Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2019; 10(20):4793-4806. doi:10.7150/jca.32475 This issue Cite
Review
Glioblastoma Treatment Modalities besides Surgery
Hao Zhang1*, Ruizhe Wang2*, Yuanqiang Yu1, Jinfang Liu1, Tianmeng Luo3, Fan Fan1,4 ![]()
1. Department of Neurosurgery, Xiangya Hospital, Central South University, Changsha, Hunan Province, China
2. Department of Urology, Xiangya Hospital, Central South University, Changsha, Hunan Province, China
3. Department of Medical Affairs, Xiangya Hospital, Central South University, Chang Sha, Hunan Province, China
4. Center for Medical Genetics & Hunan Provincial Key Laboratory of Medical Genetics, School of Life Sciences, Central South University Changsha, China
*Equal contribution
Received 2018-12-21; Accepted 2019-7-4; Published 2019-8-27
Abstract
Glioblastoma multiforme (GBM) is commonly known as the most aggressive primary CNS tumor in adults. The mean survival of it is 14 to 15 months, following the standard therapy from surgery, chemotherapy, to radiotherapy. Efforts in recent decades have brought many novel therapies to light, however, with limitations. In this paper, authors reviewed current treatments for GBM besides surgery. In the past decades, only radiotherapy, temozolomide (TMZ), and tumor treating field (TTF) were approved by FDA. Though promising in preclinical experiments, therapeutic effects of other novel treatments including BNCT, anti-angiogenic therapy, immunotherapy, epigenetic therapy, oncolytic virus therapy, and gene therapy are still either uncertain or discouraging in clinical results. In this review, we went through current clinical trials, underlying causes, and future therapy designs to present neurosurgeons and researchers a sketch of this field.
Keywords: glioblastoma, novel treatment, therapy
Introduction
Glioblastoma (GBM) is one of the most challenging tumors for physicians in oncology field [1-3]. While many researchers devoted to exploring potential treatments through its molecular mechanisms [4-8], surgery remains to be the first choice for tumor de-bulking and accessing tissue sample for pathology. As long as neurological functions not compromised, maximal tumor resection may be beneficial [9]. However, surgery alone is never enough. Because of the infiltrative nature of GBM, surgical resection alone leads to median survivals of only 3 to 6 months [10]. In the past 60 years, with the development of radiotherapy, postoperative survival has been significantly improved to approximately one year by adding adjuvant radiotherapy alone[11]. Currently, concomitant radiation and oral alkylating agent temozolomide (TMZ) extend survival to 14 to 16 months [12]. In this paper, we review current therapies other than surgery to provide a scheme for neurosurgeons and other researchers of interest (Table 1).
Comparison of different treatment strategies. ND=not determined. N/A=not applicable.
| Categories | Target molecule | Clinical trial phase (Latest) | Survival advantage (OS mo.) | Other outcomes (complications) | Indication for other cancers (FDA approved) | Ref. | |
|---|---|---|---|---|---|---|---|
| Chemotherapy | Temozolomide | DNA | FDA approved | 2.5 | Bone marrow suppression; nausea; emesis | Anaplastic astrocytoma | 12-14 |
| BCNU | DNA crosslink | FDA approved | 2.2 | Bone marrow suppression; nausea; emesis | Medulloblastoma; astrocytoma; multiple myeloma | 17-19 | |
| Lomustine | DNA crosslink | phase III | 16.7 | Bone marrow suppression; nausea; emesis | lymphoma; gastric cancer | 20-26 | |
| Cyclophosphamide | DNA | phase II | ND | Bone marrow suppression; nausea; emesis | lymphoma; multiple myeloma; leukemia | 27-31 | |
| Radiotherapy | N/A | DNA | FDA approved | 5.4-7.7 | Nausea; Emesis; Cognitive defect | Common type | 32-51 |
| TTF | N/A | Mitosis | FDA approved | 2.8 | topical skin rashes | ND | 52-59 |
| BNCT | N/A | GBM cell | phase II | 2.2 | ND | ND | 60-64 |
| Anti-angiogenic therapy | Bevacizumab | VEGF ligand | FDA approved | -0.4 | Hypertension; thromboembolic | Colorectal cancer; Lung cancer; Renal cell cancer | 65-71 |
| Nimotuzumab | EGFR | phase III | 5.2 | Chills; fever | Squamous carcinoma; Pancreatic cancer; Nasopharyngeal cancer | 72-75 | |
| ABT-414 | EGFR | phase I | ND | Blurred vision; Keratitis | ND | 76-77 | |
| Immunotherapy | Vaccination | EGFRvIII | phase III | 2 | ND | ND | 78-84 |
| Adoptive cell therapy | EGFRvIII | phase II | 5.9 | ND | ND | 85-86 | |
| check-point inhibition | PD-1 | phase II | 6.2 | Hypophysitis; encephalitis | Melanoma; lung/kidney cancer | 87-91 | |
| Immunostimulant | Immunity | phase II | 6.4 | toxicity | ND | 92-98 | |
| Epigenetic therapy | Vorinostat(deacetylase) | Histone | phase II | -1.44 | diarrhea | CTCL | 101-105 |
| VPA ( deacetylase) | phase II | 15 | Nausea; emesis | ND | 106-109 | ||
| Histone methyltransferase and demethylase | Histone | ND | ND | ND | ND | 114-115 | |
| Oncolytic virus therapy | G47Δ | GBM cell | phase II | ND | ND | ND | 116 |
| ZIKV | GBM cell | Pre-clinic | ND | ND | ND | 123 | |
| Oncolytic H-1 Parvovirus | GBM cell | phase II | ND | ND | ND | 124 | |
| Poliovirus | GBM cell | Phase I | ND | ND | ND | 125-130 | |
| Vaccinia | GBM cell | Pre-clinic | ND | ND | ND | 131-134 | |
| NDV | GBM cell | Pre-clinic | ND | ND | ND | 131-134 | |
| Gene therapy | Suicide gene therapy | gene | phase II | 0.4 | ND | ND | 138-144 |
| Tumor-suppressor Gene Therapy | gene | Pre-clinic | ND | ND | ND | 145-149 | |
| Immune-modulatory gene therapy | gene | Pre-clinic | ND | local inflammation | ND | 150-153 | |
| Affecting the tumor microenvironment | gene | Pre-clinic | ND | ND | ND | 154-157 |
1. Chemotherapy
1.1. Temozolomide
Temozolomide (TMZ) is an orally active alkylating agent approved by the United States Food and Drug Administration (FDA) to treat newly diagnosed GBM in March 2005. The therapeutic benefit of temozolomide depends on its ability to alkylate/methylate DNA, which most often occurs at the N-7 or O-6 positions of guanine residues (Fig. 1). Concomitant adjuvant TMZ chemotherapy and radiotherapy is the current standard of care for GBM patients [13]. In a large randomized trial, TMZ combined with radiation significantly improved (TMZ/RT →TMZ) median, 2- and 5- year survival for GBM patients with a median OS of 14.6 months (compared with 12.1 months using radiotherapy alone) [14]. As recommended in ESMO guideline, TMZ is administered daily (7 days a week) during radiotherapy for 5 days every 4 weeks for six cycles after radiation [13]. A Phase III Clinical Trial demonstrates no improved efficacy for DD (dose-dense) temozolomide (days 1 through 21 of a 28-day cycle) for newly diagnosed GBM (NCT00304031) [12].
Temozolomide, lomustine, carmustine, and cyclophosphamide inhibit the tumor growth by alkylating/methylating DNAs and impeding DNA crosslinking
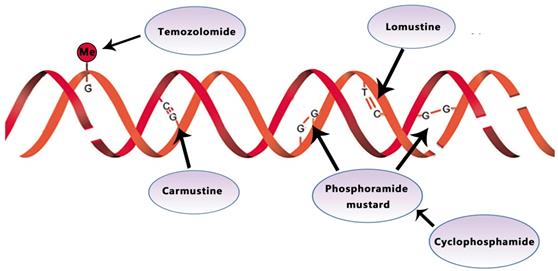
(O6-methylguanine-DNA methyl-transferase, MGMT), a DNA repair enzyme, plays a significant role in TMZ resistance. MGMT promoter methylation was found in approximately 45% of GBM. By silencing the gene on the epigenetic level, MGMT methylation decreased tumors' DNA repair capacity, increasing temozolomide susceptibility [15]. For patients without MGMT promoter methylation, O6-benzylguanine, another inhibitor of MGMT, and RNA interference-mediated MGMT silencing offer promising avenues to increase TMZ efficacy [16]. MGMT methylated tumors were associated with improved OS (21.2 v 14 months) and PFS (8.7 v 5.7 months) compared with un-methylated ones [12].
1.2. BCNU
BCNU (carmustine)-polymer wafers (Gliadel) were nitrosoureas approved by the FDA in 2002. As an alkylating agent, carmustine forms inter-strand crosslinks in DNAs to prevent DNAs from replication or transcription (Fig. 1). For newly diagnosed GBM, one recent research shows that Carmustine-impregnated wafers significantly improve survival [17]. However, Gliadel wafers also were reported to associate with increased cerebrospinal fluid (CSF) leakage and increased intracranial pressure due to brain edema, despite the moderate increase in median survival (13.8 vs. 11.6) [18]. In recurrent GBM, Jungk C proves that BCNU was rarely associated with severe side effects, particularly pulmonary toxicity and conferred favorable outcomes [19].
1.3. Lomustine
Lomustine is an alkylating nitrosoureas compound that influences DNA crosslinking and causes the methylation of the amino group (Fig. 1). It is particularly effective to treat tumors of the central nervous system due to its high lipid solubility to penetrate the BBB (blood-brain barrier) [20, 21].
The median OS after lomustine treatment is 9.1 months in primary glioblastoma [22] compared with the median OS of 7.5 months in recurrent glioblastoma [23] according to a phase II study (NCT01562197). In a recent phase III trial (NCT01149109), the median OS was significantly improved from 31.4 months with temozolomide alone to 48.1 months combining with lomustine-temozolomide [24].
Besides, lomustine was used as a comparator. In the first phase III trial (2005-004627-18) conducted in patients with recurrent glioblastoma, kinase C inhibitor Enzastaurin failed to demonstrate superior PFS to lomustine (6-month PFS 11.1% vs 19.0%) [25]. A subsequent phase III monotherapy trial in the same patient selection (2007-000383-24) also demonstrate few significant PFS improvement with oral cediranib, which supposedly functioned as pan-vascular endothelial growth factor (pan-VEGF) receptor tyrosine kinase inhibitor[26].
1.4. Cyclophosphamide
Cyclophosphamide (CPA) works against tumor through its metabolite phosphoramide mustard. This metabolite can alkylate DNA and form a cross connection to affect DNA function, thereby inhibiting the growth and reproduction of tumor cells and giving play to its anticancer effect (Fig. 1). In phase II studies of CPA, one with recurrent temozolomide-refractory glioblastoma showed 6-month PFS of 20%, the other with anaplastic astrocytoma showed 30% respectively[27, 28].
One recent research suggested that CPA improves survival in orthotopic GL261 GBM in mice by metronomic administration (every 6 days) [29], and similar results were then described by several other studies [30, 31]. So, more researches are guaranteed to futher evaluate the potential effect of CPA.
2. Radiotherapy
The post-surgery care standard for patients 70 years old or younger is partial-brain fractionated radiotherapy with concomitant TMZ. For better treatment, radiation should start as soon as safety assured. In clinical trials, researchers typically initiate radiation in 3 to 6 weeks after surgery [32]. Despite the infiltrative nature of GBM, partial-brain radiation, recommended in the guideline, leads to no worse survival than whole-brain radiotherapy.
For patients under 70 years old, optimal dose fractionation schedule for EBRT (external beam radiation) after surgery is 60 Gy in 2-Gy fractions administered over 6 weeks [32], while other plans have not provided more benefits. Two early reports demonstrated no benefit in treating GBM with EBRT when doses exceeded 60Gy in standard fractionation [33, 34]. As a result, most subsequent studies focused on doses equal to or less than 60Gy.
For patients over 70 years old, HFRT (hypofractionated radiotherapy) is recommended. Several recent studies suggested that the median OS of patients with HFRT was increased to 20 months, compared with conventional RT [35-40]. HFRT benefit patients in limiting tumor repopulation, increasing cell kill, and reducing the overall treatment time [41, 42].
A prospective study with 60 Gy delivered in 20 fractions demonstrated an OS of 9.5 months with a 1-year OS of 40 % and a PFS of 5.2 months under the condition that post-operative tumor volume <110 cm3, and acute toxicity in this study was limited to brain edema in 2 patients [43]. More recently, in a phase II trial(NCT01702610) TMZ was administered for 2 weeks before HFRT of 60 Gy in 20 fractions with concurrent and adjuvant TMZ. Results in this trial revealed that the median OS and PFS were 22.3 months and 13.7 months respectively [44]. Besides, The University of Colorado trials (NCT01209442) delivered a dose of 60 Gy in 10 fractions with concomitant and adjuvant TMZ and BEV and reported a median OS of 16.3 months [45] with RN (radio-necrosis) in 50% patients.
Other commonly used hypo-fractionations include 40Gy in 15 fractions and 25Gy in 5 fractions. However, 25Gy in 5 fractions showed non-inferiority to 40Gy in 15 fractions (7.9 vs 6.4 months) [46] in a phase III trial (NCT01450449), and 40Gy in 15 fractions presented non-inferiority to 60Gy in 30 fractions (5.6 vs. 5.1 months) [47].
Although HFRT is associated with RN, patients who developed RN had improved survival compared with those who did not [40, 45, 48], and their quality of life was not affected by RN [49]. Some studies supported that the use of Bevacizumab can potentially decrease the risk of RN [45, 50] or even treat RN [51].
In sum, the best practice of HFRT is still unclear, and further studies on HFRT are needed to better balance doses, fractions, RN, and life quality of patients.
3. Tumor treating field
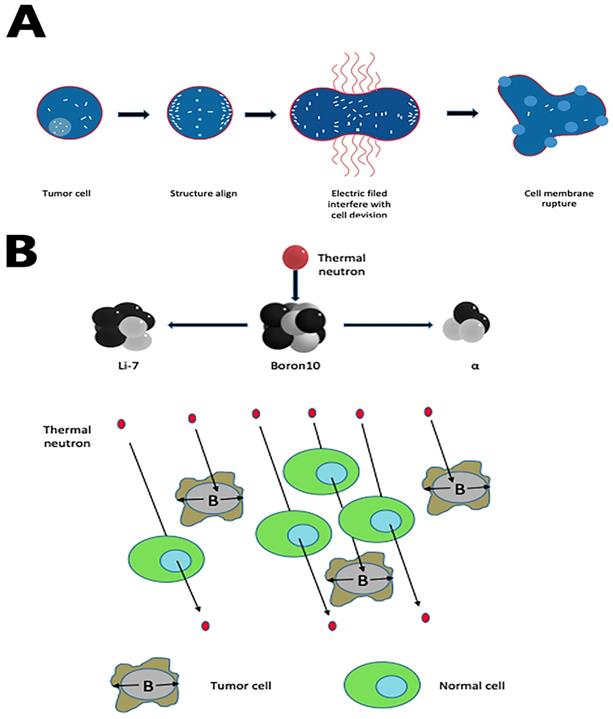
Tumor treating fields (TT-Fields) are a new modality to treat newly diagnosed and recurrent GBM. Optune, a portable medical device, uses low-intensity, intermediate- frequency, alternating electric fields (Tumor-treating filed, TTF) to disturb highly orchestrated dividing processes in GBM cells, sparing quiescent ones [52-54]. The mechanism of TT-Fields has two fundamental steps:(1) during the formation of mitotic spindles, the microtubule assembly deforms; mitosis of tumor cells remains in the interdivision stage for a long time [55]; (2) when cleavage furrow forms in mid to late mitosis, all polar molecules and dipoles in cells undergo di-electrophoresis under the action of TT-Fields, which accumulates in the cleavage furrow and eventually causes the cell membrane to rupture[54] (Fig. 2A).
(A) TTF rupture tumor cell membrane by accumulating all polar molecules and dipoles in the cleavage furrow during the interdivision stage. (B) Boron-10 in tumor cells, radiated with thermal neutrons, releases high linear energy transfer (LET) α and 7Li particles. Both alpha particles and the lithium ions produce closely spaced ionizations in the immediate vicinity of the reaction, leading to a selective tumor cell killing.
TTFs are generated via electrodes on the scalp with unique array placement based on individual's MRI results. Optune, or the NovoTTF-100A System, was approved by FDA to treat recurrent GBM in 2011 and for newly diagnosed GBM in October 2015 [56]. The decision was based on a 700 patient phase 3 clinical trial, in which patients treated with TMZ plus standard treatment was associated with a median OS and PFS of 19.4 and 7.1 months respectively, compared to 16.6 (p=0.0222) and 4.2 months (p=0.0010) for those had only standard treatment [57]. Those results were reported at ASCO 2015, and experts concluded that TT-Fields should be considered for GBM patients if not contraindicated.
Also, one recent phase 3 clinical trial (NCT00916409) demonstrates that health-related quality of life (HRQoL), cognitive, and functional status is not adversely affected by the continuous use of TT-Fields [58]. Median OS in the TT-Fields plus chemotherapy group was significantly longer versus chemotherapy alone (11.8 vs. 9.2 months)[59], supporting the therapeutic effect of TT-Fields.
4. Boron neutron capture therapy (BNCT)
Boron neutron capture therapy depends on the nuclear capture and fission reactions after boron-10 radiated with thermal neutrons from nuclear reactors. This reaction produces high linear energy transfer alpha particles (He-4) and lithium-7 since these alpha particles have a shorter path-length in tissues (5-9μm) than tumor cell diameters (about 10μm). In theory, if we use boron compound to deliver boron-10 precisely to tumor cells, this reaction can lead a selective tumor cell killing without damaging adjacent healthy cells (Fig. 2B). In the past decades, as the new boron compound boronophenylalanine (BPA) is designed, epithermal neutron beam introduced, and the proper computational system created, BNCT has evolved from intraoperative BNCT (IO-BNCT) to non-operative BNCT (NO-BNCT)[60]. The results from pre-clinic studies are prospecting. One recent study found a new dual-targeting strategy using c(RGDyC)-LP to improve BNCT for glioblastoma [61]. Yoshiya Iguchi et al. created a compound called BSH-3R, which can efficiently increase boron uptake in cells [62].
In a Japanese clinical trial (NCT00974987) of 23 patients with newly diagnosed GBM, BNCT alone after surgery provided a mean survival time of 19.5 months [63]. In a Swedish clinical trial of 29 newly diagnosed GBM patients, the mean survival time of BNCT alone group was 17.7 months compared to 15.5 months in the standard treatment group [64]. In both trials, no adjuvant TMZ therapy or radiotherapy was added to BNCT therapy; hence, researchers had initiated phase II clinical trials of combining radiation and TMZ with BNCT to treat newly diagnosed GBM patients. However, designing more tumor-selective boron compound and replacing nuclear reactors with particle accelerators remain challenging.
5. Anti-angiogenic therapy
5.1. Bevacizumab
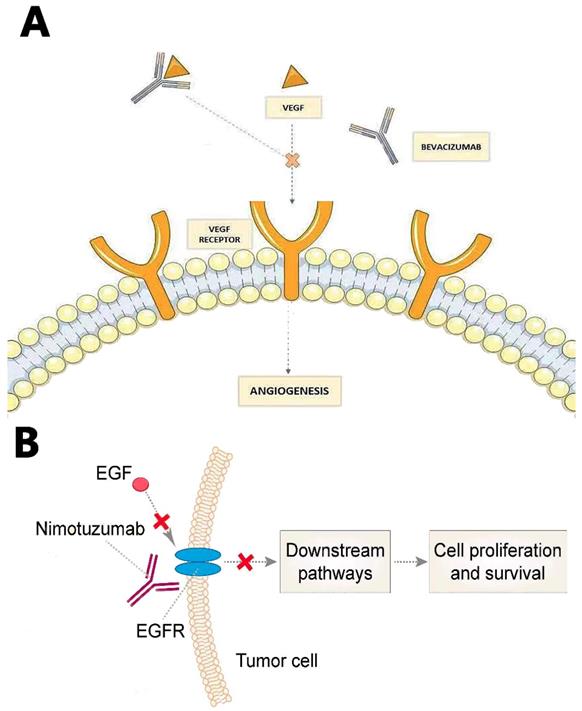
Recurrent GBMs were also treated with Bevacizumab, which is an IgG1 humanized monoclonal antibody that binds to VEGF ligand (Fig. 3A). It was approved by FDA in 2009 based on the success of two Phase II clinical trials [65, 66]. Bevacizumab inhibits VEGF to bind its receptor flt-1 and KDR on endothelial cells to reduce tumor angiogenesis and tumor growth by inactivating VEGF. Since angiogenesis is inhibited by Bevacizumab, BBB will be less disrupted. Therefore, patients may show excellent radio-graphics such as decreasing tumor enhancement and FLAIR (fast fluid-attenuated inversion recovery) hyper-intensity. However, most patients will have non-enhancing tumor progress after 3-5 months [67]. Some researchers believe Bevacizumab may increase the incidence of distant and diffuse tumor recurrence, turning GBM to a more aggressive phenotype; however, there is no convincing evidence on this debate [68].
(A) Bevacizumab prevents VEGF from binding its receptor on endothelial cells to reduce tumor angiogenesis and tumor growth. (B) Nimotuzumab binds to EGFR, blocking consequential downstream pathways to inhibit angiogenesie and GBM growth
In a recent clinical trial (NCT00884741), for newly diagnosed glioblastoma, duration of median OS between the Bevacizumab group and the placebo group is 15.7 and 16.1 months respectively, which showed no significant difference. Nevertheless, the median PFS was longer in the Bevacizumab group (10.7 months vs. 7.3 months) [69]. One recent research recommended the use of Bevacizumab to prolong PFS and OS in the recurrent setting either alone or in combination with a cytotoxic agent, yet not in the primary setting. Unfortunately, the absolute survival advantage is limited to 4 months [70].
For newly diagnosed glioblastoma, adding Bevacizumab to standard therapy does not improve overall survival and is associated with a higher chance of early adverse events such as hypertension, thromboembolic events, etc. [71]. Bevacizumab may be beneficial in prolonging progression-free survival, but still, Bevacizumab's routine addition to standard therapy for newly diagnosed GBM is not recommended in clinical practice [13].
5.2. Nimotuzumab
Nimotuzumab, Nimotuzumab is a humanized monoclonal antibody that binds to epidermal growth factor receptor (EGFR) and alters cell division. EGFR belongs to the ErbB family, which is related to many downstream pathways whose mutation or activation would facilitate angiogenesis and GBM growth [72] (Fig. 3B).
A phase II trial provided evidence of improved median OS in patients with high-grade glioma when treated with Nimotuzumab and RT compared to RT alone (17.8 vs12.6 months) [73]. But the results are not directly applicable to GBM patients because most patients in this trial have anaplastic astrocytoma other than GBM. A German phase III trial (NCT00753246) showed no significant PFS or OS improvement in GBM patients treated with standard chemo-radiation (temozolomide and RT) with or without nimotuzumab [74]. Interestingly, Nimotuzumab showed a trend of improved efficacy when administered to MGMT non-methylated GBM patients in both studies.
In a recent study conducted in Chinese patients, the median PFS and OS of Nimotuzumab in combination with TMZ and RT were 10.0 and 15.9 months, respectively, showing favorable safety and tolerability profiles in newly diagnosed GBM [75].
Whether Nimotuzumab should be used as front-line therapy was still debated, more researches are needed.
5.3. Depatuxizumab mafodotin
Depatuxizumab mafodotin (ABT-414) is monoclonal antibody-drug conjugate that binds to EGFR amplified on GBM and works against tumor through EGFR blockage and tubulin polymerization by conjugated tubulin inhibitor monomethyl auristatin F. Recently, ABT-414 demonstrated a median OS of 10.7 months in recurrent GBM patients [76]. Besides, a subsequent phase I study (NCT01800695) proved the efficacy and safety of ABT-414 and temozolomide in recurrent GBM patients with a median OS of 7.4 months [77]. Further studies are ongoing to evaluate its efficacy in newly diagnosed (NCT02573324) and recurrent glioblastoma (NCT02343406).
6. Immunotherapy
The brain is less immune privileged as researchers once thought; however, it is still immunologically specialized and provides tumors a sanctuary from systemic immunotherapy chemotherapy [78]. Current immunotherapy for brain tumors can be categorized into vaccination, adoptive cell therapy, checkpoint inhibition, and immunostimulant [79]. Open clinical trials of immunotherapy mainly focus on DC (dendritic cell) vaccination and antibodies aiming at checkpoint inhibitors, with promising, but not durable or sustainable clinical responses [80].
6.1. Vaccination
In 2008, PEP-3-KLH vaccine was first reported to induce newly diagnosed GBM patients to produce an EGFRvIII-specific antibody [81].EGFRvIII (type III epidermal growth factor receptor mutation), which is present in 24-67% of patients with GBM, allowing the continuous activation of EGFR [82].
Although some phase I/II clinical trials about PEP-3-KLH vaccine have encouraging results [83], the failure of phase III clinical trial (NCT02546102) investigating PEP-3-KLH vaccine set back the prospects for EGFRvIII-targeted peptide vaccine [84].
6.2. Adoptive cell therapy
Introducing chimeric antigen receptors (CARs) into T cells (CAR-T) to generate tumor-specific T cells, targeting an ideal marker EGFRvIII [85], has been the most widely-used ACT approach.
Phase I and II clinical trials have demonstrated significantly higher PFS and OS (26 months vs. 14.6 months) in vaccinated patients with EGFRvIII-expressing GBM tumors [86]. The accuracy of ACT has demonstrated great potential in treating GBM. Two clinical trials, (NCT02209376) and (NCT01454596) are underway, expected to have positive outcomes.
6.3. Check-point inhibition
The agents act by blocking the immunosuppressive check-points that inhibit cytotoxic T cells, leading to intensified anti-tumor immune responses. In this field, PD-1/PDL1 pathway is drawing most attention of researchers [87]. PD-1 reduces T-cell activity, inducing tolerance, and decreasing autoimmunity. One of its ligand, PD-L1, is highly expressed in GBM [88]. In a preclinical study using GL261 glioma mouse model, anti-PD-1 therapy combined with radiotherapy doubled median survival in 15-40% of mice compared with either treatment alone[89]. This promising strategy is recently supported by a randomized clinical trial, in which anti-PD-1 immunotherapy provided significant survival benefits (median OS=13.7 months, PFS=3.3months) [90].
Another pathway, CTLA-4, was also associated with improved survival when used to enhance tumor-lysate vaccines in GBM mouse model [91].
6.4. Immunostimulant
Though associated with significant CNS toxicity [92-94], IL-2, the best studied Th1 cytokine for GBM, has been frequently used in vitro to activate lymphokine-activated killer cells (LAK) with broad-spectrum antitumor effect. One encouraging report of IL-2 treatment in recurrent GBM was with the median OS of 12.2 months [95]. 75% 1-year survival in GBM [96] and 34% 1-year survival in recurrent GBM [97] shows the possible clinical benefits of using LAK stimulated by IL-2. One recently completed clinical trial (NCT01144247) of low dose IL-2 in the treatment of recurrent GBM has not been reported yet.
Although promising, immunotherapy can induce a high frequency of immune-related adverse effects that underlie the need for non-immunosuppressive and/or anti-inflammatory approaches [98].
7. Epigenetic therapy
Epigenetic changes regulate the cell phenotype through changes in gene expression without altering the DNA sequence [99]. Epigenetic drugs commonly target histone methyl-transferase, demethylases, and deacetylases to change transcriptomic profiles in order to treat tumors [100].
7.1. Histone deacetylase
Among kinds of epigenetic drugs against GBM, histone deacetylase (HDAC) inhibitors have drawn the most attention due to its broad mechanisms including cell-cycle arrest induction, differentiation, senescence, intrinsic and extrinsic apoptosis, mitotic cell death, autophagic cell death, inhibition of angiogenesis and metastasis, generation of reactive oxygen species, and enhancement in tumor immunity [101, 102]. Several preclinical studies revealed HDAC inhibitors' potential to radio-sensitize cancers, including GBM [103]. Currently, many clinical trials, mostly phase I and phase II are focusing on safety and efficacy profiles of HDAC inhibitors on GBM [104].
Vorinostat, the most advanced HDAC inhibitor, showed modest monotherapy activity (PFS=1.9 months, 6-month PFS=17%, median OS=5.7 months), while the combination of Vorinostat and other therapeutic agents is under research in multiple ongoing phase II trials [105]. Notably, one recently completed phase I/II trial (NCT01266031) demonstrated that the median OS of Vorinostat in combination with Bevacizumab was shorter than that of Bevacizumab monotherapy (7.80 vs. 9.24 months).
Also, valproic acid (VPA), well known as an antiepileptic drug, is also an HDAC inhibitor, exhibiting impressive preclinical efficacy to radio-sensitize glioma cells [106]. On the other hand, VPA also protects normal brain tissue and hippocampal neurons from radiotherapy [107]. A phase II trial of VPA, TMZ, and concurrent radiotherapy for GBM patients presented promising results (median OS=29.6 months in newly diagnosed GBM patients) [108]. Although promising, prospective data for VPA are still limited; to evaluate its efficacy and clarify optimal treating modality, further researches on VPA are in need[109].
Other two HDAC inhibitors, Panobinostat and Romidepsin, both were proved to inhibit proliferation of GBM cells in vitro and in animal studies [110, 111]. However, both of them showed disappointing results in phase II studies (NCT00859222 [112], NCT00085540 [113]).
7.2. Histone methyltransferase and demethylase
As for the other two targets, studies on histone methyltransferase and demethylases have not made great achievement on GBM. Histone demethylases play a significant role in various malignant tumors, while its role in GBM is unclear [114]. Azacytidine and Decitabine, two FDA-approved histone methyltransferase inhibitors [115], have not undergone clinically tests to evaluate its effect on GBM. Both histone methyltransferase and demethylases need to be illuminated in further details to develop as anti-GBM agents.
8. Oncolytic virus therapy
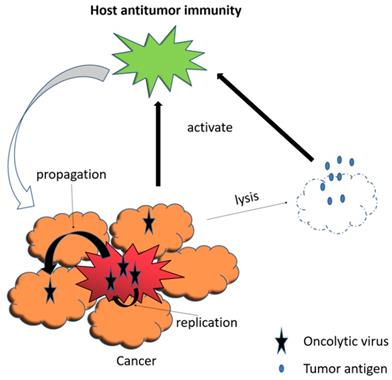
An oncolytic virus, naturally occurring or genetically engineered, can selectively replicate in and kill cancer cells while sparing the normal ones [116]. Viral infection and replication usually induce cellular stress, causing cell lysis [117, 118]. Besides, oncolytic viruses can infect tumor vessel endothelium and inhibit tumor-related angiogenesis, resulting in additional cell death of tumor due to the lack of oxygen and nutrients [117, 119]. It is worth to note that oncolytic virus therapy also remarkably induces systemic anti-tumor immunity as a result of the release of tumor-associated antigens, so as to prolong cancer patients' survival [120-122] (Fig. 4).
Viral infection and replication lyse tumor cells. The release of tumor-associated antigens induce systemic anti-tumor immunity.
So far, two genetically engineered oncolytic viruses have been approved to be drugs: Oncorine for head/neck/esophagus cancer in China and T-Vec for melanoma. G47Δ, developed by Todo et al., is a promising third generation oncolytic HSV-1 to be tested in patients with recurrent or residual GBM in a phase II study (UMIN000015995) started in 2015 Japan [116].
The prospecting effect of oncolytic virus therapy has led to studies on many other engineered oncolytic viruses, including reovirus, ZIKV, parvovirus, poliovirus, vaccinia, and NDV (Newcastle disease virus).
One recent study suggests that ZIKV, an oncolytic virus, can preferentially target GSCs (glioblastoma stem cells) [123], possessing potential efficacy for GBM patients.
In a First Phase I/IIa Glioblastoma Trial, Oncolytic H-1 Parvovirus treatment showed a PFS of 15.9 weeks and an OS of 15.4 months [124].
Poliovirus infects cancer cells via binding CD-155, a cell adhesion molecule widely expressed in GBM [125]. It causes the death of cancer cells and induces strong immunity against the tumor without severe side effects [126-129]. These encouraging characteristics of poliovirus have led to an ongoing Phase I trial (NCT01491893) for recurrent GBM patients. The results demonstrated that PVSRIPO (polio-rhinovirus chimera) immunotherapy significantly improved the survival rate at 24 and 36 months compared with that of historical controls, with two patients alive more than 69 months [130].
Vaccinia and NDV (Newcastle disease virus) both are cytotoxic to GBM cells in vitro and induced tumor regression in vivo [131-134]. However, they both lack satisfactory results from clinical trials.
Given the fact that oncolytic virus therapy is not yet an established approach to treat cancer, it may lead us to new paths if combined with immunotherapy or integrated with functional transgenes.
9. Gene therapy
Gene therapy is the treatment of disease through the introduction of therapeutic genes or manipulation of disease-related genes within target cells [135]. Current gene therapy strategies for GBM can be roughly categorized into suicide gene therapy (Fig. 5B), tumor-suppressor gene therapy (Fig. 5D), oncolytic viral gene therapy, immune-modulatory gene therapy (Fig. 5C), and affecting the tumor microenvironment [136, 137] (Fig. 5A). The corresponding delivery systems can be categorized into three basic types: direct delivery (virus-mediated and non-virus mediated), tumor-tropic cell carriers, and other carriers aiming at the unique physiochemical environment of tumors [137].
Gene therapy strategies. (A) Targeting the tumor microenvironment: viruses carry enzymes that degrade ECM components or anti-angiogenic factors that reduce vascular support of tumor. (B) GBM cells receive suicide genes from local injection of a carrier, together with systemic delivery of a prodrug. The suicide gene converts the prodrug into cytotoxic agents that kill the recipient cell and non-transduced bystander tumor cells. (C) The gene for an immunomodulatory cytokine is delivered to the tumor cells by viruses. Cytokine expression increases tumor cell apoptosis and activates immune cells such as macrophages, natural killer cells, and T-cell lymphocytes. (D) Tumor cells receive the functional copy of a tumor suppressor gene, which subsequently induces apoptosis.
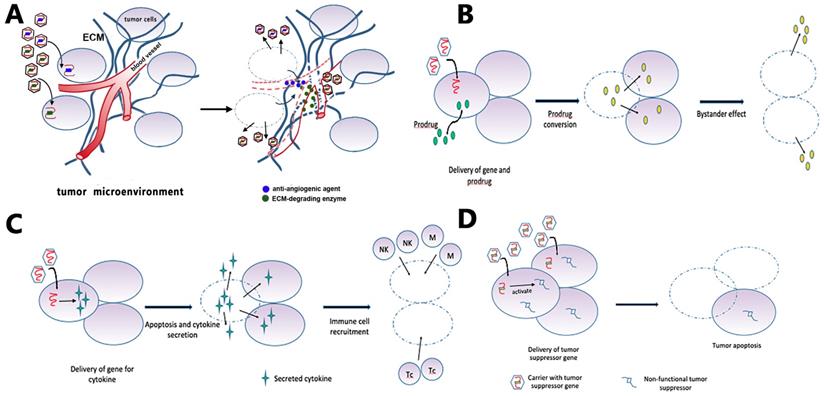
9.1. Suicide gene therapy
Two well-studied suicide gene therapies are HSV (herpes simplex virus)-derived enzyme Thymidine Kinase (HSV-TK) and bacterial enzyme Cytosine Deaminase (CDA) [138-140].HSV-TK has been proved safe in multiple phase I and II clinical trials [141-143]. However, the results from a phase III clinical trial for newly-diagnosed GBM patients were less exciting, with no significant improvements in PFS or OS [144].
CDA is currently under a phase I/II clinical trial (NCT01156584) for patients with recurrent high-grade glioma.
9.2. Tumor-suppressor Gene Therapy
p53 [145], p16 [146], p27 [147, 148], and Phosphatase and Tensin Homologue (PTEN) [149] were proved effective to inhibit the growth and invasion of GBM cells. However, there have not been any promising clinical trials for them.
9.3. Immune-modulatory gene therapy
One excellent example was the expression of the gene for IFN-beta (interferon beta) in mice. In these animal studies, IFN-β resulted in potent immune responses against tumor and improved animal survival [150]. A subsequent phase I trial examining IFN-β for recurrent malignant glioma demonstrated local inflammation and tumor necrosis [151]. Other investigations on cytokines such as IL-12 and TNF-alpha have also received exciting results in animals [152, 153].
9.4. Affecting the tumor microenvironment
Researchers employed anti-angiogenic genes or genes that remodel the tumor extracellular matrix to manipulate the tumor microenvironment. The intra-tumor injection of anti-angiogenic factor angiostatin in some studies effectively inhibited tumor vascularization and tumor growth [154-156]. Hence, anti-angiogenic gene therapy may hold future promise by affecting tumor microenvironment.
One study demonstrated increased efficacy of HSV against glioma when carried with chondroitinase ABC-I, a bacterial enzyme that degrades the glioma ECM (extracellular matrix). Surprisingly, degradation of ECM in this study did not lead to enhanced invasiveness of the remaining tumor cells [157].
Though promising in preclinical and phase I trials, anti-glioma gene therapies showed no significant benefits for patients in phase II and III trials [158-160]. Notably, the blood-brain barrier has impeded intracranial tumor treatments for decades, while intracranial injection of vectors to circumvent blood-brain barrier could be an answer [158]. Hence, besides exploring or optimizing gene therapy approaches, development of better viral or non-viral vectors to penetrate BBB, and precisely targeting tumor cells are both of great importance in order to treat GBM.
Discussion
After many years of research, Glioblastoma, hiding behind the blood-brain barrier, continues to be the most devastating brain tumor. Due to its invasive nature, surgery alone can never cut out the whole lesion. Currently, radiotherapy and TMZ chemotherapy followed by surgery is the clinical standard, providing a mean survival time only about 14 to 16 months, not to mention the poor living quality of GBM patients. Although there had been a variety of treatment strategies under research, only TMZ and tumor treatment field were approved by FDA in the past 15 years.
To date, novel treatments including BNCT, anti-angiogenic therapy, immunotherapy, epigenetic therapy, oncolytic virus therapy, and gene therapy are still having either uncertain or discouraging clinical results. Reasons for the lack of progress in GBM treatment are many folds. First, the blood-brain barrier impedes most blood-borne drugs to target tumor cells. Although GBM always disturbs this barrier due to its malignancy, not all BBB near tumor cells is broken enough for drugs to penetrate. Second, GBM stem cells are responsible for tumor resistance to radiotherapy, and hence possibly many other novel therapies. Third, most of these clinical results derive from trials on recurrent GBM patients, so it is possible that recurred tumors are more refractory to any treatment, and hence the unsatisfying clinical trial results. Therefore, we might see more encouraging results from novel therapies if we could recruit newly diagnosed GBM patients though subject to ethical review. Also, in each treatment modality lies their own challenges. For example, in BNCT the challenge is to design more effective tumor-targeting boron compound.
So far, no monotherapy for GBM is enough. Developing novel therapies and exploring new combinations of therapies are the most challenging missions for physicians and researchers. For instance, Lomustine-temozolomide significantly improves the median OS versus temozolomide alone (48.1 vs.31.4 months) [24]. Importantly, while exploring new combinations, a sound theoretical rationale should precede any random attempts.
Notably, many of the studies mentioned in this article have been conducted in vitro or in animals, requiring numerous subsequent clinical trials and tremendous effort to prove the feasibility and validity of studies. Though not satisfying to date, treatments for GBM shall have a brighter future with in-depth understanding of BBB, further understanding of tumor mechanisms, and the development of optimally combined treatment modalities.
Acknowledgements
Funding: We declare that there is no specific funding support.
Ethical approval: This article does not contain any studies with human participants or animals performed by any of the authors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Guo Y, Yan K, Fang J, Qu Q, Zhou M, Chen F. Let-7b expression determines response to chemotherapy through the regulation of cyclin D1 in glioblastoma. J Exp Clin Cancer Res. 2013;32:41
2. Zhang M, Song T, Yang L, Chen R, Wu L, Yang Z. et al. Nestin and CD133: valuable stem cell-specific markers for determining clinical outcome of glioma patients. J Exp Clin Cancer Res. 2008;27:85
3. Jiang YG, Peng Y, Koussougbo KS. Necroptosis: a novel therapeutic target for glioblastoma. Med Hypotheses. 2011;76:350-2
4. Liu L, Liu Z, Wang H, Chen L, Ruan F, Zhang J. et al. 14-3-3beta exerts glioma-promoting effects and is associated with malignant progression and poor prognosis in patients with glioma. Exp Ther Med. 2018;15:2381-7
5. Liu H, Liu Z, Jiang B, Huo L, Liu J, Lu J. Synthetic miR-145 Mimic Enhances the Cytotoxic Effect of the Antiangiogenic Drug Sunitinib in Glioblastoma. Cell Biochem Biophys. 2015;72:551-7
6. Zhao WH, Wu SQ, Zhang YD. Downregulation of miR-124 promotes the growth and invasiveness of glioblastoma cells involving upregulation of PPP1R13L. Int J Mol Med. 2013;32:101-7
7. Huang BS, Luo QZ, Han Y, Li XB, Cao LJ, Wu LX. microRNA-223 promotes the growth and invasion of glioblastoma cells by targeting tumor suppressor PAX6. Oncol Rep. 2013;30:2263-9
8. Li Q, Cheng Q, Chen Z, Peng R, Chen R, Ma Z. et al. MicroRNA-663 inhibits the proliferation, migration and invasion of glioblastoma cells via targeting TGF-beta1. Oncol Rep. 2016;35:1125-34
9. Lacroix M, Abi-Said D, Fourney DR, Gokaslan ZL, Shi W, DeMonte F. et al. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg. 2001;95:190-8
10. Davis FG, Freels S, Grutsch J, Barlas S, Brem S. Survival rates in patients with primary malignant brain tumors stratified by patient age and tumor histological type: an analysis based on Surveillance, Epidemiology, and End Results (SEER) data, 1973-1991. J Neurosurg. 1998;88:1-10
11. Walker MD, Strike TA, Sheline GE. An analysis of dose-effect relationship in the radiotherapy of malignant gliomas. Int J Radiat Oncol Biol Phys. 1979;5:1725-31
12. Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA. et al. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 2013;31:4085-91
13. Stupp R, Brada M, van den Bent MJ, Tonn JC, Pentheroudakis G, Group EGW. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):iii93-101
14. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-96
15. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M. et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997-1003
16. Warren KE, Gururangan S, Geyer JR, McLendon RE, Poussaint TY, Wallace D. et al. A phase II study of O6-benzylguanine and temozolomide in pediatric patients with recurrent or progressive high-grade gliomas and brainstem gliomas: a Pediatric Brain Tumor Consortium study. J Neurooncol. 2012;106:643-9
17. Xing WK, Shao C, Qi ZY, Yang C, Wang Z. The role of Gliadel wafers in the treatment of newly diagnosed GBM: a meta-analysis. Drug Des Devel Ther. 2015;9:3341-8
18. Westphal M, Ram Z, Riddle V, Hilt D, Bortey E, Executive Committee of the Gliadel Study G. Gliadel wafer in initial surgery for malignant glioma: long-term follow-up of a multicenter controlled trial. Acta Neurochir (Wien). 2006;148:269-75 discussion 75
19. Jungk C, Chatziaslanidou D, Ahmadi R, Capper D, Bermejo JL, Exner J. et al. Chemotherapy with BCNU in recurrent glioma: Analysis of clinical outcome and side effects in chemotherapy-naive patients. BMC Cancer. 2016;16:81
20. Cheng CJ, Fujimura S, Grunberger D, Weinstein IB. Interaction of 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea (NSC 79037) with nucleic acids and proteins in vivo and in vitro. Cancer Res. 1972;32:22-7
21. Baer JC, Freeman AA, Newlands ES, Watson AJ, Rafferty JA, Margison GP. Depletion of O6-alkylguanine-DNA alkyltransferase correlates with potentiation of temozolomide and CCNU toxicity in human tumour cells. Br J Cancer. 1993;67:1299-302
22. Wick W, Gorlia T, Bendszus M, Taphoorn M, Sahm F, Harting I. et al. Lomustine and Bevacizumab in Progressive Glioblastoma. The New England journal of medicine. 2017;377:1954-63
23. Brandes AA, Carpentier AF, Kesari S, Sepulveda-Sanchez JM, Wheeler HR, Chinot O. et al. A Phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 2016;18:1146-56
24. Herrlinger U, Tzaridis T, Mack F, Steinbach JP, Schlegel U, Sabel M. et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): a randomised, open-label, phase 3 trial. Lancet (London, England). 2019;393:678-88
25. Wick W, Puduvalli VK, Chamberlain MC, van den Bent MJ, Carpentier AF, Cher LM. et al. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol. 2010;28:1168-74
26. Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A. et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31:3212-8
27. Chamberlain MC, Tsao-Wei DD. Salvage chemotherapy with cyclophosphamide for recurrent, temozolomide-refractory glioblastoma multiforme. Cancer. 2004;100:1213-20
28. Chamberlain MC, Tsao-Wei DD, Groshen S. Salvage chemotherapy with cyclophosphamide for recurrent temozolomide-refractory anaplastic astrocytoma. Cancer. 2006;106:172-9
29. Ferrer-Font L, Arias-Ramos N, Lope-Piedrafita S, Julia-Sape M, Pumarola M, Arus C. et al. Metronomic treatment in immunocompetent preclinical GL261 glioblastoma: effects of cyclophosphamide and temozolomide. NMR Biomed. 2017:30
30. Hanahan D, Bergers G, Bergsland E. Less is more, regularly: metronomic dosing of cytotoxic drugs can target tumor angiogenesis in mice. J Clin Invest. 2000;105:1045-7
31. Gnoni A, Silvestris N, Licchetta A, Santini D, Scartozzi M, Ria R. et al. Metronomic chemotherapy from rationale to clinical studies: a dream or reality? Crit Rev Oncol Hematol. 2015;95:46-61
32. Sulman EP, Ismaila N, Armstrong TS, Tsien C, Batchelor TT, Cloughesy T. et al. Radiation Therapy for Glioblastoma: American Society of Clinical Oncology Clinical Practice Guideline Endorsement of the American Society for Radiation Oncology Guideline. J Clin Oncol. 2017;35:361-9
33. Chang CH, Horton J, Schoenfeld D, Salazer O, Perez-Tamayo R, Kramer S. et al. Comparison of postoperative radiotherapy and combined postoperative radiotherapy and chemotherapy in the multidisciplinary management of malignant gliomas. A joint Radiation Therapy Oncology Group and Eastern Cooperative Oncology Group study. Cancer. 1983;52:997-1007
34. Salazar OM, Rubin P, Feldstein ML, Pizzutiello R. High dose radiation therapy in the treatment of malignant gliomas: final report. Int J Radiat Oncol Biol Phys. 1979;5:1733-40
35. Ciammella P, Galeandro M, D'Abbiero N, Podgornii A, Pisanello A, Botti A. et al. Hypo-fractionated IMRT for patients with newly diagnosed glioblastoma multiforme: a 6 year single institutional experience. Clin Neurol Neurosurg. 2013;115:1609-14
36. Reddy K, Damek D, Gaspar LE, Ney D, Waziri A, Lillehei K. et al. Phase II trial of hypofractionated IMRT with temozolomide for patients with newly diagnosed glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2012;84:655-60
37. Floyd NS, Woo SY, Teh BS, Prado C, Mai WY, Trask T. et al. Hypofractionated intensity-modulated radiotherapy for primary glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2004;58:721-6
38. Jastaniyah N, Murtha A, Pervez N, Le D, Roa W, Patel S. et al. Phase I study of hypofractionated intensity modulated radiation therapy with concurrent and adjuvant temozolomide in patients with glioblastoma multiforme. Radiat Oncol. 2013;8:38
39. Chen C, Damek D, Gaspar LE, Waziri A, Lillehei K, Kleinschmidt-DeMasters BK. et al. Phase I trial of hypofractionated intensity-modulated radiotherapy with temozolomide chemotherapy for patients with newly diagnosed glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 2011;81:1066-74
40. Iuchi T, Hatano K, Kodama T, Sakaida T, Yokoi S, Kawasaki K. et al. Phase 2 trial of hypofractionated high-dose intensity modulated radiation therapy with concurrent and adjuvant temozolomide for newly diagnosed glioblastoma. Int J Radiat Oncol Biol Phys. 2014;88:793-800
41. Budach W, Gioioso D, Taghian A, Stuschke M, Suit HD. Repopulation capacity during fractionated irradiation of squamous cell carcinomas and glioblastomas in vitro. Int J Radiat Oncol Biol Phys. 1997;39:743-50
42. Hingorani M, Colley WP, Dixit S, Beavis AM. Hypofractionated radiotherapy for glioblastoma: strategy for poor-risk patients or hope for the future? Br J Radiol. 2012;85:e770-81
43. Sultanem K, Patrocinio H, Lambert C, Corns R, Leblanc R, Parker W. et al. The use of hypofractionated intensity-modulated irradiation in the treatment of glioblastoma multiforme: preliminary results of a prospective trial. Int J Radiat Oncol Biol Phys. 2004;58:247-52
44. Shenouda G, Souhami L, Petrecca K, Owen S, Panet-Raymond V, Guiot MC. et al. A Phase 2 Trial of Neoadjuvant Temozolomide Followed by Hypofractionated Accelerated Radiation Therapy With Concurrent and Adjuvant Temozolomide for Patients With Glioblastoma. Int J Radiat Oncol Biol Phys. 2017;97:487-94
45. Ney DE, Carlson JA, Damek DM, Gaspar LE, Kavanagh BD, Kleinschmidt-DeMasters BK. et al. Phase II trial of hypofractionated intensity-modulated radiation therapy combined with temozolomide and bevacizumab for patients with newly diagnosed glioblastoma. J Neurooncol. 2015;122:135-43
46. Roa W, Kepka L, Kumar N, Sinaika V, Matiello J, Lomidze D. et al. International Atomic Energy Agency Randomized Phase III Study of Radiation Therapy in Elderly and/or Frail Patients With Newly Diagnosed Glioblastoma Multiforme. J Clin Oncol. 2015;33:4145-50
47. Terasaki M, Eto T, Nakashima S, Okada Y, Ogo E, Sugita Y. et al. A pilot study of hypofractionated radiation therapy with temozolomide for adults with glioblastoma multiforme. J Neurooncol. 2011;102:247-53
48. Azoulay M, Ho CK, Fujimoto DK, Modlin LA, Gibbs IC, Hancock SL. et al. A Phase I/II Trial of 5 Fraction Stereotactic Radiosurgery With 5-mm Margins With Concurrent and Adjuvant Temozolomide in Newly Diagnosed Supratentorial Glioblastoma Multiforme. Int J Radiat Oncol Biol Phys. 2016;96:E131-E2
49. Pollom EL, Fujimoto D, Wynne J, Seiger K, Modlin LA, Jacobs LR. et al. Phase 1/2 Trial of 5-Fraction Stereotactic Radiosurgery With 5-mm Margins With Concurrent and Adjuvant Temozolomide in Newly Diagnosed Supratentorial Glioblastoma: Health-Related Quality of Life Results. Int J Radiat Oncol Biol Phys. 2017;98:123-30
50. Omuro A, Beal K, Gutin P, Karimi S, Correa DD, Kaley TJ. et al. Phase II study of bevacizumab, temozolomide, and hypofractionated stereotactic radiotherapy for newly diagnosed glioblastoma. Clin Cancer Res. 2014;20:5023-31
51. Levin VA, Bidaut L, Hou P, Kumar AJ, Wefel JS, Bekele BN. et al. Randomized double-blind placebo-controlled trial of bevacizumab therapy for radiation necrosis of the central nervous system. Int J Radiat Oncol Biol Phys. 2011;79:1487-95
52. Rehman AA, Elmore KB, Mattei TA. The effects of alternating electric fields in glioblastoma: current evidence on therapeutic mechanisms and clinical outcomes. Neurosurg Focus. 2015;38:E14
53. Trusheim J, Dunbar E, Battiste J, Iwamoto F, Mohile N, Damek D. et al. A state-of-the-art review and guidelines for tumor treating fields treatment planning and patient follow-up in glioblastoma. CNS Oncol. 2017;6:29-43
54. Kirson ED, Dbaly V, Tovarys F, Vymazal J, Soustiel JF, Itzhaki A. et al. Alternating electric fields arrest cell proliferation in animal tumor models and human brain tumors. Proc Natl Acad Sci U S A. 2007;104:10152-7
55. Kirson ED, Gurvich Z, Schneiderman R, Dekel E, Itzhaki A, Wasserman Y. et al. Disruption of cancer cell replication by alternating electric fields. Cancer Res. 2004;64:3288-95
56. Mehta M, Wen P, Nishikawa R, Reardon D, Peters K. Critical review of the addition of tumor treating fields (TTFields) to the existing standard of care for newly diagnosed glioblastoma patients. Crit Rev Oncol Hematol. 2017;111:60-5
57. Stupp R, Taillibert S, Kanner AA, Kesari S, Steinberg DM, Toms SA. et al. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA. 2015;314:2535-43
58. Zhu JJ, Demireva P, Kanner AA, Pannullo S, Mehdorn M, Avgeropoulos N. et al. Health-related quality of life, cognitive screening, and functional status in a randomized phase III trial (EF-14) of tumor treating fields with temozolomide compared to temozolomide alone in newly diagnosed glioblastoma. J Neurooncol. 2017;135:545-52
59. Kesari S, Ram Z, Investigators EFT. Tumor-treating fields plus chemotherapy versus chemotherapy alone for glioblastoma at first recurrence: a post hoc analysis of the EF-14 trial. CNS Oncol. 2017;6:185-93
60. Kageji T, Nagahiro S, Mizobuchi Y, Matsuzaki K, Nakagawa Y, Kumada H. Boron neutron capture therapy (BNCT) for newly-diagnosed glioblastoma: comparison of clinical results obtained with BNCT and conventional treatment. J Med Invest. 2014;61:254-63
61. Kang W, Svirskis D, Sarojini V, McGregor AL, Bevitt J, Wu Z. Cyclic-RGDyC functionalized liposomes for dual-targeting of tumor vasculature and cancer cells in glioblastoma: An in vitro boron neutron capture therapy study. Oncotarget. 2017;8:36614-27
62. Iguchi Y, Michiue H, Kitamatsu M, Hayashi Y, Takenaka F, Nishiki T. et al. Tumor-specific delivery of BSH-3R for boron neutron capture therapy and positron emission tomography imaging in a mouse brain tumor model. Biomaterials. 2015;56:10-7
63. Kawabata S, Miyatake S, Nonoguchi N, Hiramatsu R, Iida K, Miyata S. et al. Survival benefit from boron neutron capture therapy for the newly diagnosed glioblastoma patients. Appl Radiat Isot. 2009;67:S15-8
64. Henriksson R, Capala J, Michanek A, Lindahl SA, Salford LG, Franzen L. et al. Boron neutron capture therapy (BNCT) for glioblastoma multiforme: a phase II study evaluating a prolonged high-dose of boronophenylalanine (BPA). Radiother Oncol. 2008;88:183-91
65. Kreisl TN, Kim L, Moore K, Duic P, Royce C, Stroud I. et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J Clin Oncol. 2009;27:740-5
66. Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE. et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27:4733-40
67. Pope WB, Xia Q, Paton VE, Das A, Hambleton J, Kim HJ. et al. Patterns of progression in patients with recurrent glioblastoma treated with bevacizumab. Neurology. 2011;76:432-7
68. Nowosielski M, Wiestler B, Goebel G, Hutterer M, Schlemmer HP, Stockhammer G. et al. Progression types after antiangiogenic therapy are related to outcome in recurrent glioblastoma. Neurology. 2014;82:1684-92
69. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA. et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. The New England journal of medicine. 2014;370:699-708
70. Diaz RJ, Ali S, Qadir MG, De La Fuente MI, Ivan ME, Komotar RJ. The role of bevacizumab in the treatment of glioblastoma. J Neurooncol. 2017;133:455-67
71. Poulsen HS, Urup T, Michaelsen SR, Staberg M, Villingshoj M, Lassen U. The impact of bevacizumab treatment on survival and quality of life in newly diagnosed glioblastoma patients. Cancer Manag Res. 2014;6:373-87
72. Bai H, Harmanci AS, Erson-Omay EZ, Li J, Coskun S, Simon M. et al. Integrated genomic characterization of IDH1-mutant glioma malignant progression. Nat Genet. 2016;48:59-66
73. Solomon MT, Selva JC, Figueredo J, Vaquer J, Toledo C, Quintanal N. et al. Radiotherapy plus nimotuzumab or placebo in the treatment of high grade glioma patients: results from a randomized, double blind trial. BMC Cancer. 2013;13:299
74. Westphal M, Heese O, Steinbach JP, Schnell O, Schackert G, Mehdorn M. et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. Eur J Cancer. 2015;51:522-32
75. Wang Y, Pan L, Sheng XF, Chen S, Dai JZ. Nimotuzumab, a humanized monoclonal antibody specific for the EGFR, in combination with temozolomide and radiation therapy for newly diagnosed glioblastoma multiforme: First results in Chinese patients. Asia Pac J Clin Oncol. 2016;12:e23-9
76. Gan HK, Reardon DA, Lassman AB, Merrell R, van den Bent M, Butowski N. et al. Safety, pharmacokinetics, and antitumor response of depatuxizumab mafodotin as monotherapy or in combination with temozolomide in patients with glioblastoma. Neuro Oncol. 2018;20:838-47
77. Lassman AB, van den Bent MJ, Gan HK, Reardon DA, Kumthekar P, Butowski N. et al. Safety and efficacy of depatuxizumab mafodotin + temozolomide in patients with EGFR-amplified, recurrent glioblastoma: results from an international phase I multicenter trial. Neuro Oncol. 2019;21:106-14
78. Mitchell MS. Relapse in the central nervous system in melanoma patients successfully treated with biomodulators. J Clin Oncol. 1989;7:1701-9
79. McGranahan T, Li G, Nagpal S. History and current state of immunotherapy in glioma and brain metastasis. Ther Adv Med Oncol. 2017;9:347-68
80. Huang B, Zhang H, Gu L, Ye B, Jian Z, Stary C. et al. Advances in Immunotherapy for Glioblastoma Multiforme. J Immunol Res. 2017;2017:3597613
81. Schmittling RJ, Archer GE, Mitchell DA, Heimberger A, Pegram C, Herndon JE 2nd. et al. Detection of humoral response in patients with glioblastoma receiving EGFRvIII-KLH vaccines. J Immunol Methods. 2008;339:74-81
82. Heimberger AB, Suki D, Yang D, Shi W, Aldape K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J Transl Med. 2005;3:38
83. Kong Z, Wang Y, Ma W. Vaccination in the immunotherapy of glioblastoma. Hum Vaccin Immunother. 2018;14:255-68
84. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H. et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18:1373-85
85. Del Vecchio CA, Li G, Wong AJ. Targeting EGF receptor variant III: tumor-specific peptide vaccination for malignant gliomas. Expert Rev Vaccines. 2012;11:133-44
86. Swartz AM, Li QJ, Sampson JH. Rindopepimut: a promising immunotherapeutic for the treatment of glioblastoma multiforme. Immunotherapy. 2014;6:679-90
87. Huang J, Liu F, Liu Z, Tang H, Wu H, Gong Q. et al. Immune Checkpoint in Glioblastoma: Promising and Challenging. Front Pharmacol. 2017;8:242
88. Berghoff AS, Kiesel B, Widhalm G, Rajky O, Ricken G, Wohrer A. et al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. 2015;17:1064-75
89. Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J. et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343-9
90. Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB. et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25:477-86
91. Agarwalla P, Barnard Z, Fecci P, Dranoff G, Curry WT Jr. Sequential immunotherapy by vaccination with GM-CSF-expressing glioma cells and CTLA-4 blockade effectively treats established murine intracranial tumors. J Immunother. 2012;35:385-9
92. Meyers CA, Yung WK. Delayed neurotoxicity of intraventricular interleukin-2: a case report. J Neurooncol. 1993;15:265-7
93. Vecht CJ, Keohane C, Menon RS, Punt CJ, Stoter G. Acute fatal leukoencephalopathy after interleukin-2 therapy. N Engl J Med. 1990;323:1146-7
94. Samlowski WE, Park KJ, Galinsky RE, Ward JH, Schumann GB. Intrathecal administration of interleukin-2 for meningeal carcinomatosis due to malignant melanoma: sequential evaluation of intracranial pressure, cerebrospinal fluid cytology, and cytokine induction. J Immunother Emphasis Tumor Immunol. 1993;13:49-54
95. Hayes RL, Koslow M, Hiesiger EM, Hymes KB, Hochster HS, Moore EJ. et al. Improved long term survival after intracavitary interleukin-2 and lymphokine-activated killer cells for adults with recurrent malignant glioma. Cancer. 1995;76:840-52
96. Dillman RO, Duma CM, Ellis RA, Cornforth AN, Schiltz PM, Sharp SL. et al. Intralesional lymphokine-activated killer cells as adjuvant therapy for primary glioblastoma. J Immunother. 2009;32:914-9
97. Dillman RO, Duma CM, Schiltz PM, DePriest C, Ellis RA, Okamoto K. et al. Intracavitary placement of autologous lymphokine-activated killer (LAK) cells after resection of recurrent glioblastoma. J Immunother. 2004;27:398-404
98. Gelao L, Criscitiello C, Esposito A, Goldhirsch A, Curigliano G. Immune checkpoint blockade in cancer treatment: a double-edged sword cross-targeting the host as an "innocent bystander". Toxins (Basel). 2014;6:914-33
99. Shabason JE, Tofilon PJ, Camphausen K. Grand rounds at the National Institutes of Health: HDAC inhibitors as radiation modifiers, from bench to clinic. J Cell Mol Med. 2011;15:2735-44
100. Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069-78
101. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673-91
102. Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009;107:600-8
103. Diss E, Nalabothula N, Nguyen D, Chang E, Kwok Y, Carrier F. VorinostatSAHA Promotes Hyper-Radiosensitivity in Wild Type p53 Human Glioblastoma Cells. J Clin Oncol Res. 2014:2
104. Lee DH, Ryu HW, Won HR, Kwon SH. Advances in epigenetic glioblastoma therapy. Oncotarget. 2017;8:18577-89
105. Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS. et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27:2052-8
106. Hosein AN, Lim YC, Day B, Stringer B, Rose S, Head R. et al. The effect of valproic acid in combination with irradiation and temozolomide on primary human glioblastoma cells. J Neurooncol. 2015;122:263-71
107. Zhou Y, Niu J, Li S, Hou H, Xu Y, Zhang W. et al. Radioprotective effects of valproic acid, a histone deacetylase inhibitor, in the rat brain. Biomed Rep. 2015;3:63-9
108. Krauze AV, Myrehaug SD, Chang MG, Holdford DJ, Smith S, Shih J. et al. A Phase 2 Study of Concurrent Radiation Therapy, Temozolomide, and the Histone Deacetylase Inhibitor Valproic Acid for Patients With Glioblastoma. Int J Radiat Oncol Biol Phys. 2015;92:986-92
109. Ochiai S, Nomoto Y, Yamashita Y, Watanabe Y, Toyomasu Y, Kawamura T. et al. Roles of Valproic Acid in Improving Radiation Therapy for Glioblastoma: a Review of Literature Focusing on Clinical Evidence. Asian Pac J Cancer Prev. 2016;17:463-6
110. Yao ZG, Li WH, Hua F, Cheng HX, Zhao MQ, Sun XC. et al. LBH589 Inhibits Glioblastoma Growth and Angiogenesis Through Suppression of HIF-1alpha Expression. J Neuropathol Exp Neurol. 2017;76:1000-7
111. Sawa H, Murakami H, Kumagai M, Nakasato M, Yamauchi S, Matsuyama N. et al. Histone deacetylase inhibitor, FK228, induces apoptosis and suppresses cell proliferation of human glioblastoma cells in vitro and in vivo. Acta Neuropathol. 2004;107:523-31
112. Lee EQ, Reardon DA, Schiff D, Drappatz J, Muzikansky A, Grimm SA. et al. Phase II study of panobinostat in combination with bevacizumab for recurrent glioblastoma and anaplastic glioma. Neuro Oncol. 2015;17:862-7
113. Iwamoto FM, Lamborn KR, Kuhn JG, Wen PY, Yung WK, Gilbert MR. et al. A phase I/II trial of the histone deacetylase inhibitor romidepsin for adults with recurrent malignant glioma: North American Brain Tumor Consortium Study 03-03. Neuro Oncol. 2011;13:509-16
114. Hoffmann I, Roatsch M, Schmitt ML, Carlino L, Pippel M, Sippl W. et al. The role of histone demethylases in cancer therapy. Mol Oncol. 2012;6:683-703
115. Derissen EJ, Beijnen JH, Schellens JH. Concise drug review: azacitidine and decitabine. Oncologist. 2013;18:619-24
116. Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373-9
117. Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y. et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. 2013;12:103
118. Cary ZD, Willingham MC, Lyles DS. Oncolytic vesicular stomatitis virus induces apoptosis in U87 glioblastoma cells by a type II death receptor mechanism and induces cell death and tumor clearance in vivo. J Virol. 2011;85:5708-17
119. Breitbach CJ, Paterson JM, Lemay CG, Falls TJ, McGuire A, Parato KA. et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol Ther. 2007;15:1686-93
120. Todo T, Rabkin SD, Sundaresan P, Wu A, Meehan KR, Herscowitz HB. et al. Systemic antitumor immunity in experimental brain tumor therapy using a multimutated, replication-competent herpes simplex virus. Hum Gene Ther. 1999;10:2741-55
121. Guo ZS, Liu Z, Bartlett DL. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immunity. Front Oncol. 2014;4:74
122. Workenhe ST, Mossman KL. Oncolytic virotherapy and immunogenic cancer cell death: sharpening the sword for improved cancer treatment strategies. Mol Ther. 2014;22:251-6
123. Lubin JA, Zhang RR, Kuo JS. Zika Virus has Oncolytic Activity Against Glioblastoma Stem Cells. Neurosurgery. 2018;82:E113-E4
124. Geletneky K, Hajda J, Angelova AL, Leuchs B, Capper D, Bartsch AJ. et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol Ther. 2017;25:2620-34
125. Merrill MK, Bernhardt G, Sampson JH, Wikstrand CJ, Bigner DD, Gromeier M. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro Oncol. 2004;6:208-17
126. Goetz C, Gromeier M. Preparing an oncolytic poliovirus recombinant for clinical application against glioblastoma multiforme. Cytokine Growth Factor Rev. 2010;21:197-203
127. Brown MC, Dobrikova EY, Dobrikov MI, Walton RW, Gemberling SL, Nair SK. et al. Oncolytic polio virotherapy of cancer. Cancer. 2014;120:3277-86
128. Gromeier M, Lachmann S, Rosenfeld MR, Gutin PH, Wimmer E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc Natl Acad Sci U S A. 2000;97:6803-8
129. Dobrikova EY, Broadt T, Poiley-Nelson J, Yang X, Soman G, Giardina S. et al. Recombinant oncolytic poliovirus eliminates glioma in vivo without genetic adaptation to a pathogenic phenotype. Mol Ther. 2008;16:1865-72
130. Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH. et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N Engl J Med. 2018;379:150-61
131. Zulkifli MM, Ibrahim R, Ali AM, Aini I, Jaafar H, Hilda SS. et al. Newcastle diseases virus strain V4UPM displayed oncolytic ability against experimental human malignant glioma. Neurol Res. 2009;31:3-10
132. Ali R, Alabsi AM, Ali AM, Ideris A, Omar AR, Yusoff K. et al. Cytolytic effects and apoptosis induction of Newcastle disease virus strain AF2240 on anaplastic astrocytoma brain tumor cell line. Neurochem Res. 2011;36:2051-62
133. Duggal R, Geissinger U, Zhang Q, Aguilar J, Chen NG, Binda E. et al. Vaccinia virus expressing bone morphogenetic protein-4 in novel glioblastoma orthotopic models facilitates enhanced tumor regression and long-term survival. J Transl Med. 2013;11:155
134. Advani SJ, Buckel L, Chen NG, Scanderbeg DJ, Geissinger U, Zhang Q. et al. Preferential replication of systemically delivered oncolytic vaccinia virus in focally irradiated glioma xenografts. Clin Cancer Res. 2012;18:2579-90
135. Bansal K, Engelhard HH. Gene therapy for brain tumors. Curr Oncol Rep. 2000;2:463-72
136. Kwiatkowska A, Nandhu MS, Behera P, Chiocca EA, Viapiano MS. Strategies in gene therapy for glioblastoma. Cancers (Basel). 2013;5:1271-305
137. Kane JR, Miska J, Young JS, Kanojia D, Kim JW, Lesniak MS. Sui generis: gene therapy and delivery systems for the treatment of glioblastoma. Neuro Oncol. 2015;17(Suppl 2):ii24-ii36
138. Fischer U, Steffens S, Frank S, Rainov NG, Schulze-Osthoff K, Kramm CM. Mechanisms of thymidine kinase/ganciclovir and cytosine deaminase/ 5-fluorocytosine suicide gene therapy-induced cell death in glioma cells. Oncogene. 2005;24:1231-43
139. Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. 1986;46:5276-81
140. Valerie K, Hawkins W, Farnsworth J, Schmidt-Ullrich R, Lin PS, Amir C. et al. Substantially improved in vivo radiosensitization of rat glioma with mutant HSV-TK and acyclovir. Cancer Gene Ther. 2001;8:3-8
141. Prados MD, McDermott M, Chang SM, Wilson CB, Fick J, Culver KW. et al. Treatment of progressive or recurrent glioblastoma multiforme in adults with herpes simplex virus thymidine kinase gene vector-producer cells followed by intravenous ganciclovir administration: a phase I/II multi-institutional trial. J Neurooncol. 2003;65:269-78
142. Germano IM, Fable J, Gultekin SH, Silvers A. Adenovirus/herpes simplex-thymidine kinase/ganciclovir complex: preliminary results of a phase I trial in patients with recurrent malignant gliomas. J Neurooncol. 2003;65:279-89
143. Assi H, Candolfi M, Baker G, Mineharu Y, Lowenstein PR, Castro MG. Gene therapy for brain tumors: basic developments and clinical implementation. Neurosci Lett. 2012;527:71-7
144. Rainov NG. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum Gene Ther. 2000;11:2389-401
145. Lang FF, Yung WK, Sawaya R, Tofilon PJ. Adenovirus-mediated p53 gene therapy for human gliomas. Neurosurgery. 1999;45:1093-104
146. Chintala SK, Fueyo J, Gomez-Manzano C, Venkaiah B, Bjerkvig R, Yung WK. et al. Adenovirus-mediated p16/CDKN2 gene transfer suppresses glioma invasion in vitro. Oncogene. 1997;15:2049-57
147. Schiappacassi M, Lovat F, Canzonieri V, Belletti B, Berton S, Di Stefano D. et al. p27Kip1 expression inhibits glioblastoma growth, invasion, and tumor-induced neoangiogenesis. Mol Cancer Ther. 2008;7:1164-75
148. Park KH, Lee J, Yoo CG, Kim YW, Han SK, Shim YS. et al. Application of p27 gene therapy for human malignant glioma potentiated by using mutant p27. J Neurosurg. 2004;101:505-10
149. Davies MA, Lu Y, Sano T, Fang X, Tang P, LaPushin R. et al. Adenoviral transgene expression of MMAC/PTEN in human glioma cells inhibits Akt activation and induces anoikis. Cancer Res. 1998;58:5285-90
150. Qin XQ, Tao N, Dergay A, Moy P, Fawell S, Davis A. et al. Interferon-beta gene therapy inhibits tumor formation and causes regression of established tumors in immune-deficient mice. Proc Natl Acad Sci U S A. 1998;95:14411-6
151. Chiocca EA, Smith KM, McKinney B, Palmer CA, Rosenfeld S, Lillehei K. et al. A Phase I Trial of Ad.hIFN-beta Gene Therapy for Glioma. Mol Ther. 2008;16:618-26
152. Vom Berg J, Vrohlings M, Haller S, Haimovici A, Kulig P, Sledzinska A. et al. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J Exp Med. 2013;210:2803-11
153. Enderlin M, Kleinmann EV, Struyf S, Buracchi C, Vecchi A, Kinscherf R. et al. TNF-alpha and the IFN-gamma-inducible protein 10 (IP-10/CXCL-10) delivered by parvoviral vectors act in synergy to induce antitumor effects in mouse glioblastoma. Cancer Gene Ther. 2009;16:149-60
154. Tanaka T, Cao Y, Folkman J, Fine HA. Viral vector-targeted antiangiogenic gene therapy utilizing an angiostatin complementary DNA. Cancer Res. 1998;58:3362-9
155. Ma HI, Lin SZ, Chiang YH, Li J, Chen SL, Tsao YP. et al. Intratumoral gene therapy of malignant brain tumor in a rat model with angiostatin delivered by adeno-associated viral (AAV) vector. Gene Ther. 2002;9:2-11
156. Szentirmai O, Baker CH, Bullain SS, Lin N, Takahashi M, Folkman J. et al. Successful inhibition of intracranial human glioblastoma multiforme xenograft growth via systemic adenoviral delivery of soluble endostatin and soluble vascular endothelial growth factor receptor-2: laboratory investigation. J Neurosurg. 2008;108:979-88
157. Dmitrieva N, Yu L, Viapiano M, Cripe TP, Chiocca EA, Glorioso JC. et al. Chondroitinase ABC I-mediated enhancement of oncolytic virus spread and antitumor efficacy. Clin Cancer Res. 2011;17:1362-72
158. Hicks MJ, Chiuchiolo MJ, Ballon D, Dyke JP, Aronowitz E, Funato K. et al. Anti-Epidermal Growth Factor Receptor Gene Therapy for Glioblastoma. PLoS One. 2016;11:e0162978
159. Kroeger KM, Muhammad AK, Baker GJ, Assi H, Wibowo MK, Xiong W. et al. Gene therapy and virotherapy: novel therapeutic approaches for brain tumors. Discov Med. 2010;10:293-304
160. Tobias A, Ahmed A, Moon KS, Lesniak MS. The art of gene therapy for glioma: a review of the challenging road to the bedside. J Neurol Neurosurg Psychiatry. 2013;84:213-22
Author contact
![]() Corresponding author: Fan Fan, Xiangya Hospital, 87 Xiangya Road, Changsha, Hunan Province, China, Tel: +8673189753537, Fax: +8673189753537, E-mail: fanfanedu.cn
Corresponding author: Fan Fan, Xiangya Hospital, 87 Xiangya Road, Changsha, Hunan Province, China, Tel: +8673189753537, Fax: +8673189753537, E-mail: fanfanedu.cn