Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(16):3778-3788. doi:10.7150/jca.30359 This issue Cite
Research Paper
Anti-tumor Drug THZ1 Suppresses TGFβ2-mediated EMT in Lens Epithelial Cells via Notch and TGFβ/Smad Signaling Pathway
Jie Ning1, Xinqi Ma1, Chongde Long1, Yuxiang Mao1, Xielan Kuang1,2, Zixin Huang1, Yuting Fan1, Han Zhang1, Qing Xia1, Renchun Wang3, Yu Liang4, Shuibin Lin4, Qingjiong Zhang1, Huangxuan Shen1,2 ![]()
1. State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, 54 Xianlie Road, Guangzhou 510060, China.
2. Biobank of Eye, State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, 54 Xianlie Road, Guangzhou 510060, China.
3. The Second Clinical Medicine School of Lanzhou University, No.199, West Donggang Road, Lanzhou, Gansu Province, 730000, China.
4. Center for Translational Medicine, the First Affiliated Hospital, Sun Yat-sen University, Guangzhou, 510080, China.
Received 2018-10-2; Accepted 2019-5-24; Published 2019-6-9
Abstract
Selective covalent CDK7 inhibitor THZ1 is a promising potential anti-tumor drug in many kinds of cancers. Epithelial-mesenchymal Transition (EMT) is highly related to cancer initiation, development, invasion and metastasis and other pathogenesis processes. We treated cancer cell line Hela229 and three retinoblastoma cell lines so-RB50, WERI-Rb-1, Y79 with gradient concentration of THZ1, and found that THZ1 could inhibit cell viability and EMT, suggesting that THZ1 may be a promising drug for human cervical cancer and retinoblastoma treatment. Our results verified the role of THZ1 in EMT for the first time, however, the mechanism needs further study. Here we report that THZ1 suppresses the TGFβ2 induced EMT in human SRA01/04 lens epithelial cells (LECs), rabbit primary lens epithelial cells, and whole rat lens culture semi-in vivo model. RNA-sequencing and KEGG analysis revealed that the THZ1 inhibits EMT by down-regulating phosphorylate Smad2 and Notch signaling pathway. On the other hand, we found that THZ1 could strongly inhibit LECs proliferation through G2/M phase arrest as well as attenuating of MAPK, PI3K/AKT signaling pathway. Our results uncovered the function and underlying mechanism of THZ1 in regulation of EMT, which provides a new perspective of the anti-tumor effect by THZ1 and may offer a novel treatment for PCO.
Keywords: anti-tumor, THZ1, epithelial-mesenchymal transition, Notch, TGFβ/Smad signaling pathway
Introduction
THZ1 is a selective covalent inhibitor of cyclin-dependent kinase CDK7 that down-regulates RNA polymerase II progressing and mRNA capping [1-3]. THZ1 has a central role in suppressing super-enhancer-linked oncogenic transcription, and can be a potential anti-tumor drug especially for Myc driven cancers [4, 5]. The function of THZ1 in inhibiting the survival of cancer cells [6, 7] has been verified in oesophageal squamous cell carcinoma, T-cell lymphomas, small cell lung cancer, melanoma, etc [8-10].
Involved in various physiological processes, EMT is highly related with the embryonic development, fibrosis, wound healing and cancer progression [11-13]. TGFβ (Transforming growth factor β) is a key factor to promote epithelial cells to undergo EMT and fibrosis [14]. Within the TGFβ family, TGFβ2 is the main subtype in eyes [15]. EMT is characterized by switched expression of typical makers in the mRNA and protein levels. Epithelial markers including E-cadherin, ZO-1 (TPJ-1) are down-regulated during EMT, while the expression of the mesenchymal markers including N-cadherin, Fibronectin, α-SMA, Collagen, ZEB1 are increased during EMT [16-18].
Posterior capsular opacification (PCO), also known as a secondary cataract, is a common long-term complication in which the eye lens lose transparency after lens implantation surgery in cataract patients[19, 20]. As the most prevalent blindness disease in the world, currently the best way to treat cataract is by means of surgery to implant intraocular lenses (IOLs) [21]. IOLs can successfully recover the vision, but unfortunately, it could potentially lead to PCO. Years after surgery, lens equator epithelial cells may undergo persistent aberrant proliferation, migration, and fibrosis. When PCO occurs, the lens cells lose epithelial characteristics and become mesenchymal-like cells[22], which is a typical EMT process in vivo. PCO is treated by Nd:YAG (neodymiumdoped yttrium aluminum garnet) laser capsulotomy [23], which brings complications such as macular edema, retinal detachment, etc [24]. Therefore, inhibition of the EMT of lens epithelial cells presents a promising way to prevent and treat PCO.
In this study, we reported for the first time that the potential anti-neoplastic compound THZ1 strongly prevents the EMT in cancer cells lines Hela229, so-RB50, WERI-Rb-1, Y79. In addition, THZ1 treatment attenuated cell proliferation and suppressed TGFβ2 induced EMT in SRA01/04 lens epithelial cells, rabbit primary lens epithelial cells and whole lens culture semi-in vivo model. These results uncover a new insight for the treatment of THZ1 in cancers, and indicate that THZ1 may function in preventing and curing PCO.
Materials and Methods
Cell Culture and Reagents
Human SRA01/04 lens epithelial cell (LEC) were cultured in high-glucose DMEM growth medium (Gibco, Thermo Fisher Scientific, USA) containing 10% FBS (Biowest, FRA), 1% penicillin (100µg/ml) and 1% streptomycin (100µg/ml) at 37°C in a 5% CO2 atmosphere. SRA01/04 cell line was authenticated by short tandem repeat (STR) analysis at Sun Yat-Sen University (Supplementary Figure S1). Rabbit primary lens epithelial cells were isolated from 7~8 weeks old New Zealand white rabbits and cultured in DMEM/F12 with 20% FBS, 1% penicillin, 1% streptomycin and 1% NEAA (Gibco, Life Technologies Corporation). All cell lines were negative for mycoplasma. Recombinant human TGFβ2 was purchased from Novoprotein. For TGFβ2 and THZ1 (Sellcek, CN) treatment, the cells were cultured in fresh culture medium with 10ng/ml recombinant human TGFβ2 and different concentrations of THZ1 for 24h.
Cell apoptosis and cell cycle analyzed by flow cytometry
For cell apoptosis LECs (2×105) were seeded in 6-well plates, after being treated with different doses of THZ1 for 24h, cells were trypsinized and washed twice by PBS and then incubated with MultiCaspase kit (Millipore) for 30 min at 37°C in dark, then 7-aminoactinomycin D (7-AAD, Millipore) were added and incubated for 5 minutes in room temperature at dark [25].
For cell cycle analysis, cells were fixed in cold 70% ethanol at 4°C overnight before incubated with staining solution (BD) at room temperature for 15 minutes following the manufacturer's instructions. After that, the fluorescence intensity was measured by MuseTM Cell Analyzer (Millipore).
Scratch assays and Cell Viability
For scratch assay, LECs (7×105) were seeded in 6cm dishes, after cell attached, a white tip was used to draw scratches and then floated cells were washed by pre-warmed PBS twice. Then the cells were cultured in DMEM with 1% FBS, and the area of wound was photographed at different time points. Cell viability was detected by the 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl-terazolium bromide (MTT) assay. LEC cells were cultured in 96-well plates at a density of 4,000 cells/well for different time points. 20µl of MTT (5mg/ml, Sigma-Aldrich) were added to the cells for incubation at 37°C for 2-4h. Then the medium in the well was removed and 200ul of dimethyl sulfoxide (DMSO, Sigma-Aldrich; Merck KGaA) solution were added. The absorbance at 490nm wavelengths was detected by micro-plate reader (BioTek Instruments, Winooski, VT) after 10 min of vibration.
Real-Time PCR analysis for gene expression
Total RNA of LEC cells were isolated by using Trizol reagent (Invitrogen), then cDNA reverse was transcribed with the PrimeScript II 1st Strand cDNA Synthesis Kit (TaKaRa, Japan). Real-Time PCR was conducted by LightCycler 96(Roche, Basel, Switzerland) with the SYBR Premix Ex TaqTM Kit (TaKaRa). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as en endogenous control [26]. The sequences of primers used in this study are presented in Supplementary Table 1.
Western Blot
Western blot was performed as previously described [27]. Primary antibodies used in this study were showed in Supplementary Table 2.
Immunofluorescence
LECs were seeded on glass slides in 24-well plates and treated with agents as indicated. After being washed by PBS, cells were fixed with 4% paraformaldehyde for 10mins, permeabilized with 0.5% Triton X-100 for 15 min, and then blocked with 5% bovine serum albumin (BSA) in PBS for 1 h. After being incubated with primary antibody at 4°C overnight, cells were treated with Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody in dark at room temperature for 1 h. Then cells were incubated with 50ng/ml DAPI for 5 min before being mounted on slides and visualized under a fluorescence microscope (Zeiss Axioplan2 Imaging, DE).
Explant culture and treatment
All experimental procedures conformed to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Eyeballs of 21~22 days Wistar rats were taken off and steeped in 0.25% povidone-iodine for 3 mins before washed by PBS containing 1% penicillin (100µg/ml) and 1% streptomycin (100µg/ml). After separated using forceps and microscissors, the lens were maintained in 4ml M199 medium containing 1% FBS, 1% NEAA, 1% penicillin and 1% streptomycin. TGFβ2 and THZ1 were added to the medium at the final concentration of 5ng/ml and 100nM. Lens were cultured for 7days and photographed using a stereoscopic microscope, culture medium were changed every three days.
Statistical analysis
All experiments showed in the figures were conducted three or more repetitions. IBM SPSS Statistics 20 software was used for statistical analysis. Data were presented as mean ± standard deviation (SD) and the significance of the difference among groups were determined by multiple LSD's multiple-comparison test (one-way ANOVA). A value of P<0.05 was considered statistically significant.
Results
THZ1 inhibits proliferation of LECs through G2/M phase cell cycle arrest
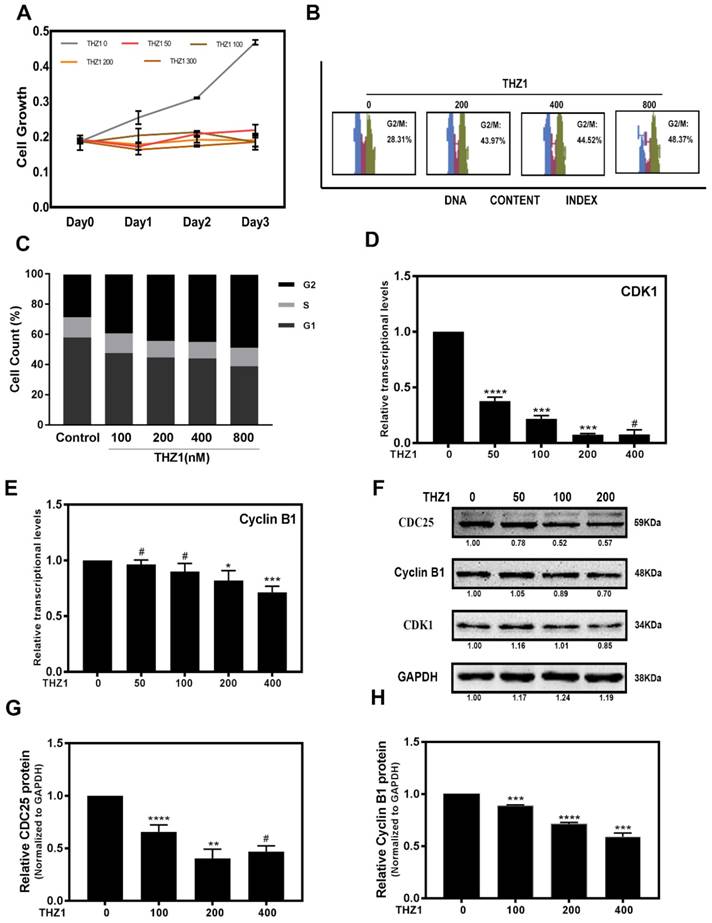
To study the role of THZ1 in regulation of LECs, we treated LECs with different concentrations of THZ1. When LECs were treated with THZ1 at a concentration higher than 400nM, the apoptotic cells increased significantly (Supplementary Figure S2A, S2B), thus we treated LECs with THZ1 below 400nM in the following experiments. In order to determine the cytotoxicity of THZ1, MTT test was carried out to determine LECs proliferation under various concentrations (0, 50, 100, 200, 400nM) of THZ1, and we found that cell proliferation were significantly suppressed by THZ1 even at very low concentration (Figure 1A). We tested cell cycle by flow cytometry to explore whether the anti-proliferative effect from THZ1 is due to cell cycle arrest. As shown in the Figure 1B and 1 C, LECs were treated with different concentrations of THZ1 for 24h and the percentage of G2/M cells was accumulated THZ1 accordingly. Through RT-PCR analysis, we found that THZ1 treatment attenuated the mRNA levels of cell cycle regulators cyclin-dependent kinase (CDK1) and Cyclin B1 (Figure 1D, 1E), and significantly decreased the protein levels of CDK1, Cyclin B1 and CDC25C (Figure 1F, 1G and 1H). Those results indicated that THZ1 inhibits the proliferation of LECs by G2/M arrest through induction of Cyclin B1/CDK1 complex.
THZ1 inhibited proliferation of LECs through G2/M phase cell cycle arrest. (A)The portion of viable LECs was determined by MTT assay after treatment of THZ1 (0, 50, 100, 200 and 400nM) for 24 h, 48 h and 72 h. (B) LECs were subjected to 0, 100, 200, 400, 800nM of THZ1. Flow cytometry results of cell cycle phase are shown. (C) Bar graphs represent the mean ± S.E.M. of three independent experiments. (D, E) The mRNA expression level of CDK1 and Cyclin B1 were detected by RT-PCR after treated by various concentrations of THZ1 for 24 h. (F) The protein expression levels of CDC25, Cyclin B1, CDK1 were detected by western blot analysis. Quantification of protein level showed in the G and H. Results are presented as the mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 vs. Control.
THZ1 inhibits EMT in cancer cells and LECs
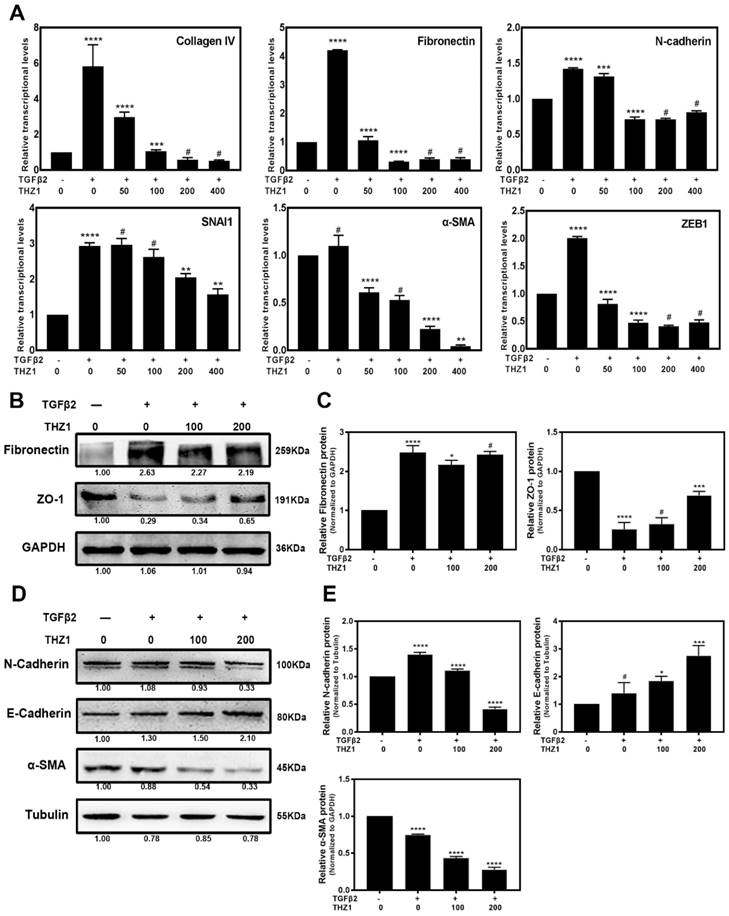
Our data revealed that THZ1 could induce cell apoptosis and inhibit cell viability in Hela 229 (Supplementary Figure S3A, S3B, S3C) and three retinoblastoma cell lines so-RB50, WERI-Rb-1, Y79 (Data unpublished). Then we treated these cancer cell lines within low concentration at which THZ1 couldn't suppress cell viability and proliferation. Interestingly we found that THZ1 inhibited EMT in Hela cells (Supplementary Figure S3D) and three retinoblastoma cell lines (Data unpublished) as shown by the decreased expression of symbolic mesenchymal makers. We next aimed to understand the mechanism of EMT inhibition by THZ1 and to determine whether THZ1 plays the same role in normal cell like LECs. Previous studies reported that LECs underwent EMT when treated with TGFβ2 [28]. Our initial experiment uncovered that 10ng/ml of TGFβ2 is the appropriate concentration to establish EMT model in LEC cells (Supplementary Figure S2C). Then we co-treated LECs with 10ng/ml TGFβ2 and various concentrations (0, 50, 100, 200, 400nM) of THZ1, and analyzed the expression levels of EMT makers by Real-Time PCR, western blot and immunofluorescence. RT-PCR analysis showed that the expression levels of mesenchymal makers including Fibronectin, N-cadherin, α-SMA, Collagen IV, SNAI1 [29], ZEB1 were increased notably after treated with TGFβ2. Interestingly, the mRNA levels of those genes decreased in a dose-dependent manner under THZ1 treatment (Figure 2A). We also found that the treatment of TGFβ2 resulted in changes of cell morphology, including elongation and membrane protrusions, nevertheless, co-treated with THZ1 retained an epithelial morphology similar to the control group (Supplementary Figure S4). Consistently, results of western blot indicated that THZ1 treatment resulted in a clear increased expression of epithelial makers E-cadherin and ZO-1 in a dose-dependent manner (Figure 2B and 2C), and a decrease of protein levels of mesenchymal makers including N-cadherin, Fibronectin, ɑ-SMA (Figure 2D, 2E). In agreement with those results, immunofluorescence staining revealed that THZ1 abrogated TGFβ2 mediated up-regulation of Fibronectin (Figure 3A).
EMT induced by TGFβ2 in LECs was suppressed after treated with THZ1. (A) After co-treated LECs with TGFβ2 (10ng/ml) and THZ1 (0, 50, 100, 200, 400nM) or DMSO for 24 hour, the mRNA expression level of mesenchymal makers include Fibronectin, ZEB1, SMA, Collagen IV, N-cadherin, SNAI1 were examined by RT-PCR. Results were normalized to GAPDH. Protein level of E-cadherin, N-cadherin, ɑ-SMA, ZO-1, Fibronectin were detected by western blot with GAPDH and β-Tubulin as the internal control (B and D). Quantification of protein level showed in the C and E. Results are presented as the mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 vs. Control.
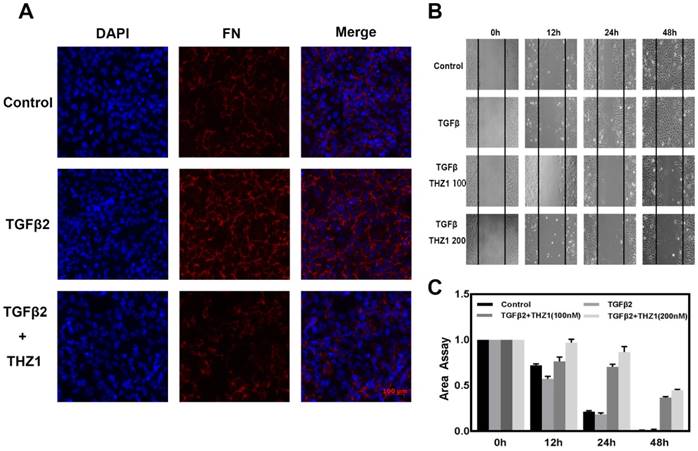
THZ1 prevents TGFβ2-induced EMT in LECs and reduced cells migration capacity. LECs were conducted with TGFβ2 (10ng/mL), THZ1 (100nM) or not for 24 h, immunofluorescence analysis of FN (red), DAPI (blue) were observed using fluorescence microscope. (B) The migratory ability of LECs were detected by scratch assay and observed using a microscope. (C) The area of scratch was measured by Image J. All experiments were repeated three times with similar results.
THZ1 inhibits migration of LEC cells, and protects rabbit primary lens epithelial cell from TGFβ2 induced EMT
Migration is a typical capacity of epithelia, to study THZ1's function in EMT, we monitored the migratory capacities of LEC cells using the in vitro scratch assay. TGFβ2 treatment stimulated the migration ability of LECs, when treated with THZ1 the migration area of the LECs was significantly inhibited in a dose-dependent manner (Figure 3B and 3C).
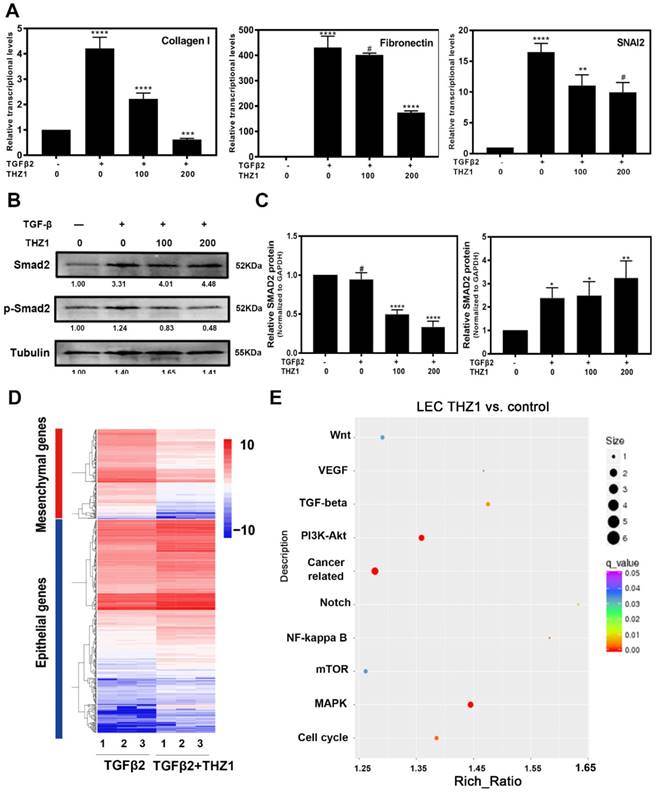
Then, we confirmed the protective role of THZ1 from EMT in primary rabbit LECs. After co-treating primary rabbit LECs with TGFβ2 (10ng/ml) and THZ1 (0, 50, 100nM), we found that THZ1 treatment decreased the expression levels of mesenchymal makers Fibronectin, Collagen I, SNAI2, further supporting the essential role of THZ1 in regulation of EMT (Figure 4A ).
THZ1 protect rabbit primary lens epithelial cell from TGFβ2 induced EMT, influenced the phosphorylation of Smad2 and EMT signatures of LECs. (A) The down-regulation of Fibronectin, SNAI2, Collagen I was examined by RT-PCR after co-treated rabbit primary lens epithelial cell with TGFβ2 and THZ1 (0, 100, 200nM). Protein level of p-Smad2, Smad2 was detected by western blot with GAPDH as the internal control (B). C is the quantification of B. (D) Heat map analysis of the EMT signature in LECs co-treated with TGFβ2 (10ng/ml) and THZ1 (100nM) or DMSO for 24 h. (E) Influenced signaling pathways based on KEGG analysis from RNA-sequence. Results are presented as the mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 vs. Control.
THZ1 suppresses phosphorylation of Smad2 to inhibit TGFβ2 signaling pathway
We next determined the molecular mechanisms underlying THZ1 inhibition of EMT. As shown in Figure 4B and 4C, the results of western blot showed that TGFβ2 significantly increased phospho-smad2 level, which was down-regulated in a dose-dependent manner by THZ1 treatment. As a control, the expression of total smad2 was up-regulated, indicating that THZ1 inhibited the canonical TGFβ2 pathway through suppression of Smad2 phosphorylation.
RNA-seq analysis shows THZ1 inhibits LECs proliferation by the MAPK, PI3K/AKT, ERK1/2 signaling pathway, and abrogates TGFβ2 induced EMT through down-regulating the Notch pathway
We next conducted RNA-Seq analysis to find out the possible signaling pathways by which THZ1 inhibits LEC EMT. RNA samples from LECs treated with TGFβ2 (10ng/ml) and THZ1 (100nM) for 24 h and control LECs were isolated and analyzed. Differential expression genes in the two groups were analyzed and enriched using the KEGG pathway database. Gene enrichment heat map revealed that mesenchymal makers were down-regulated while epithelial makers were up-regulated after THZ1 treatment (Figure 4D).
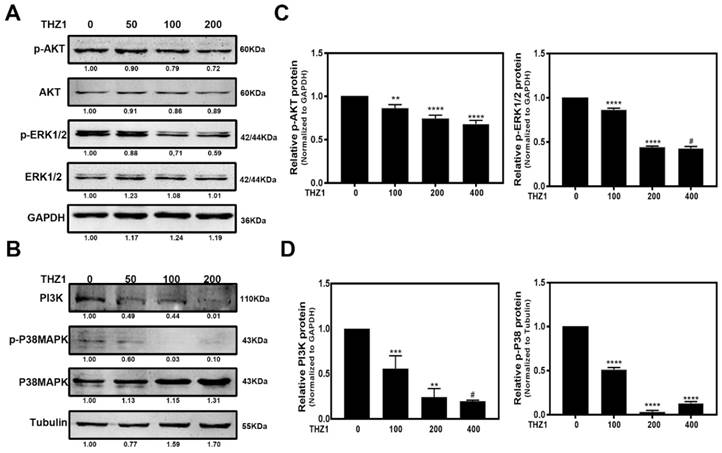
As shown in Figure 4E, the p38 MAPK signaling pathway and PI3K-AKT signaling pathway had the largest difference in gene counts. P38 MAPK signaling pathway is involved in cell cycle control, proliferation, inflammation, stress responses, tumor suppression and promotion [30]. PI3K/AKT signaling pathway also has been reported to regulate cell proliferation, differentiation, cellular metabolism and cancer cell survival [31]. In order to ensure whether these pathways are responsible for inhibiting LECs proliferation and cell cycle progression, we detected PI3K, p-AKT, AKT, p-P38, P38 and extracellular signal-regulated kinase (ERK) of MAPK by western blot after THZ1 treatment in LECs. Our result showed that THZ1 depressed the protein level of PI3K, and reduced the phosphorylation of AKT, P38 and ERK1/2 without affecting their total expression levels (Figure 5A, 5B, 5C, 5D). Thus the inhibition effects of THZ1 towards the proliferation may also be through decreasing the activation of MAPK, ERK1/2 and PI3K/AKT signaling pathways.
THZ1 inactivated the MAPK, ERK1/2 and PI3K/AKT signaling pathways. (A,B) LECs were treated with various concentrations of THZ1 (0, 50, 100 and 200nM) for 24 h, then the protein level of PI3K, p-AKT, AKT, p-P38, P38, p-ERK1/2 and ERK1/2 were detected by western blot analysis with GAPDH and Tubulin as the internal control. Quantification of protein level showed in the C and D. Results are presented as the mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 vs. Control.
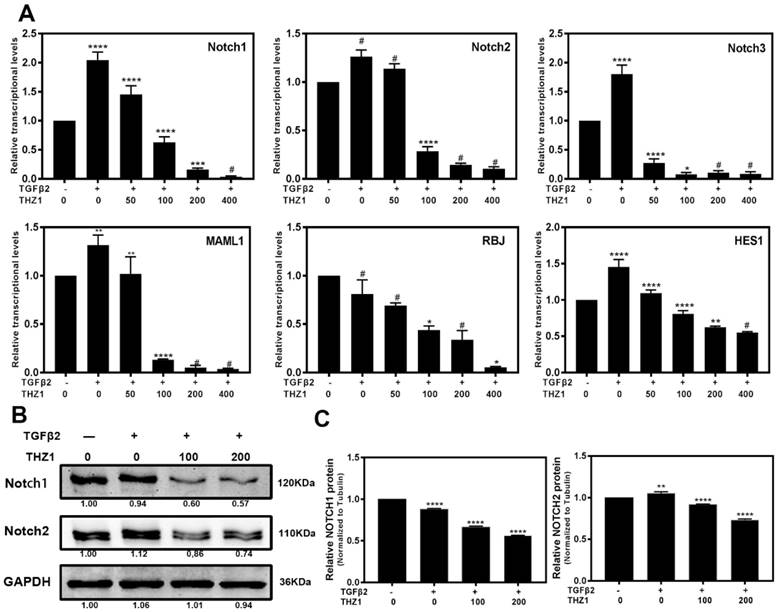
Notch signaling pathway is a crucial regulator to control the balance between cell proliferation, apoptosis, differentiation and EMT as well [32, 33]. In the LECs, Notch pathway regulates the progress of EMT via mediating the TGFβ2/Smad pathway [34]. KEGG analysis showed that Notch pathway also played an important role in LECs after THZ1 treatment, therefore we further determined the mRNA (Figure 6A) and protein (Figure 6B and 6C) levels of Notch pathway components. TGFβ2 increased the expression level of Notch1~3, MAML1, RBJ and the downstream gene Hes1 in LECs. On the other hand, THZ1 treatment abolished the up-regulation of these genes in a dose-dependent manner. Taken together, down-regulating Notch signaling pathway may be another way for THZ1 to protect LECs from TGFβ2 induced EMT.
THZ1 inhibited the Notch signaling pathway. (A) RT-PCR was conducted to detect the mRNA expression of Notch1, Notch2, Notch3, MAML1, RBJ and Hes1 in LECs after co-treated LECs with TGFβ2 (10ng/ml) and THZ1 (0, 50, 100, 200,400nM) or DMSO for 24 hour, (B) Protein level of Notch1, Notch2 was detected by western blot with GAPDH as the internal control. Quantification of protein level from three independent experiments are showed in C. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 vs. Control.
THZ1 attenuates TGFβ2 induced comparable cataractous changes in the semi-in vivo model of whole lens culture
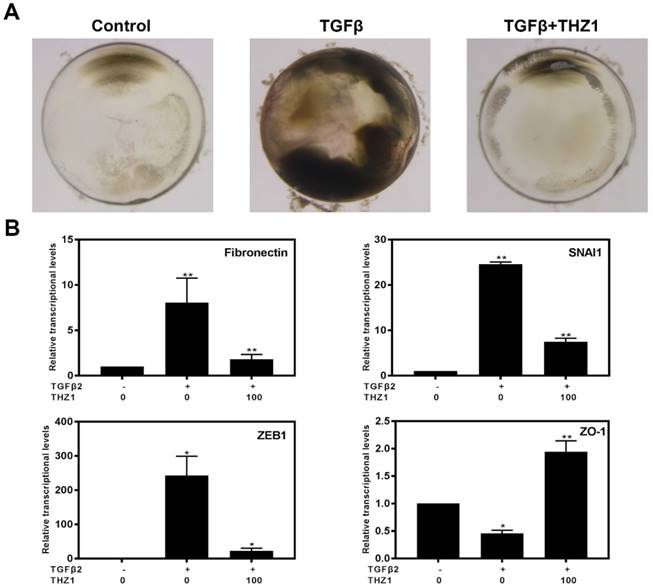
Previous study reported that TGFβ2 induces abnormal morphologic changes in lens explantation in vitro including appearance of spindle-shaped cells, capsule wrinkling and gradually become opaque, which is similar with changes found in human subcapsular cataracts and after-cataracts [35]. In order to verify whether THZ1 could reverse TGFβ2 induced EMT in a physiological system, we separated whole lens. As shown in the Figure 7A, lens cultured in TGFβ2 developed obvious anterior opacities compared to control, nevertheless, THZ1 treatment reversed those changes in lens, resulting in transparency in morphology just like control. Then we detected the mRNA levels of Fibronectin, SNAI1, ZEB1, ZO-1 found that THZ1 reversed the up-regulation of those mesenchymal makers and the down-regulation of epithelial maker (Figure 7B). To sum up, THZ1 could attenuate EMT induced by TGFβ2 in semi-in vivo model.
THZ1 suppressed EMT in whole lens culture semi-in vivo model. (A) Whole rat lenses were co-treated with TGFβ2 (5ng/ml) and THZ1 (100nM) or DMSO for 7 days and the morphology of them were photographed using a stereoscopic microscope. (B) RT-PCR was conducted to detect the mRNA expression of Fibronectin, SNAI1, ZEB1 and ZO-1 in rat lenses after co-treated with TGFβ2 (5ng/ml) and THZ1 (100nM) or DMSO for 7 days. Results are presented as the mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001 vs. Control.
Discussion
In present study, we reported that THZ1 suppressed LECs proliferation through G2/M phase cell cycle arrest and inactivating PI3K/AKT, MAPK, ERK1/2 signaling pathway. More importantly, THZ1 dramatically inhibited the TGFβ2-induced EMT in lens epithelial cell line, primary rabbit lens epithelial cell and whole lens culture semi-in vivo model through attenuated classical TGFβ/Smad pathway and Notch signaling pathway. As a result, our study uncovered that THZ1 can be a new strategy to prevent and treat PCO and brought a new perspective of the anti-tumor effect by THZ1.
Cell cycle is highly controlled by cycle checkpoints to ensure DNA damage repairing before the beginning of mitosis, these checkpoints are regulated by CDKs (cyclin-dependent kinases) and their related pathways. For the G2/M phase, the activated CDC25 (cell division cycle 25 homolog) activates the CDK1 via de-phosphorylation, and then the activated CDK1 forms a complex with Cyclin B1 in the nucleus, which promotes cell into mitosis. Our result showed that THZ1 induces an obvious G2/M phase arrest in LECs. In addition, those important regulators of G2/M transition including CDC25, CDK1, Cyclin B1 were down-regulated both in mRNA and protein levels by THZ1 in a dose-dependent manner. These results indicated that THZ1 induces cell cycle arrest in G2/M phase via inactivating CDK1-Cyclin B1 complex, which might result in the growth inhibition of LECs.
Recently, many studies reported that THZ1, a cyclin-dependent kinase CDK7 inhibitor, maybe a promising antitumor drug [36]. THZ1 has been reported to inhibit cancer growth in oesophageal squamous cancer, T-cell lymphomas, small cell lung cancer, melanoma, Ewing sarcoma, breast cancer, nasopharyngeal carcinoma, high-grade glioma, ovarian cancer, etc. [37-39]. In the progression of cancer, EMT is activated for in invasion into blood vessels and separation from the primary tumor [40-42]. Nevertheless, there is no research discussing whether the compound THZ1 has an influence on the progress of EMT. We proved that THZ1 could inhibit EMT in cancer cells including Hela229 and retinoblastoma cell lines. We further explored broader function of THZ1 and revealed that THZ1 could also inhibit EMT in normal cells. Our study showed that TGFβ2-induced EMT in LECs could also inhibit by THZ1. After co-treating LECs and rabbit primary lens epithelial cells with TGFβ2 and THZ1, we found that the typical mesenchymal markers were down-regulated while epithelial markers were up-regulated compared to the cell only treated by TGFβ2. On the other hand, scratch assay also indicated that THZ1 inhibited cell migration. The whole rat lens culture semi-in vivo model also indicated THZ1 can keep lens transparent and protect it from TGFβ2 induced EMT. Our data also showed that the expression of p-smad2 was obviously down-regulated while the total expression level of smad2 was not changed, which means the TGFβ/Smad signaling pathway that triggers EMT in LECs was inhibited.
In order to study the mechanism of THZ1 in suppression of EMT on LECs, we executed RNA sequencing and KEGG assay, and found the TGFβ signaling pathway was affected, which is consistent with our previous result. MAPK, PI3K/AKT as well as Notch signaling pathway were also affected during the process. The MAPK, PI3K/AKT pathway are key regulators for the control of cell cycle, proliferation and differentiation, as the extracellular signal-regulated kinase (ERK) of MAPK, ERK1/2 also plays an important role on cell adhesion, survival and proliferation. In our research, THZ1 inactivated the p38 MAPK, AKT and ERK and down-regulated PI3K, which is consistent with our RNA-seq result and implied an additional explanation for proliferation inhibition of THZ1 in LECs.
Involved in cell apoptosis, differentiation and survival, Notch signaling pathway is a key regulator in the induction of EMT [27, 43]. In addition, the Notch pathway is also involved in the EMT of LECs. Our data showed that TGFβ2 activated the Notch pathway, however, THZ1 reversed the up-regulation of Notch1~3, MAML1, RBPJ, and their downstream gene Hes1 in a dose-dependent manner. The inhibition of Notch signaling pathway may be another mechanism accounting for the THZ1 mediated inhibition of EMT.
Overall, our result provided, for the first time, the evidence that THZ1 attenuated TGFβ2 induced EMT and inhibited LECs proliferation. Our data not only provided a novel treatment strategy for EMT related diseases such as PCO in eyes, but also contributed to revealing a probably mechanism in THZ1 to cure cancers.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (81670874), the Natural Science Foundation of Guangdong Province, China (2017A030313613) and the Fundamental Research Funds of the State Key Laboratory of Ophthalmology.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB. et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616-20
2. Franco HL, Kraus WL. No driver behind the wheel? Targeting transcription in cancer. Cell. 2015;163:28-30
3. Nilson KA, Guo J, Turek ME, Brogie JE, Delaney E, Luse DS. et al. THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Molecular cell. 2015;59:576-87
4. Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM. et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159:1126-39
5. Coin F, Egly JM. Revisiting the Function of CDK7 in Transcription by Virtue of a Recently Described TFIIH Kinase Inhibitor. Molecular cell. 2015;59:513-4
6. Cao K, Shilatifard A. Inhibit globally, act locally: CDK7 inhibitors in cancer therapy. Cancer cell. 2014;26:158-9
7. Sugai T, Uesugi N, Kitada Y, Yamada N, Osakabe M, Eizuka M. et al. Analysis of the expression of cancer-associated fibroblast- and EMT-related proteins in submucosal invasive colorectal cancer. Journal of Cancer. 2018;9:2702-12
8. Jiang YY, Lin DC, Mayakonda A, Hazawa M, Ding LW, Chien WW. et al. Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut. 2017;66:1358-68
9. Cayrol F, Praditsuktavorn P, Fernando TM, Kwiatkowski N, Marullo R, Calvo-Vidal MN. et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nature communications. 2017;8:14290
10. Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T. et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer cell. 2014;26:909-22
11. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21-45
12. Xue H, Tian GY. MiR-429 regulates the metastasis and EMT of HCC cells through targeting RAB23. Archives of biochemistry and biophysics. 2018;637:48-55
13. Li J, Xia L, Zhou Z, Zuo Z, Xu C, Song H. et al. MiR-186-5p upregulation inhibits proliferation, metastasis and epithelial-to-mesenchymal transition of colorectal cancer cell by targeting ZEB1. Archives of biochemistry and biophysics. 2018;640:53-60
14. Shen SJ, Zhang YH, Gu XX, Jiang SJ, Xu LJ. Yangfei Kongliu Formula, a compound Chinese herbal medicine, combined with cisplatin, inhibits growth of lung cancer cells through transforming growth factor-beta1 signaling pathway. Journal of integrative medicine. 2017;15:242-51
15. Lovicu FJ, Schulz MW, Hales AM, Vincent LN, Overbeek PA, Chamberlain CG. et al. TGFbeta induces morphological and molecular changes similar to human anterior subcapsular cataract. The British journal of ophthalmology. 2002;86:220-6
16. Shu DY, Lovicu FJ. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Progress in retinal and eye research. 2017;60:44-65
17. Horejs CM. Basement membrane fragments in the context of the epithelial-to-mesenchymal transition. European journal of cell biology. 2016;95:427-40
18. Zhao K, He J, Wang YF, Jin SD, Fan Y, Fang N. et al. EZH2-mediated epigenetic suppression of EphB3 inhibits gastric cancer proliferation and metastasis by affecting E-cadherin and vimentin expression. Gene. 2018;686:118-24
19. Zhao Y, Yang K, Li J, Huang Y, Zhu S. Comparison of hydrophobic and hydrophilic intraocular lens in preventing posterior capsule opacification after cataract surgery: An updated meta-analysis. Medicine (Baltimore). 2017;96:e8301
20. Garcia-Medina JJ, Del Rio-Vellosillo M, Zanon-Moreno V, Santos-Bueso E, Gallego-Pinazo R, Ferreras A. et al. Does Posterior Capsule Opacification Affect the Results of Diagnostic Technologies to Evaluate the Retina and the Optic Disc? BioMed research international. 2015;2015:813242
21. Lovicu FJ, Shin EH, McAvoy JW. Fibrosis in the lens. Sprouty regulation of TGFbeta-signaling prevents lens EMT leading to cataract. Experimental eye research. 2016;142:92-101
22. Wormstone IM, Eldred JA. Experimental models for posterior capsule opacification research. Experimental eye research. 2016;142:2-12
23. Singh H, Kwan SL, Tam D, Mamalis N, MacLean K, Ahmed II. Fracture and dislocation of a glass intraocular lens optic as a complication of neodymium:YAG laser posterior capsulotomy: Case report and literature review. Journal of cataract and refractive surgery. 2015;41:2323-8
24. Milazzo S, Grenot M, Benzerroug M. [Posterior capsule opacification]. Journal francais d'ophtalmologie. 2014;37:825-30
25. Fan Y, Huang Z, Long C, Ning J, Zhang H, Kuang X. et al. ID2 protects retinal pigment epithelium cells from oxidative damage through p-ERK1/2/ID2/NRF2. Archives of biochemistry and biophysics. 2018;650:1-13
26. Huang Z, Lin S, Long C, Zhou X, Fan Y, Kuang X. et al. Notch signaling pathway mediates Doxorubicin-driven apoptosis in cancers. Cancer management and research. 2018;10:1439-48
27. Shen H, McElhinny AS, Cao Y, Gao P, Liu J, Bronson R. et al. The Notch coactivator, MAML1, functions as a novel coactivator for MEF2C-mediated transcription and is required for normal myogenesis. Genes & development. 2006;20:675-88
28. Zhang G, Kang L, Chen J, Xue Y, Yang M, Qin B. et al. CtBP2 Regulates TGFbeta2-Induced Epithelial-Mesenchymal Transition Through Notch Signaling Pathway in Lens Epithelial Cells. Current eye research. 2016;41:1057-63
29. Xiong Y, Wang Y, Wang L, Huang Y, Xu Y, Xu L. et al. MicroRNA-30b targets Snail to impede epithelial-mesenchymal transition in pancreatic cancer stem cells. Journal of Cancer. 2018;9:2147-59
30. Xu Y, Li N, Xiang R, Sun P. Emerging roles of the p38 MAPK and PI3K/AKT/mTOR pathways in oncogene-induced senescence. Trends in biochemical sciences. 2014;39:268-76
31. Polivka J Jr, Janku F. Molecular targets for cancer therapy in the PI3K/AKT/mTOR pathway. Pharmacology & therapeutics. 2014;142:164-75
32. Wang Z, Li Y, Kong D, Sarkar FH. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Current drug targets. 2010;11:745-51
33. Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer letters. 2013;341:41-5
34. Chen X, Xiao W, Chen W, Luo L, Ye S, Liu Y. The epigenetic modifier trichostatin A, a histone deacetylase inhibitor, suppresses proliferation and epithelial-mesenchymal transition of lens epithelial cells. Cell death & disease. 2013;4:e884
35. Hales AM, Chamberlain CG, McAvoy JW. Cataract induction in lenses cultured with transforming growth factor-beta. Investigative ophthalmology & visual science. 1995;36:1709-13
36. Ma X, Kuang X, Xia Q, Huang Z, Fan Y, Ning J. et al. Covalent CDK7 Inhibitor THZ1 Inhibits Myogenic Differentiation. Journal of Cancer. 2018;9:3149-55
37. Greenall SA, Lim YC, Mitchell CB, Ensbey KS, Stringer BW, Wilding AL. et al. Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis. 2017;6:e336
38. Sahni JM, Keri RA. Targeting bromodomain and extraterminal proteins in breast cancer. Pharmacological research. 2018;129:156-76
39. Zhang Z, Peng H, Wang X, Yin X, Ma P, Jing Y. et al. Preclinical Efficacy and Molecular Mechanism of Targeting CDK7-Dependent Transcriptional Addiction in Ovarian Cancer. Molecular cancer therapeutics. 2017;16:1739-50
40. D'Amico S, Shi J, Martin BL, Crawford HC, Petrenko O, Reich NC. STAT3 is a master regulator of epithelial identity and KRAS-driven tumorigenesis. Genes & development. 2018;32:1175-87
41. Zhang W, Zhai Y, Wang W, Cao M, Ma C. Enhanced expression of lncRNA TP73-AS1 predicts unfavorable prognosis for gastric cancer and promotes cell migration and invasion by induction of EMT. Gene. 2018;678:377-83
42. M JR, S V. BMI1 and PTEN are key determinants of breast cancer therapy: A plausible therapeutic target in breast cancer. Gene. 2018;678:302-11
43. Kabashima-Niibe A, Higuchi H, Takaishi H, Masugi Y, Matsuzaki Y, Mabuchi Y. et al. Mesenchymal stem cells regulate epithelial-mesenchymal transition and tumor progression of pancreatic cancer cells. Cancer science. 2013;104:157-64
Author contact
![]() Corresponding author: Huangxuan Shen, Ph.D., Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China. Phone: (+8620)-8733-5261; FAX: (+8620)-8733-3721; Email: shenhxsysu.edu.cn
Corresponding author: Huangxuan Shen, Ph.D., Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou, China. Phone: (+8620)-8733-5261; FAX: (+8620)-8733-3721; Email: shenhxsysu.edu.cn