Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(16):3819-3829. doi:10.7150/jca.30018 This issue Cite
Research Paper
PGC1α downregulation and glycolytic phenotype in thyroid cancer
Chien-Liang Liu1,2, Po-Sheng Yang1, Tao-Yeuan Wang3, Shih-Yuan Huang1,2, Yi-Hue Kuo1,2, Shih-Ping Cheng1,4 ![]()
1. Department of Surgery, MacKay Memorial Hospital and Mackay Medical College, Taipei, Taiwan
2. Department of Medical Research, MacKay Memorial Hospital, Taipei, Taiwan
3. Department of Pathology, MacKay Memorial Hospital and Mackay Medical College, Taipei, Taiwan
4. Department of Pharmacology, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan
Received 2018-9-17; Accepted 2019-5-25; Published 2019-6-9
Abstract
Increased aerobic glycolysis portends an unfavorable prognosis in thyroid cancer. The metabolic reprogramming likely results from altered mitochondrial activity and may promote cancer progression. Peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) plays a pivotal role in mitochondrial biogenesis and function. In the present study, we aimed to evaluate the clinicopathological significance of PGC1α expression and the potential effects of PGC1α modulation. Firstly, the expression of PGC1α in thyroid cancer samples was evaluated using western blot analysis and immunohistochemical staining. Compared with normal thyroid tissue, PGC1α expression was downregulated in thyroid cancer. PGC1α-negative papillary cancer was associated with BRAF V600E mutation, large tumor size, extrathyroidal or lymphovascular invasion, lymph node metastasis, and advanced stage. The results were consistent with the analysis of The Cancer Genome Atlas data. PGC1α expression correlated with oxygen consumption in thyroid cancer cells and was inversely related to AKT activity. The biologic relevance of PGC1α was further investigated by gain- and loss-of-function experimental studies. PGC1α overexpression led to augmented oxidative metabolism and accelerated tumor growth, whereas PGC1α knockdown induced a glycolytic phenotype but reduced tumor growth in vivo. In conclusion, PGC1α downregulation is associated with glycolytic metabolism and advanced disease in thyroid cancer. Nonetheless, manipulating PGC1α expression and metabolic phenotype does not necessarily translate into beneficial effects. It suggests that the metabolic phenotype is likely the consequence rather than the cause of disease progression in thyroid cancer.
Keywords: PGC1α, Glycolysis, Thyroid cancer
Introduction
The phenomenon of high rates of aerobic glycolysis in cancer cells was described as early as in the late 1920s by Otto Warburg. Subsequently, metabolic reprogramming has been found to be one of the core hallmarks of cancer, which might enable cancer cells to survive a hostile microenvironment and sustain rapid growth [1]. The high rate of glucose utilization by such tumors can be visualized clinically by 18fluorine-labeled-fluorodeoxyglucose positron emission tomography/computed tomography (FDG-PET/CT). In differentiated thyroid cancer, FDG uptake has important prognostic implications. Positive FDG-PET/CT scan was associated with a short thyroglobulin doubling time, and patients with FDG-avid lesions might have shorter survival [2, 3]. These observations suggest that a glycolytic state links to aggressive tumor biology of thyroid cancer.
Age is a major determining factor for overall survival in patients with thyroid cancer. We have shown that older age at diagnosis is associated with an upregulation of glycolysis signature in tumor tissue [4]. It is well recognized that mitochondrial alterations are involved in a diverse range of acquired disorders, including aging and neurodegenerative diseases [5]. There is compelling evidence that bioenergetic changes in mitochondria are actively participating in the metabolic reprogramming during tumorigenesis [6]. Among metabolic regulators, peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) is a transcription factor coactivator which plays a pivotal role in mitochondrial biogenesis and function [7]. Cellular PGC1α is regulated by stress sensors such as AMP-activated protein kinase (AMPK). Experimental studies suggest that PGC1α is also important in the metabolic reprogramming of cancer cells and is aberrantly regulated by several oncogenes and signaling pathways [8].
Recently, we employed next-generation sequencing technology to profile the changes in gene expression during tumor progression. The functional enrichment analysis indicated that the oxidation-reduction pathway was downregulated, and the PGC1α expression was significantly decreased from stage I to stage IV in papillary thyroid cancer [9]. Because of the crucial importance of interactions between PGC1α and metabolic reprogramming, we sought to investigate the clinicopathological significance of PGC1α expression in thyroid cancer. Furthermore, we aimed to assess whether manipulation of PGC1α expression would affect the phenotype of thyroid cancer and whether targeting PGC1α could be utilized as a therapeutic strategy in the management of thyroid cancer.
Materials and Methods
Patient cohort
The study (14MMHIS091e) was approved by the Institutional Review Board of MacKay Memorial Hospital. Consecutive patients who underwent thyroidectomy for thyroid cancer between 2001 and 2012 were randomly selected for tissue analysis. The details of the selection process have been described in our previous study, and the scanned pathologic images were publicly available for automatic quantitative evaluation [10, 11]. The criteria of specific pathologic features were described elsewhere [12, 13]. In the present study, a total of 121 patients with papillary thyroid cancer were included for the PGC1α expression analysis in tumor samples and adjacent normal thyroid tissues. The tumor-node-metastasis (TNM) staging was based on the seventh edition of the American Joint Committee on Cancer staging classification.
Immunohistochemistry
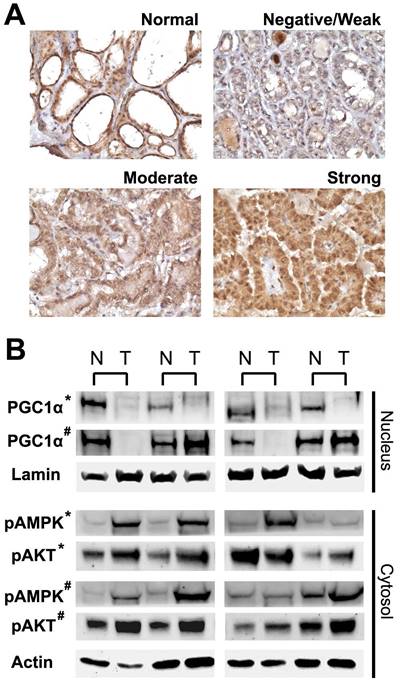
The expression level of PGC1α was investigated in paraffin sections using immunohistochemistry with a polyclonal antibody to PGC1α (ab54481; Abcam, Cambridge, UK). Immunohistochemical staining was performed using standard techniques as previously described [14]. We observed that the expression of PGC1α in thyroid follicular cells exhibited characteristic nuclear staining and homogeneous cytoplasmic staining (Figure 1A). Accordingly, the presence of any areas with ≥50% follicular cells showing definitive nuclear staining were considered PGC1α-positive.
Expression of peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) in normal thyroid tissue and papillary thyroid cancer (negative/weak, moderate, or strong expression). (A) Representative microscopic photographs of immunohistochemical staining. Original magnification, 400x. (B) Western blot analysis of nuclear and cytoplasmic protein extracts from paired samples (N, normal; T, tumor). Lamin and actin were used as nuclear and cytoplasmic loading controls, respectively. *, papillary thyroid cancer; #, follicular thyroid cancer.
The Cancer Genome Atlas (TCGA) data analysis
The transcriptome sequencing data of primary tumor samples of TCGA papillary thyroid cancer (THCA) dataset were downloaded from the Genomic Data Commons of the National Cancer Institute as previously reported [15]. The PGC1α expression level was measured as log-transformed RNA-Seq by Expectation-Maximization (RSEM) values.
Cell culture and reagents
Follicular cell-derived thyroid cancer cell lines K1, FTC-133, and 8505C were obtained from the European Collection of Authenticated Cell Cultures (ECACC), Salisbury, UK, and ML-1 from the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ), Braunschweig, Germany. PGC1α-expressing lentivirus vector, control lentivirus vector, PGC1α-targeting siRNA, and control scramble siRNA were purchased from Santa Cruz Biotechnology, Dallas, TX. AICAR, a cell-permeable AMPK activator, and LY294002, a selective phosphatidylinositol 3-kinase (PI3K) inhibitor, were obtained from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). PLX4032 (vemurafenib), a BRAF inhibitor, was obtained from Selleck Chemicals, Houston, TX.
Cell growth and clonogenic assay
Cell viability and growth were determined by trypan blue exclusion. Cells were harvested, stained with 0.4% trypan blue solution (Sigma-Aldrich), and counted in hemocytometer chambers. For the colony-forming assay, 500 thyroid cancer cells were seeded into six-well plates. After 10-14 days, colonies were stained with 3% crystal violet and counted [16].
Real-time quantitative polymerase chain reaction
Real-time quantitative polymerase chain reaction (qRT-PCR) was performed to determine the relative mRNA expression of specific genes as previously described [17]. The list of primer sequences is provided in Supplementary Table S1.
Western blot analysis
Nuclear and cytoplasmic protein extracts were prepared using the CelLytic NuCLEAR Extraction Kit (Sigma-Aldrich). Immunoblotting was performed with antibodies against PGC1α (ab54481; Abcam), lamin B1 (ab16048; Abcam), phospho-AMPKα (ab133448; Abcam), AMPKα (ab80039; Abcam), phospho-AKT (#4056; Cell Signaling Technology, Danvers, MA), AKT (#4685; Cell Signaling), phospho-ERK (#4376; Cell Signaling), ERK (#4695; Cell Signaling), or β-actin (A5441; Sigma-Aldrich) as indicated [18].
Metabolic analyses
Oxygen consumption rate (OCR) was determined using the XF24 Extracellular Flux Analyzer (Seahorse Bioscience; Agilent Technologies, North Billerica, MA) [19]. Glucose consumption, ATP content, and lactate production were evaluated using the Glucose Uptake Assay Kit, ATP Assay Kit, and Lactate Assay Kit (all from Sigma-Aldrich), respectively, according to the manufacturer's recommendations. To visualize mitochondria, cells were stained with MitoTracker Red CMXRos (Thermo Fisher Scientific) for 30 min. Nuclear counterstaining was performed using 4',6-diamidino-2-phenylindole (DAPI). Fluorescent images were acquired with a fluorescence microscope.
Xenograft model of thyroid cancer
All animal care and experiments were approved by the Institutional Animal Care and Use Committee of MacKay Memorial Hospital. Establishment of the murine xenograft tumor model was performed as previously described [20]. Six-week-old female BALB/c nude mice purchased from the National Laboratory Animal Center, Taipei, Taiwan, received subcutaneous injections of 1 x 107 thyroid cancer cells into their bilateral flanks. Tumor volume was measured twice weekly.
Statistical analysis
Patient data were analyzed using the chi-square test, Fisher's exact test, the Cochran-Armitage trend test, or the Mann-Whitney U test, as appropriate. Statistical comparisons of independent experiments were performed using two-tailed Student's t tests. A P value < 0.05 was considered statistically significant.
Results
PGC1α is downregulation in thyroid cancer
We started with the immunohistochemical analysis of PGC1α expression in papillary thyroid cancer. As shown in Figure 1A, normal thyroid tissues exhibited strong nuclear immunoreactivity for PGC1α with little staining in the intervening stroma. The expression of PGC1α was diminished in some of the papillary thyroid cancers. Western blotting was performed using proteins isolated from additional paired samples, and the results were in agreement with the immunohistochemical data (Figure 1B). PGC1α expression was consistently low in malignant thyroid tissue except that a slight increase in PGC1α expression was observed in some follicular thyroid cancers. Phosphorylated AMPK and phosphorylated AKT levels were overexpressed in the malignant counterparts.
The translational relevance of PGC1α downregulation in thyroid cancer was investigated by examining the clinicopathological correlation. Based on our predefined criteria, about 56% of papillary cancer was interpreted as negative for PGC1α expression. We found that patients with PGC1α-negative papillary cancer had large tumor size, more frequent extrathyroidal or lymphovascular invasion, higher incidence of lymph node metastasis, and more advanced TNM stage (Table 1). Of note, the majority of PGC1α-negative cancer harbored the BRAF V600E mutation.
Associations between peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) expression and clinicopathological features in papillary thyroid cancer (n = 121)
| PGC1α-negative (n = 68) | PGC1α-positive (n = 53) | P value | |
|---|---|---|---|
| Age (years) a | 45 (33-58) | 42 (37-49) | 0.481 |
| Sex | 0.276 | ||
| Male | 8 (12%) | 10 (19%) | |
| Female | 60 (88%) | 43 (81%) | |
| Body mass index (kg/m2) a | 24.0 (21.8-26.5) | 23.1 (22.8-24.5) | 0.661 |
| Tumor size (cm) a | 2.5 (1.8-3.1) | 1.7 (1.2-2.8) | 0.003 |
| Extrathyroidal invasion b | < 0.001 | ||
| No | 23 (34%) | 41 (77%) | |
| Microscopic | 32 (47%) | 11 (21%) | |
| Macroscopic | 13 (19%) | 1 (2%) | |
| Multifocality | 0.364 | ||
| Present | 24 (35%) | 23 (43%) | |
| Absent | 44 (65%) | 30 (57%) | |
| Lymphovascular invasion | 0.012 | ||
| Present | 19 (28%) | 5 (9%) | |
| Absent | 49 (72%) | 48 (91%) | |
| BRAF V600E mutation | < 0.001 | ||
| Present | 61 (90%) | 23 (43%) | |
| Absent | 7 (10%) | 30 (57%) | |
| Lymph node metastasis b | 0.009 | ||
| N0 | 27 (40%) | 33 (62%) | |
| N1a | 28 (41%) | 16 (30%) | |
| N1b | 13 (19%) | 4 (8%) | |
| Distant metastasis | 0.255 | ||
| Present | 3 (4%) | 0 (0%) | |
| Absent | 65 (96%) | 53 (100%) | |
| TNM stage b | 0.001 | ||
| Stage I | 36 (53%) | 42 (79%) | |
| Stage II | 2 (3%) | 2 (4%) | |
| Stage III | 17 (25%) | 8 (15%) | |
| Stage IV | 13 (19%) | 1 (2%) |
Data are expressed as number (percentage) or median (interquartile range). a Mann-Whitney test, b Cochran-Armitage trend test.
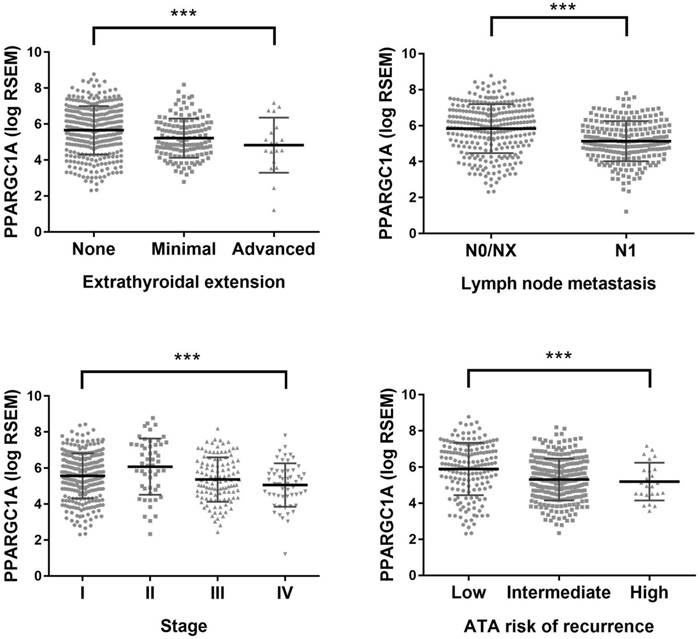
For external validation, TCGA data were evaluated for the association between PGC1α expression and clinicopathological factors. The level of PGC1α expression in papillary thyroid cancer was lower than that in the matched normal sample (n = 59; median RSEM, 269 versus 1828; Wilcoxon signed-rank P < 0.0001). PGC1α expression was progressively downregulated along with the increasing disease stage or the increasing recurrence risk defined by the American Thyroid Association (Figure 2). The expression of PGC1α tends to further downregulated in the distant metastatic sites than the primary tumor (n = 8; median RSEM, 142 versus 303; P = 0.069). Moreover, tumors harboring the TERT promoter mutation tend to have lower PGC1α expression (P = 0.069), but the marginal difference was lost after propensity score matching for the TNM stage and other clinicopathological parameters (P = 0.54) [21]. Overall, the analysis of TCGA data confirmed our observation that PGC1α expression was downregulated in parallel to disease progression of papillary thyroid cancer.
Expression of peroxisome proliferator-activated receptor γ coactivator 1α (PPARGC1A) in papillary thyroid cancer (THCA) dataset of The Cancer Genome Atlas (TCGA). ***, P < 0.001.
PGC1α correlates with oxidative metabolism and is upregulated by AKT inhibition
We investigated cellular respiration and metabolism by Seahorse assay and correlated the measured OCR with PGC1α expression in a panel of thyroid cancer cell lines. As shown in Figure 3A, the oxygen consumption of thyroid cancer cells was modestly related to the PGC1α expression level (R2 = 0.52, P < 0.0001).
(A) The correlation of oxygen consumption rate (OCR) and peroxisome proliferator-activated receptor γ coactivator 1α (PGC1α) expression in thyroid cancer cell lines. (B) Western blot analysis of nuclear and cytoplasmic protein extracts from FTC-133 and K1 cells treated with vehicle control, AICAR 1 mM, LY294002 20 μM, or PLX4032 20 μM for 24 h. Lamin and actin were used as nuclear and cytoplasmic loading controls, respectively.
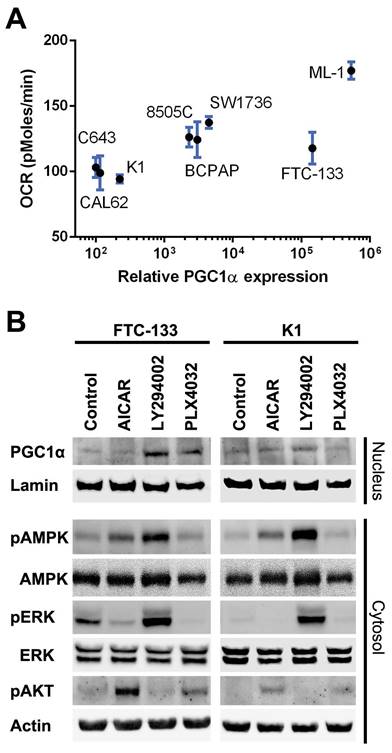
Following treatment with AICAR, phosphorylated AMPK was increased in FTC-133 and K1 cells, but there was no significant change in PGC1α expression (Figure 3B). Treatment with LY294002 suppressed AKT activity but upregulated AMPK and ERK phosphorylation. Interestingly, a remarkable increase in PGC1α expression was observed following LY294002 administration. Treatment with PLX4032 upregulated PGC1α expression in FTC-133 but not K1 cells. Taken together, PGC1α downregulation in malignant thyroid tissue probably results from the activation of the AKT pathway.
PGC1α overexpression augments oxidative metabolism
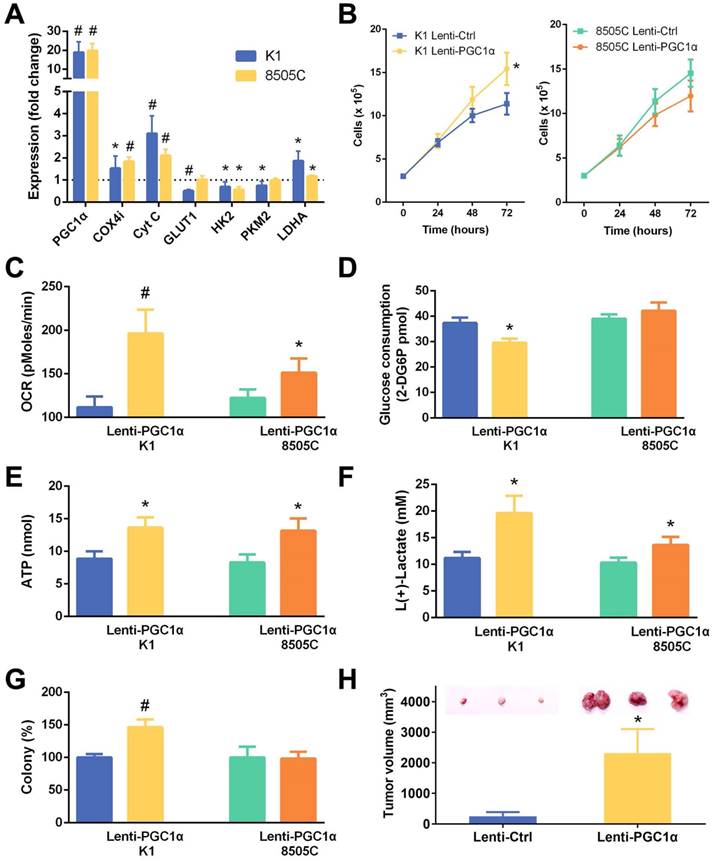
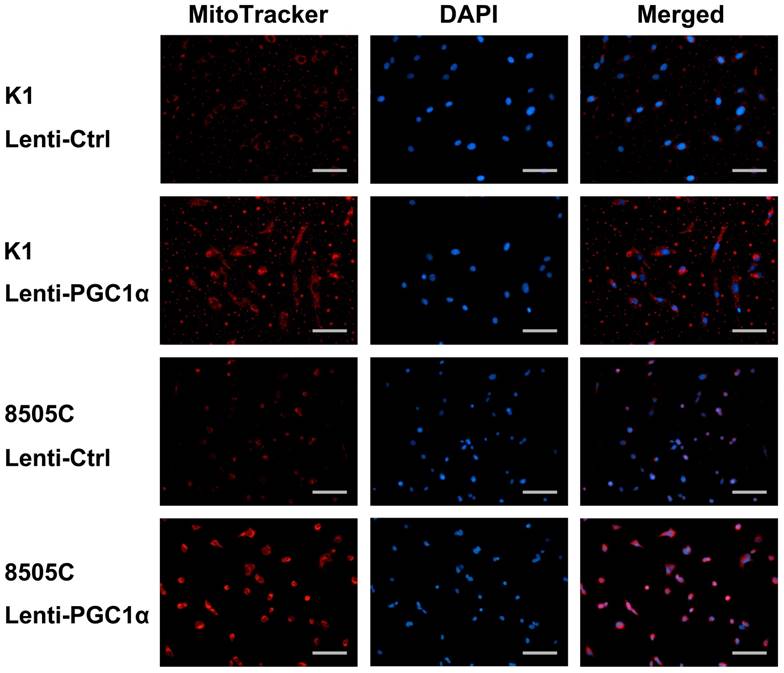
To gain an insight into the therapeutic potential of the restoration of PGC1α expression, K1 and 8505C cells which had low PGC1α expression levels were transfected with a PGC1α-expressing lentivirus vector. As expected, the expression of mitochondrial gene targets of PGC1α involved in oxidative phosphorylation (COX4i and cytochrome c) were upregulated (Figure 4A). Most glycolysis regulators, including glucose transporter (GLUT1), hexokinase 2 (HK2), and pyruvate kinase muscle isozyme 2 (PKM2), were downregulated. Nonetheless, the expression of lactate dehydrogenase (LDHA) was increased along with PGC1α overexpression. Consistent with the role of PGC1α in association with mitochondrial biogenesis, PGC1α-overexpressing thyroid cancer cells showed an increase in MitoTracker red fluorescence, an indicator of mitochondrial mass (Figure 5).
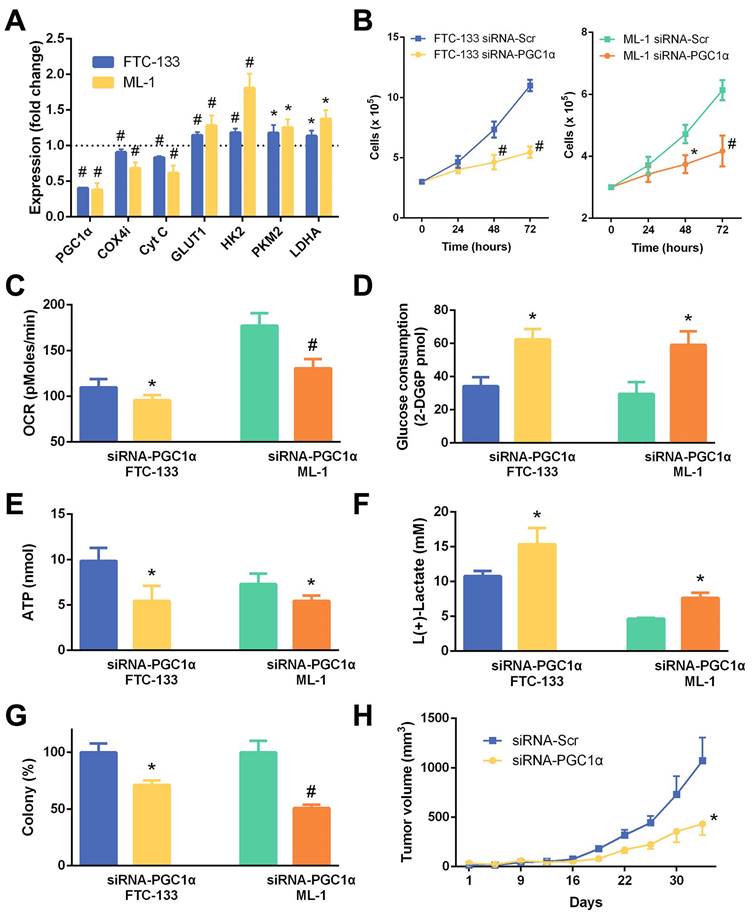
Phenotypic characteristics of thyroid cancer cell lines (K1 and 8505C) transfected with a PGC1α-expressing lentivirus vector (Lenti-PGC1α) versus control lentivirus vector (Lenti-Ctrl). (A) Relative changes in mRNA expression of metabolic genes. (B) Cell growth. (C) Oxygen consumption rate (OCR). (D) Glucose consumption. (E) ATP content. (F) Lactate production. (G) Colony formation. (H) Xenograft tumor growth of K1 thyroid cancer cells in nude mice (n = 4 per group). *, P < 0.05; #, P < 0.001.
MitoTracker staining of K1 and 8505C thyroid cancer cells transfected with a control lentivirus vector (Lenti-Ctrl) or PGC1α-expressing lentivirus vector (Lenti-PGC1α). Nuclear counterstaining was performed using 4',6-diamidino-2-phenylindole (DAPI). Scale bar, 100 μm.
In accordance with the changes in mRNA expression of corresponding genes, PGC1α-overexpressing thyroid cancer cells had an increase in the OCR and ATP levels (Figure 4C and 4E). PGC1α-overexpressing K1 cells had lower glucose consumption (Figure 4D). Furthermore, lactate production was increased following PGC1α overexpression (Figure 4F), consistent with an upregulation of LDHA. Taken together, the restoration of PGC1α expression led to a relatively augmented oxidative metabolism and glycolysis in thyroid cancer cells.
Accompanied by alterations in metabolic phenotype after PGC1α overexpression, the cell growth and clonogenic ability of K1 cells were significantly increased (Figure 4B and 4G). There was no change in growth or colony formation in 8505C cells transfected with a PGC1α-expressing lentivirus vector. Our xenograft tumor model supports the in vitro results. Subcutaneous transplantation of PGC1α-overexpressing K1 cells in nude mice formed a significantly higher tumor burden compared to control cells (Figure 4H).
PGC1α silencing induces a glycolytic phenotype
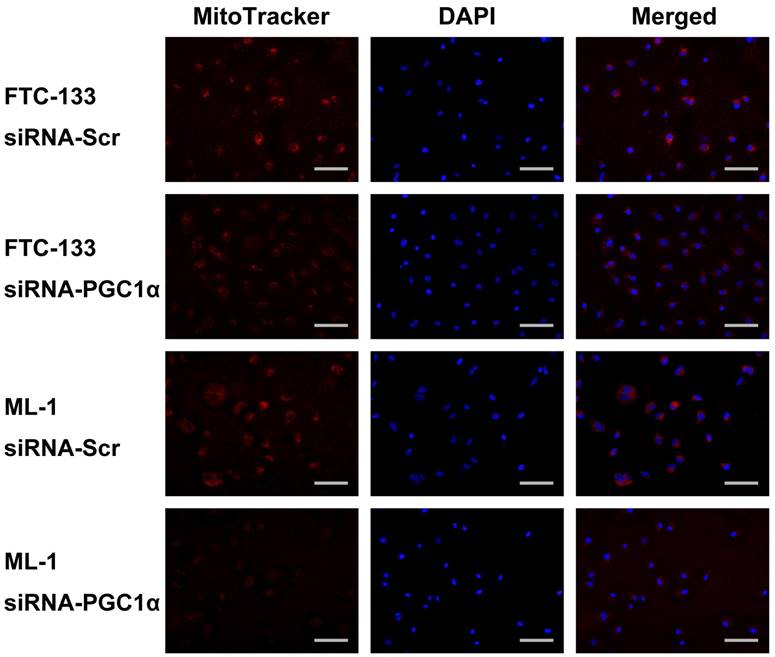
We further examined the effects of PGC1α knockdown in thyroid cancer cells with relatively high PGC1α expression levels. FTC-133 and ML-1 cells were transfected with PGC1α-targeting siRNA, and the expression of COX4i and cytochrome c was consistently reduced (Figure 6A). MitoTracker fluorescent staining supports a decrease in mitochondrial activity (Figure 7). Interestingly, the expression of all glycolytic genes (GLUT1, HK2, PKM2, and LDHA) was significantly upregulated. Markedly, the OCR and ATP levels were decreased following PGC1α knockdown (Figure 6C and 6E). By contrast, glucose consumption and lactate production were increased (Figure 6D and 6F), consistent with a shift to the glycolytic phenotype.
Phenotypic characteristics of thyroid cancer cell lines (FTC-133 and ML-1) transfected with a PGC1α-targeting siRNA (siRNA-PGC1α) versus control scramble siRNA (siRNA-Scr). (A) Relative changes in mRNA expression of metabolic genes. (B) Cell growth. (C) Oxygen consumption rate (OCR). (D) Glucose consumption. (E) ATP content. (F) Lactate production. (G) Colony formation. (H) Xenograft tumor growth of FTC-133 thyroid cancer cells in nude mice (n = 6 per group). *, P < 0.05; #, P < 0.001.
MitoTracker staining of FTC-133 and ML-1 thyroid cancer cells transfected with a control scramble siRNA (siRNA-Scr) or PGC1α-targeting siRNA (siRNA-PGC1α). Nuclear counterstaining was performed using 4',6-diamidino-2-phenylindole (DAPI). Scale bar, 100 μm.
The change in metabolic phenotype after PGC1α knockdown did not enhance the aggressiveness of thyroid cancer cells. On the contrary, the cell growth and clonogenic ability of FTC-133 and ML-1 cells reduced (Figure 6B and 6G). In addition, the xenograft tumor growth in nude mice of FTC-133 cells transfected with PGC1α-targeting siRNA was significantly slower (Figure 6H). Taken together, although PGC1α silencing induced a glycolytic phenotype in thyroid cancer cells, it did not promote cell proliferation or tumor growth.
Discussion
Energy supply in normal thyrocytes mostly relies on mitochondrial oxidative phosphorylation. In thyroid malignancies, oncogene activation and/or tumor suppressor inactivation leads to a variety of metabolic modifications including an accelerated glycolytic activity [22]. The metabolic ecology may, in turn, support tumor progression [1]. In this regard, BRAF V600E, the most prevalent somatic oncogenic mutation in papillary thyroid cancer, confers a higher likelihood of FDG-PET avidity and is associated with higher standardized uptake values (SUVs) [23]. This is in line with the findings of the present study. We showed that the PGC1α expression was downregulated in advanced disease and was closely related to the occurrence of BRAF V600E mutation. The downregulation of PGC1α in thyroid cancer likely reflects compromised mitochondrial integrity, reduced respiration, and compensatory enhancement in glycolysis during tumor progression. It is consistent with the observed relationship between the FDG-PET avidity and patient outcomes [3].
PGC1α acts as a key conduit for AMPK to control the expression of metabolic genes [24]. In agreement with our observations, Vidal and colleagues demonstrated that phosphorylated AMPK was upregulated in papillary thyroid cancer [25]. However, the relationship of phosphorylated AMPK and nuclear PGC1α expression was in the opposite direction. Furthermore, AICAR treatment in thyroid cancer cells increased AMPK phosphorylation but not PGC1α expression. Therefore, it is unlikely that AMPK is the main regulator of PGC1α expression in thyroid cancer. Previous studies indicated that phosphorylation of PGC1α by AKT could attenuate PGC1α activity [7, 26]. Aberrant activation of the PI3K/AKT pathway is involved in the progression and dedifferentiation of thyroid cancer [27, 28]. It is also worth noting that FDG uptake in tumors may correlate with AKT pathway activity [29, 30]. Taken together, our results suggest that PGC1α expression and metabolic phenotype were largely regulated by the PI3K/AKT pathway rather than the AMPK pathway in thyroid cancer.
Considering the clinical implications of the glycolytic shift in thyroid cancer, it would be interesting to evaluate the therapeutic potential of manipulating PGC1α expression levels with accompanying changes in metabolic phenotypes. As expected, cells transfected with a PGC1α-expressing vector had increased oxidative metabolism and aerobic glycolysis through the induction of metabolic genes. Strikingly, cell growth and tumor formation of the PGC1α-overexpressing cells in immunocompromised mice were also enhanced. On the other hand, although glycolysis was upregulated following siRNA-mediated silencing of PGC1α, mice injected with PGC1α-knockdown FTC-133 cells showed a significant delay in tumor growth. A reasonable interpretation of our findings is that the metabolic characteristics might be cell-context dependent intrinsic properties acquired during disease progression. In this sense, simply modifying the balance between glycolysis and oxidative metabolism would not translate into beneficial effects.
In conclusion, the expression of PGC1α in thyroid cancer samples is negatively regulated by AKT activity, and the PGC1α expression is reduced at a later stage of the disease or in BRAF-mutant tumors. We observed a positive correlation between the PGC1α expression and oxygen consumption in thyroid cancer cells. PGC1α overexpression augmented oxidative metabolism as well as glycolysis and promoted colony formation and xenograft tumor growth. Silencing PGC1α induced a glycolytic phenotype and delayed the tumor growth. These findings suggest the metabolic phenotype is likely the consequence rather than the cause of disease progression in thyroid cancer.
Supplementary Material
Supplementary table.
Acknowledgements
This research was supported by research grants from the Ministry of Science and Technology of Taiwan (MOST-104-2314-B-195-004-MY3 and MOST-107-2314-B-195-001-MY3) and MacKay Memorial Hospital (MMH-10712, MMH-E-108-10, and MMH-10813).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Martinez-Outschoorn UE, Peiris-Pages M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol. 2017;14:11-31
2. Giovanella L, Trimboli P, Verburg FA, Treglia G, Piccardo A, Foppiani L, Ceriani L. Thyroglobulin levels and thyroglobulin doubling time independently predict a positive 18F-FDG PET/CT scan in patients with biochemical recurrence of differentiated thyroid carcinoma. Eur J Nucl Med Mol Imaging. 2013;40:874-80
3. Salvatori M, Biondi B, Rufini V. Imaging in endocrinology: 2-[18F]-fluoro-2-deoxy-D-glucose positron emission tomography/computed tomography in differentiated thyroid carcinoma: clinical indications and controversies in diagnosis and follow-up. Eur J Endocrinol. 2015;173:R115-30
4. Hsu YC, Liu CL, Yang PS, Tsai CH, Lee JJ, Cheng SP. Interaction of age at diagnosis with transcriptional profiling in papillary thyroid cancer. World J Surg. 2016;40:2922-9
5. Andreux PA, Houtkooper RH, Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov. 2013;12:465-83
6. Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell. 2016;166:555-66
7. Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015;282:647-72
8. Tan Z, Luo X, Xiao L, Tang M, Bode AM, Dong Z, Cao Y. The role of PGC1α in cancer metabolism and its therapeutic implications. Mol Cancer Ther. 2016;15:774-82
9. Cheng SP, Chen MJ, Chien MN, Lin CH, Lee JJ, Liu CL. Overexpression of teneurin transmembrane protein 1 is a potential marker of disease progression in papillary thyroid carcinoma. Clin Exp Med. 2017;17:555-64
10. Wang TY, Liu CL, Chen MJ, Lee JJ, Pun PC, Cheng SP. Expression of haem oxygenase-1 correlates with tumour aggressiveness and BRAF V600E expression in thyroid cancer. Histopathology. 2015;66:447-56
11. Wang CW, Lee YC, Calista E, Zhou F, Zhu H, Suzuki R, Komura D, Ishikawa S, Cheng SP. A benchmark for comparing precision medicine methods in thyroid cancer diagnosis using tissue microarrays. Bioinformatics. 2018;34:1767-73
12. Lee F, Yang PS, Chien MN, Lee JJ, Leung CH, Cheng SP. An increased neutrophil-to-lymphocyte ratio predicts incomplete response to therapy in differentiated thyroid cancer. Int J Med Sci. 2018;15:1757-63
13. Cheng SP, Chien MN, Wang TY, Lee JJ, Lee CC, Liu CL. Reconsideration of tumor size threshold for total thyroidectomy in differentiated thyroid cancer. Surgery. 2018;164:504-10
14. Liu CL, Yang PS, Chien MN, Chang YC, Lin CH, Cheng SP. Expression of serine peptidase inhibitor Kunitz type 1 in differentiated thyroid cancer. Histochem Cell Biol. 2018;149:635-44
15. Chien MN, Yang PS, Lee JJ, Wang TY, Hsu YC, Cheng SP. Recurrence-associated genes in papillary thyroid cancer: An analysis of data from The Cancer Genome Atlas. Surgery. 2017;161:1642-50
16. Chang YC, Chen CK, Chen MJ, Lin JC, Lin CH, Huang WC, Cheng SP, Chen SN, Liu CL. Expression of 3β-hydroxysteroid dehydrogenase type 1 in breast cancer is associated with poor prognosis independent of estrogen receptor status. Ann Surg Oncol. 2017;24:4033-41
17. Zhou J, Zhong X, Lin J, Hong Z. Qianliening capsule promotes mitochondrial pathway mediated the apoptosis of benign prostatic hyperplasia epithelial-1 cells by regulating the miRNA-181a. Int J Gerontol. 2018;12:244-50
18. Cheng SP, Liu CL, Chen MJ, Chien MN, Leung CH, Lin CH, Hsu YC, Lee JJ. CD74 expression and its therapeutic potential in thyroid carcinoma. Endocr Relat Cancer. 2015;22:179-90
19. Yang PS, Hsu YC, Lee JJ, Chen MJ, Huang SY, Cheng SP. Heme oxygenase-1 inhibitors induce cell cycle arrest and suppress tumor growth in thyroid cancer cells. Int J Mol Sci. 2018;19:2502
20. Lee JJ, Wang TY, Liu CL, Chien MN, Chen MJ, Hsu YC, Leung CH, Cheng SP. Dipeptidyl peptidase IV as a prognostic marker and therapeutic target in papillary thyroid carcinoma. J Clin Endocrinol Metab. 2017;102:2930-40
21. Chien MN, Yang PS, Hsu YC, Liu TP, Lee JJ, Cheng SP. Transcriptome analysis of papillary thyroid cancer harboring telomerase reverse transcriptase promoter mutation. Head Neck. 2018;40:2528-37
22. Ciavardelli D, Bellomo M, Consalvo A, Crescimanno C, Vella V. Metabolic alterations of thyroid cancer as potential therapeutic targets. Biomed Res Int. 2017;2017:2545031
23. Santhanam P, Khthir R, Solnes LB, Ladenson PW. The relationship of BRAFV600E mutation status to FDG PET/CT avidity in thyroid cancer: a review and meta-analysis. Endocr Pract. 2018;24:21-6
24. Antico Arciuch VG, Russo MA, Kang KS, Di Cristofano A. Inhibition of AMPK and Krebs cycle gene expression drives metabolic remodeling of Pten-deficient preneoplastic thyroid cells. Cancer Res. 2013;73:5459-72
25. Vidal AP, Andrade BM, Vaisman F, Cazarin J, Pinto LF, Breitenbach MM, Corbo R, Caroli-Bottino A, Soares F, Vaisman M, Carvalho DP. AMP-activated protein kinase signaling is upregulated in papillary thyroid cancer. Eur J Endocrinol. 2013;169:521-8
26. Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012-6
27. Saji M, Ringel MD. The PI3K-Akt-mTOR pathway in initiation and progression of thyroid tumors. Mol Cell Endocrinol. 2010;321:20-8
28. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13:184-99
29. Ma WW, Jacene H, Song D, Vilardell F, Messersmith WA, Laheru D, Wahl R, Endres C, Jimeno A, Pomper MG, Hidalgo M. [18F]fluorodeoxyglucose positron emission tomography correlates with Akt pathway activity but is not predictive of clinical outcome during mTOR inhibitor therapy. J Clin Oncol. 2009;27:2697-704
30. Mizuno T, Kamai T, Abe H, Sakamoto S, Kitajima K, Nishihara D, Yuki H, Kambara T, Betsunoh H, Yashi M, Fukabori Y, Kaji Y, Yoshida K. Clinically significant association between the maximum standardized uptake value on 18F-FDG PET and expression of phosphorylated Akt and S6 kinase for prediction of the biological characteristics of renal cell cancer. BMC Cancer. 2015;15:1097
Author contact
![]() Corresponding author: Shih-Ping Cheng, MD, PhD, Department of Surgery, MacKay Memorial Hospital, 92, Section 2, Chung-Shan North Road, Taipei 10449, Taiwan. Phone: +886 2 2543 3535; Fax: +886 2 2723 3897; E-mail: surg.mmhcom
Corresponding author: Shih-Ping Cheng, MD, PhD, Department of Surgery, MacKay Memorial Hospital, 92, Section 2, Chung-Shan North Road, Taipei 10449, Taiwan. Phone: +886 2 2543 3535; Fax: +886 2 2723 3897; E-mail: surg.mmhcom