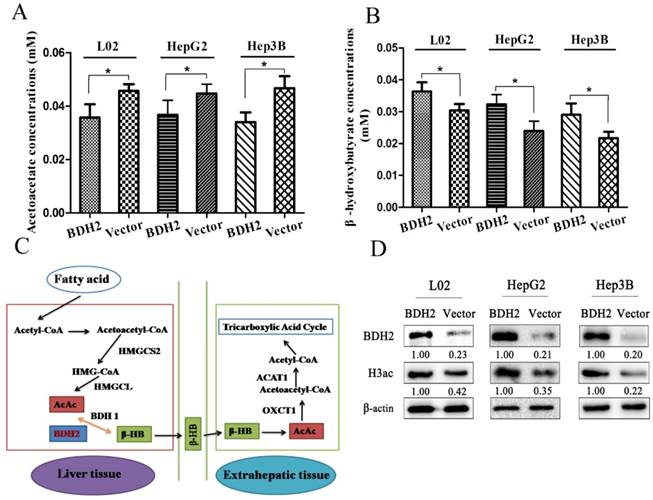
BDH2 expression affects H3 histone acetylation in the cell
Previous studies reported that β-HB inhibited histone deacetylase and increased histone acetylation [20, 21], so we determined whether the level of H3 histone acetylation changed in BDH2-overexpressing cells through western blot. The results showed that the level of H3 histone acetylation was upregulated in BDH2-overexpressing cells (Fig 2D).
BDH2 expression was downregulated in HCC cell lines
BDH2 expression was tested in six HCC cell lines. Real-time PCR was carried out to quantify BDH2 mRNA expression. The results showed that BDH2 expression was downregulated in HepG2, Hep3B, QGY-7703, HuH-7, MHCC-97L and MHCC-97H cell lines compared with that of the L02 cell line (a normal liver cell line) (Fig 3A).
BDH2 expression in different HCC cell lines and validation of BDH2 overexpression in transfected HCC cell lines. A. Lower expression levels of BDH2 mRNA were detected in HCC cell lines (HepG2, Hep3B, QGY-7703, HuH-7, MHCC-97L and MHCC-97H) than in normal L02 cells, according to the qRT-PCR assay. GAPDH was used as an internal quantitative control. B. BDH2 overexpression was successfully verified in transfected HCC cell lines (HepG2 and Hep3B) by qRT-PCR. C. BDH2 overexpression was successfully verified in transfected HCC cell lines (HepG2 and Hep3B) by western blot. All experiments were performed using vector-transfected cells as a control. Asterisks indicate statistically significant differences (*P<0.05, **P<0.01).
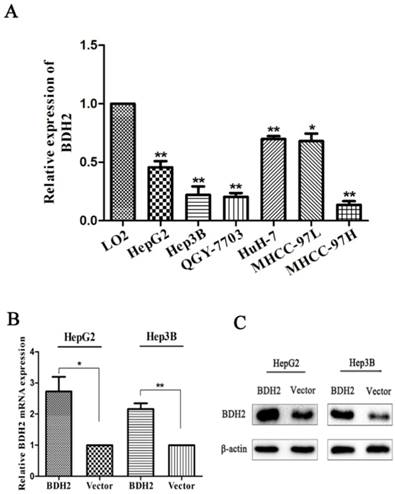
To investigate the biofunction of BHD2, we transfected a plasmid to construct BDH2-expressing cell lines (HepG2-BDH2, Hep3B-BDH2). Meanwhile, control vector plasmids were transfected to construct control cells (HepG2-Vector, Hep3B-Vector). Before conducting the experiments for the biological function of BDH2, the expression of BDH2 in these transfected cell lines was confirmed by qRT-PCR and western blot (Fig 3B, C).
BDH2 inhibited the growth and proliferation of HCC cells
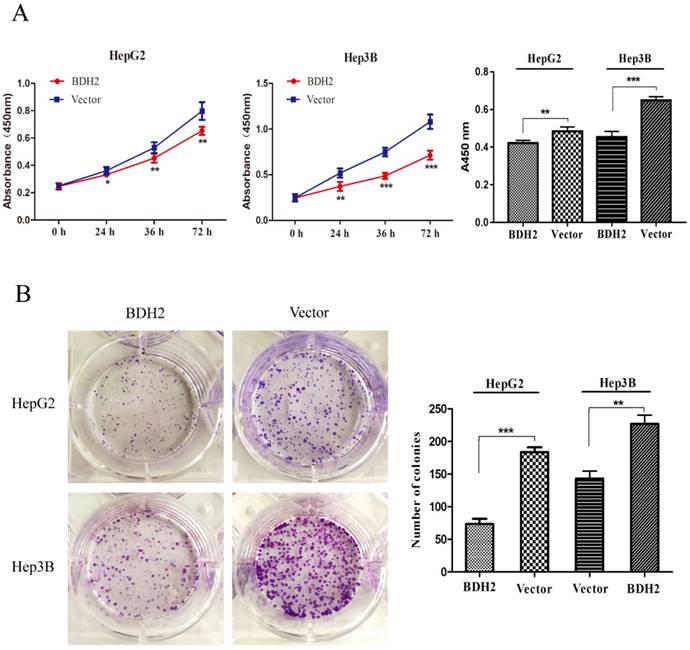
To explore the function of BDH2 on the growth of HCC cells, cell viability assays and colony formation assays were conducted with HepG2 and Hep3B HCC cells. Cell viability assays revealed that an overexpression of BDH2 inhibited the cell proliferation of HCC cells (Fig 4A). In the colony formation assays, we found that the colony formation efficiency was suppressed in HCC cells that overexpressed BDH2 (Fig 4B). The above observations suggest that BHD2 suppressed HCC cell growth.
Overexpression of BDH2 suppresses HCC cell viability and proliferation. A. The results of the cell viability assay showed that the overexpression of BDH2 suppressed the cell viability of HCC cells (HepG2 and Hep3B). B. A colony formation assay was performed to verify that upregulation of BDH2 inhibited the colony-forming ability of cells (*P<0.05, **P<0.01, ***P<0.001). All of the experiments were conducted using vector-transfected cells as a control.
BDH2 suppressed the migration of HCC cells
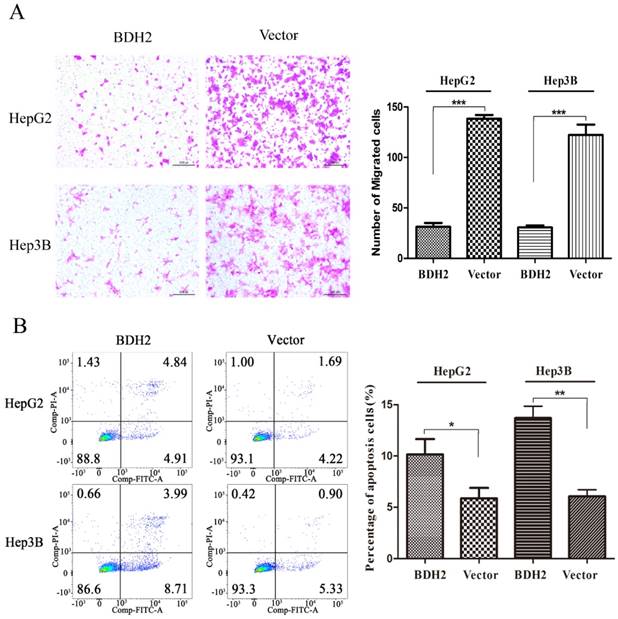
Transwell migration assays were performed to investigate the functions of BHD2 on the migration of HCC cells. The results revealed that the BDH2-transfected HCC cells had a weaker migration than the control cells (Fig 5A).
Overexpression of BDH2 suppresses HCC cell migration and induces HCC cell apoptosis. A. The results of transwell assays showed that the overexpression of BDH2 inhibited the migration of HCC cells (HepG2 and Hep3B). B. The results of annexin V-FITC/PI staining and quantitative analysis revealed that BDH2 overexpression induced apoptosis in HCC cells. Asterisks indicates statistically significant difference (*P<0.05, **P<0.01, ***P<0.001). All experiments were performed using vector-transfected cells as a control.
BDH2 promoted apoptosis of HCC cells
A cell apoptosis analysis was conducted to determine whether the inhibition of HCC cell growth and proliferation by BHD2 was relevant to apoptosis. In the cell apoptosis analysis, we found that the percentage of apoptotic cells was significantly higher in HCC cells transfected with BDH2 than in control cells (Fig 5B). These results confirmed that BDH2 induced apoptosis in HCC cells.
BDH2 promoted mitochondrial apoptosis
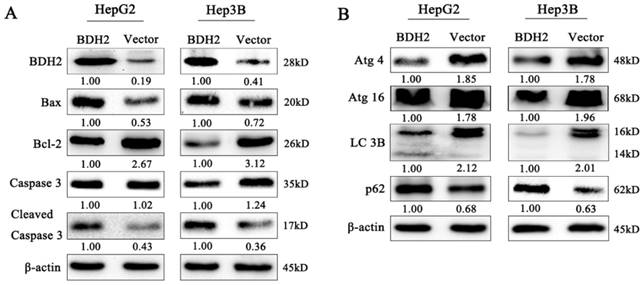
To further determine whether BDH2 affected the process of mitochondrial apoptosis, which is involved in HCC cell growth and migration, the expression of apoptosis-related proteins was detected by western blot analysis. Our results revealed that the proapoptotic protein Bax was upregulated and the expression of the antiapoptotic protein Bcl-2 was decreased in BDH2-transfected HCC cells (Fig 6A). Moreover, we also found that BDH2 induced HCC cell apoptosis via a caspase-3-independent pathway. Compared with that of negative control cells, the expression of cleaved caspase-3 was upregulated in the HCC cells transfected with BDH2 (Fig 6A). These results demonstrate that BDH2 expression regulates mitochondrial apoptosis in HCC cells via a caspase-3-independent pathway.
Effects of BDH2 on HCC cell apoptosis, autophagy and their relevant proteins. A. The expression of several key cell apoptosis proteins was examined by western blot. B. Determination of the expression of key autophagy regulators by western blot. All of the relative quantitative values are shown below the corresponding blots. All experiments were performed using vector-transfected cells as a control.
Effect of BDH2 on the autophagy of HCC cells
Autophagy plays critical physiological and pathological roles in the pathogenesis of various diseases, especially cancers [22]. We detected the expression of autophagy-related proteins by western blot analysis to determine the effect of BDH2 on the autophagy of HCC cells. The results revealed that BDH2 upregulation suppressed autophagy and inhibited the expression of LC3, Atg4 and Atg16 and increased p62 protein expression in HCC cells (Fig 6B). These results suggested that BDH2 expression contributed to autophagy inhibition in HCC cells.
Discussion
Although surgical and nonsurgical treatments for hepatocellular cancer have undergone tremendous development, the overall survival of patients with HCC is still poor because of disease metastasis and recurrence [23-25]. As the largest chemical factory in the body, the liver participates in different kinds of complicated biochemical reactions, and the pathogenesis of HCC becomes more intricate, which leads to more challenges in the treatment of HCC. Recently, Huang et al. reported that hepatocellular carcinoma could reset the process of ketolysis and make it possible for cancer cells to employ ketone bodies as an energy supply and progress under nutritional deprivation stress [16]. It is well-known that the normal liver cannot utilize ketones as an energy supply and thus the finding of a novel metabolic adaptation of HCC may contribute to an unprecedented direction for targeted therapy. Mitochondrial R-β -hydroxybutyrate dehydrogenase (BDH)2, which belongs to the short-chain dehydrogenase/reductase superfamily, has been extensively studied and constitutes a paradigm of lipid and ketolysis regulation of enzymatic function [18, 19, 26-31]. BDH2 (DHRS6) is one of the gene families involved in ketone metabolism [32, 33] as a “backup” or lower affinity system for utilizing hydroxybutyrate [10]. We proved that BDH2 may catalyze AcAc and convert it into β-HB in ketone metabolism, which upregulated the level of β-HB in liver cells. Moreover, previous studies reported that β-HB inhibited histone deacetylase and increased histone acetylation [20, 21], and our results showed that the level of H3 histone acetylation was upregulated in BDH2-overexpressing cells. Furthermore, previous studies proved that the inhibition of histone deacetylase could inhibit cancer progression and that histone deacetylase inhibitors acted as therapeutic strategies for cancer [34, 35]. Therefore, we hypothesize that BDH2 may upregulate the level of β-HB, which inhibits histone deacetylase and H3 histone acetylation in HCC cells. However, until now, there has been no study to report the association between BDH2 expression and HCC progression. Therefore, we performed the first study to demonstrate the mechanism of BDH2 in HCC.
In the present study, we first compared the expression of BDH2 mRNA and protein levels between HCC tissues and normal liver tissue using RT-PCR and immunohistochemistry. The results showed that the expression of BDH2 was significantly lower in HCC tissues than that in normal liver tissue. Then, we analyzed the association between BDH2 expression and the clinicopathological parameters of patients. We found that the pooled results demonstrated that low BDH2 expression was significantly correlated with poor prognosis in patients with HCC. To explore the underlying mechanisms of BDH2 expression regulation in HCC progression, we performed related experiments in HCC cell lines. The pooled outcomes indicated that BDH2 was downregulated in HCC cell lines and inhibited tumor cell growth, proliferation and migration. These results suggest that BDH2 may act as a tumor suppressor in HCC.
Previous studies reported that BDH2 could participate in the regulation of immune system cells, which may contribute to the DNA demethylation of CD4+ T cells in systemic lupus erythematosus and dysregulate intracellular iron in macrophages [13, 36]. In neoplasm studies, BDH2 may influence the progression of acute myeloid leukemia and esophageal squamous cell carcinoma through the regulation of cell apoptosis [14, 15]. However, the mechanisms of how BHD2 inhibited tumor cell growth, proliferation and migration in HCC were not verified. In the in vitro studies, we also found that BHD2 regulates cell apoptosis in HCC. Proteins in the Bcl-2 family, an intracellular protein group, play an important role in programmed cell death. The intrinsic or mitochondrial apoptotic cascade follows the Bcl-2 pathway [37, 38]. Our study revealed that BDH2 could downregulate the expression of Bcl-2 and cause an increase in the level of Bax and cleaved caspase-3, thereby inducing apoptosis in HCC cells. These results suggested that BDH2 inhibited HCC cell growth, proliferation and migration by inducing apoptosis through the Bcl-2 pathway.
Recently, relevant studies have paid considerable attention to autophagy in regards to tumor progression, particularly in cell apoptosis [39, 40]. As a conserved cellular process, autophagy plays a vital role in maintaining intercellular homeostasis by degrading the broken proteins and aging organelles [41-43]. Recent studies investigated the association between autophagy and apoptosis [44]. Autophagy could be regulated by apoptosis-regulating genes, such as Bcl-2 family members [45]. Furthermore, autophagy may be an upstream initiator for apoptosis [40]. Some autophagy-associated proteins were involved in cell apoptosis, such as Atg5, beclin 1 and Atg4D [45]. Autophagy may inhibit the progression of apoptosis through the degradation of the caspase family and Bcl-2 family proteins [46, 47]. To investigate whether BDH2 induced cell apoptosis through the regulation of the autophagy process, we detected the expression of autophagy-related proteins by western blot. The results showed that when BHD2 induced cell apoptosis and inhibited the expression of antiapoptosis protein Bcl-2, autophagy-related proteins (LC3B, Atg4, and Atg16) were downregulated and the p62 protein was upregulated. When cell apoptosis was inhibited by BDH2, autophagy-related proteins (LC3B, Atg4, and Atg16) were upregulated, and the p62 protein was downregulated. Therefore, BDH2 may inhibit HCC cell growth, proliferation and migration by inducing apoptosis, which is regulated by autophagy.
In conclusion, we first revealed that BDH2 was downregulated in HCC tissues and associated with poor prognosis in patients with HCC. BDH2 acted as a tumor suppressor regulating mitochondrial apoptosis and autophagy in HCC. The functional and mechanistic analyses of BHD2 suggested that BDH2 may be a prognostic marker and provided a more effective management strategy for patients with HCC.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81702375, 81572726); the Science and Technology Project of Guangdong Province, China (2017b020247057); the Science and Technology Project of Guangzhou City, China (201704020175); the Natural Science Foundation of Guangdong Province, China (2016A030313200, 2018A030313641, 2016A030313848); the Science and Technology Planning Project of Guangzhou city, China (201804010211); and the Medical Research Foundation of Guangdong Province, China (A2016312).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2018
2. Rahbari NN, Mehrabi A, Mollberg NM. et al. Hepatocellular carcinoma: current management and perspectives for the future. Annals of surgery. 2011;253:453-69
3. Yamamoto J, Kosuge T, Takayama T. et al. Recurrence of hepatocellular carcinoma after surgery. The British journal of surgery. 1996;83:1219-22
4. Chen XP, Qiu FZ, Wu ZD. et al. Long-term outcome of resection of large hepatocellular carcinoma. The British journal of surgery. 2006;93:600-6
5. Forner A, Llovet JM, Bruix J. Chemoembolization for intermediate HCC: is there proof of survival benefit? Journal of hepatology. 2012;56:984-6
6. Armengol C, Boix L, Bachs O. et al. p27(Kip1) is an independent predictor of recurrence after surgical resection in patients with small hepatocellular carcinoma. Journal of hepatology. 2003;38:591-7
7. Dhir M, Melin AA, Douaiher J. et al. A Review and Update of Treatment Options and Controversies in the Management of Hepatocellular Carcinoma. Annals of surgery. 2016;263:1112-25
8. Poon RT, Cheung TT, Kwok PC. et al. Hong Kong consensus recommendations on the management of hepatocellular carcinoma. Liver cancer. 2015;4:51-69
9. Bruix J, Reig M, Sherman M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology. 2016;150:835-53
10. Guo K, Lukacik P, Papagrigoriou E. et al. Characterization of human DHRS6, an orphan short chain dehydrogenase/reductase enzyme: a novel, cytosolic type 2 R-beta-hydroxybutyrate dehydrogenase. The Journal of biological chemistry. 2006;281:10291-7
11. Devireddy LR, Hart DO, Goetz DH. et al. A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell. 2010;141:1006-17
12. Zughaier SM, Kandler JL, Shafer WM. Neisseria gonorrhoeae modulates iron-limiting innate immune defenses in macrophages. PloS one. 2014;9:e87688
13. Zughaier SM, Stauffer BB, McCarty NA. Inflammation and ER stress downregulate BDH2 expression and dysregulate intracellular iron in macrophages. Journal of immunology research. 2014;2014:140728
14. Yang WC, Tsai WC, Lin PM. et al. Human BDH2, an anti-apoptosis factor, is a novel poor prognostic factor for de novo cytogenetically normal acute myeloid leukemia. Journal of biomedical science. 2013;20:58
15. Zang W, Wang T, Wang Y. et al. Knockdown of long non-coding RNA TP73-AS1 inhibits cell proliferation and induces apoptosis in esophageal squamous cell carcinoma. Oncotarget. 2016;7:19960-74
16. Huang Li T, Wang L Zhang L. et al. Hepatocellular carcinoma redirects to ketolysis for progression under nutrition deprivation stress. Cell research. 2016;26:1112-30
17. Newman JC, Verdin E. Ketone bodies as signaling metabolites. Trends in endocrinology and metabolism: TEM. 2014;25:42-52
18. Chelius D, Loeb-Hennard C, Fleischer S. et al. Phosphatidylcholine activation of human heart (R)-3-hydroxybutyrate dehydrogenase mutants lacking active center sulfhydryls: site-directed mutagenesis of a new recombinant fusion protein. Biochemistry. 2000;39:9687-97
19. Green D, Marks AR, Fleischer S. et al. Wild type and mutant human heart (R)-3-hydroxybutyrate dehydrogenase expressed in insect cells. Biochemistry. 1996;35:8158-65
20. Shimazu T, Hirschey MD, Newman J. et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science (New York, NY). 2013;339:211-4
21. Huang C, Wang P, Xu X. et al. The ketone body metabolite beta-hydroxybutyrate induces an antidepression-associated ramification of microglia via HDACs inhibition-triggered Akt-small RhoGTPase activation. Glia. 2018;66:256-78
22. Jiang P, Mizushima N. Autophagy and human diseases. Cell research. 2014;24:69-79
23. Bruix J, Llovet JM. Major achievements in hepatocellular carcinoma. Lancet (London, England). 2009;373:614-6
24. Iiizumi M, Liu W, Pai SK. et al. Drug development against metastasis-related genes and their pathways: a rationale for cancer therapy. Biochimica et biophysica acta. 2008;1786:87-104
25. Forner A, Reig ME, de Lope CR. et al. Current strategy for staging and treatment: the BCLC update and future prospects. Seminars in liver disease. 2010;30:61-74
26. Churchill P, Hempel J, Romovacek H. et al. Primary structure of rat liver D-beta-hydroxybutyrate dehydrogenase from cDNA and protein analyses: a short-chain alcohol dehydrogenase. Biochemistry. 1992;31:3793-9
27. Dalton LA, McIntyre JO, Fleischer S. Effect of selective thiol-group derivatization on enzyme kinetics of (R)-3-hydroxybutyrate dehydrogenase. The Biochemical journal. 1993;296( Pt 3):563-9
28. Marks AR, McIntyre JO, Duncan TM. et al. Molecular cloning and characterization of (R)-3-hydroxybutyrate dehydrogenase from human heart. The Journal of biological chemistry. 1992;267:15459-63
29. Jornvall H, Persson B, Krook M. et al. Short-chain dehydrogenases/reductases (SDR). Biochemistry. 1995;34:6003-13
30. Kallberg Y, Oppermann U, Jornvall H. et al. Short-chain dehydrogenase/reductase (SDR) relationships: a large family with eight clusters common to human, animal, and plant genomes. Protein science: a publication of the Protein Society. 2002;11:636-41
31. Oppermann U, Filling C, Hult M. et al. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chemico-biological interactions. 2003;143-144:247-53
32. Maurer GD, Brucker DP, Bahr O. et al. Differential utilization of ketone bodies by neurons and glioma cell lines: a rationale for ketogenic diet as experimental glioma therapy. BMC cancer. 2011;11:315
33. Pavlides S, Tsirigos A, Migneco G. et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell cycle (Georgetown, Tex). 2010;9:3485-505
34. Wawruszak A, Kalafut J, Okon E. et al. Histone Deacetylase Inhibitors and Phenotypical Transformation of Cancer Cells. Cancers. 2019:11
35. Eckschlager T, Plch J, Stiborova M. et al. Histone Deacetylase Inhibitors as Anticancer Drugs. International journal of molecular sciences. 2017:18
36. Zhao M, Li MY, Gao XF. et al. Downregulation of BDH2 modulates iron homeostasis and promotes DNA demethylation in CD4(+) T cells of systemic lupus erythematosus. Clinical immunology (Orlando, Fla). 2018;187:113-21
37. Zhan N, Xiong YY, Lan J. et al. [Relationship between Helicobacter pylori infection and expression of c-myc, Bcl-2, and Bax protein in different gastric mucosa lesions]. Ai zheng = Aizheng = Chinese journal of cancer. 2003;22:1034-7
38. Strasser A, Vaux DL. Viewing BCL2 and cell death control from an evolutionary perspective. Cell death and differentiation. 2018;25:13-20
39. Ravegnini G, Sammarini G, Nannini M. et al. Gastrointestinal stromal tumors (GIST): Facing cell death between autophagy and apoptosis. Autophagy. 2017;13:452-63
40. Hwang KE, Kim YS, Jung JW. et al. Inhibition of autophagy potentiates pemetrexed and simvastatin-induced apoptotic cell death in malignant mesothelioma and non-small cell lung cancer cells. Oncotarget. 2015;6:29482-96
41. Galluzzi L, Pietrocola F, Levine B. et al. Metabolic control of autophagy. Cell. 2014;159:1263-76
42. Mizushima N. Autophagy: process and function. Genes & development. 2007;21:2861-73
43. Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Current topics in microbiology and immunology. 2009;335:1-32
44. Oh BS, Shin EA, Jung JH. et al. Apoptotic Effect of Galbanic Acid via Activation of Caspases and Inhibition of Mcl-1 in H460 Non-Small Lung Carcinoma Cells. Phytotherapy research: PTR. 2015;29:844-9
45. Li YR, Li S, Ho CT. et al. Tangeretin derivative, 5-acetyloxy-6,7,8,4'-tetramethoxyflavone induces G2/M arrest, apoptosis and autophagy in human non-small cell lung cancer cells in vitro and in vivo. Cancer biology & therapy. 2016;17:48-64
46. Li J, Hou N, Faried A. et al. Inhibition of autophagy by 3-MA enhances the effect of 5-FU-induced apoptosis in colon cancer cells. Annals of surgical oncology. 2009;16:761-71
47. Abedin MJ, Wang D, McDonnell MA. et al. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell death and differentiation. 2007;14:500-10
Author contact
![]() Corresponding authors: Meihai Deng, Department of Hepatobiliary Surgery, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou 510000, China. E-mail: dengmeihsysu.edu.cn. Zhicheng Yao, Department of General Surgery, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou 510000, China. E-mail: yaozhch2sysu.edu.cn.
Corresponding authors: Meihai Deng, Department of Hepatobiliary Surgery, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou 510000, China. E-mail: dengmeihsysu.edu.cn. Zhicheng Yao, Department of General Surgery, The Third Affiliated Hospital, Sun Yat-sen University, Guangzhou 510000, China. E-mail: yaozhch2sysu.edu.cn.