Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(2):334-344. doi:10.7150/jca.32989 This issue Cite
Research Paper
SFI Enhances Therapeutic Efficiency of Gefitinib: An Insight into Reversal of Resistance to Targeted Therapy in Non-small Cell Lung Cancer Cells
Zhenzhen Pan1*, Kai Wang1*, Qiufang Chen2, Xiulan Zheng1, Zhengyu Song1, Xuansheng Ding1 ![]()
1. China Pharmaceutical University, School of Basic Medicine and Clinical Pharmacy, Nanjing, 211198, China
2. Xiamen Maternity and Child Health Care Hospital, Xiamen, 361000, China
*Both authors contributed equally to this work.
Received 2019-1-10; Accepted 2019-9-1; Published 2020-1-1
Abstract
Background: The clinical application of EGFR tyrosine kinase inhibitors is always accompanied by inevitable drug resistance. However, the mechanism remains elusive. In the present study, we investigate the involvement of MAPK/SREBP1 pathway in NSCLC gefitinib resistance and evaluate the synergistic effects of shenqi fuzheng injection (SFI) and gefitinib on NSCLC cells.
Methods: To investigate the MAPK/SREBP1 pathway involved in gefitinib resistance, Western blotting was used to examine p-MEK, p-ERK and SREBP1 expression in PC-9 and PC-9/GR cells, MTT was used on cell proliferation, wound healing assay was used on cell migration. To detect the cooperative effects of SFI and gefitinib, clonogenic assay was used on cell proliferation. Apoptosis assay was analyzed by flow cytometry. Immunofluorescence was used to detect gefitinib binding to EGFR. Western blotting was used to detect whether SFI regulate the resistance to gefitinib via the suppression of MAPK/SREBP1 pathway.
Results: Our results showed that MAPK/SREBP1 pathway mediated resistance to gefitinib in NSCLC cells. MAPK pathway was found to directly target SREBP1 and inhibition of SREBP1 increased gefitinib sensitivity. In addition, SFI showed cooperative anti-proliferation and pro-apoptosis impacts on gefitinib resistant cells via down-regulating MAPK/SREBP1 pathway. Moreover, the combination of SFI and gefitinib enhanced gefitinib binding to EGFR resulting in the restoration of sensitivity to gefitinib.
Conclusions: Taken together, MAPK/SREBP1 pathway could be regarded as the potential treatment target for overcoming resistance to EGFR-TKIs in NSCLC and adjuvant therapy of SFI could be a potential therapeutic strategy for gefitinib resistant treatment.
Keywords: non-small cell lung cancer, EGFR-TKI, drug resistance, SREBP1, shenqi fuzheng injection
Introduction
Lung cancer is one of the most common types of cancer diagnosed worldwide and leads to high mortality [1, 2]. Non-small cell lung cancer (NSCLC) accounts for 85% of all cases [3]. Approximately 64% of patients with NSCLC harbor an oncogenic driver mutation, such as epidermal growth factor receptor (EGFR) and Kirsten rat sarcoma 2 viral oncogene homolog (KRAS), which leads to improvements in survival and safety compared with conventional chemotherapy [4]. EGFR tyrosine kinase inhibitors (EGFR-TKIs) are effective in approximately 70% of NSCLC with EGFR activating mutation [5]. Inevitably, the most of patients develop acquired resistance after 1 year treatment with EGFR-TKIs on average due to a variety of mechanisms [6].
The possible mechanisms of EGFR-TKIs resistance include second-site mutation of EGFR kinase domain, histological transformation, bypass signaling pathways activation including MAPK pathway and molecular changes to promote cell survival and inhibition of apoptosis [7]. For instance, AZD9291 was discovered to overcome secondary resistance mutations, resistant cells can develop a bypass pathway to reactivate downstream proliferation and survival signal [8]. It is necessary to figure out the complex and ambiguous resistant mechanisms and develop alternative therapeutic methods to overcome EGFR-TKIs resistance.
Our team has demonstrated that high intracellular level of cholesterol was the leading cause of gefitinib resistance in non-small cell lung cancer [9]. Evidence showed that the expression of sterol regulatory element binding protein 1 (SREBP1) was high in tumor tissue [10].SREBP1 is a key transcription factor of lipid homeostasis and activates genes required for the synthesis of cholesterol [11]. Inhibiting SREBP1 expression decreased the tumor growth in vivo [12]. The signal transduction pathways, such as MAPK pathway, have been identified to regulate SREBP1 expression in cancer cells [13, 14]. However, the role of MAPK/SREBP1 playing in EGFR mutation gefitinib resistant NSCLC cells has not been clarified.
Many Traditional Chinese medicine (TCM) have anticancer effects and can enhance the efficacy of EGFR-TKIs in NSCLC [15, 16]. It is a promising strategy to combine TCM and EGFR-TKIs as anticancer treatment to overcome drug resistance. Shenqi fuzheng injection (SFI) is a modern TCM commonly used in clinic as an antitumor injection. Two Chinese medicine herbs, codonopsis and astragali are the main resources of SFI [17]. It has been reported that combination of SFI and chemotherapy could improve the quality of life, reduce toxicity and exhibit synergistic antitumor effects in NSCLC patients [18, 19]. However, the synergistic effects of SFI and gefitinib on gefitinib resistant NSCLC cells, which may become a promising strategy to overcome EGFR-TKIs resistance, and underlying mechanisms are poorly understood.
In the current study, the role of MAPK/SREBP1 pathway in NSCLC with resistance to gefitinib was assessed for the first time, and the potential therapeutic effect of targeting MAPK/SREBP1 pathway was examined in NSCLC cells.
Materials and methods
Chemicals and reagents
Shengqi fuzheng injection (Z19990065) was provided by livzon Pharmaceutics ltd. (Zhuhai, China). For cell culture, SFI was dissolved in DMEM to different concentration gradients. Gefitinib was purchased from Aladdin Industrial Corporation (Shanghai, China). The 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) was purchased from Biosharp (Shanghai, China). Dulbecco's modified eagle medium (DMEM), DMSO, Penicillin- Streptomycin Solution, Annexin V-FITC kit were purchased from KeyGen (KeyGen, Nanjing, China). DAPI staining solution, EGF, BCA protein assay kit, Crystal Violet Staining Solution were purchased from Beyotime Biotechnology (Shanghai, China). Anti-p- EGFR(Tyr 1172), anti-EGFR, anti-MEK1/2, anti-p- ERK1/2(thr202/tyr204), anti-ERK1/2, anti-β-actin, goat anti-rabbit IgG H&L (HRP), anti-cleaved-caspase 3, anti-cleaved-caspase 9, anti-Bcl-2, anti-Bax antibodies were purchased from Wanlei Bio. (Shenyang, China). Anti-p-MEK1/2 (Ser217/221) antibody was purchased from Cell Signaling Technology (Cell Signaling Technology, Danvers, MA, USA).
Cell culture
Human NSCLC H1650 and H1975 cells were obtained from 3D Medicines. Human NSCLC PC-9 and PC-9/GR cells were given by Dr. Zhou Caicun. Cells were cultured in DMEM containing 12% Fetal bovine serum (Biological Industries), Penicillin- Streptomycin Solution (1X) at 37℃ in an atmosphere of 5% CO2.
Cell proliferation assay
The cell proliferation assay of gefitinib and SFI was measured by MTT. Cells in 96-well plates were treated with indicated drugs. Then, each well added 150 μL MTT solution incubated at 37℃ for 4 h. Absorbance was determined at 570 nm using a microplate reader (Thermo Multiskan_FC, American).
Drug synergy analysis
The data from the PC-9/GR, H1975 and H1650 cell proliferation assays was analyzed by CompuSyn software (Biosoft, Cambridge, UK) to investigate the synergistic effects of SFI and gefitinib on cells in vitro. The combination index (CI)-isobologram equation was described as previously [20].
Clonogenic assay
500 cells per well were seeded into 12-well plates and cultured in DMEM supplemented with no drug solution, SFI (1:10), Gefitinib 4 μM or in combination at 37˚C. After 14 days, the cells were fixed with 4% paraformaldehyde and stained with crystal violet solution. Finally, the plates were washed dried at room temperature and photographed.
Wound healing assay
To evaluate the migration ability of cells, a wound healing assay was performed. The cells were seeded in 96-well plates and then, as its growth to 80% confluence, the wounds were scratched with pipette tip across the center of the well. After having been washed, the cells were incubated in humid atmosphere. The migrated cells were observed via the optical microscope at 0 and 12 h, respectively.
Apoptosis assay
To test apoptosis, cells were planted in 6-well plates incubated with indicated drugs for 24 h before cell dissociation and assemblage. The cells were harvested and re-suspended in 500 µL binding buffer. The cells were then stained with 5 µL Annexin V-FITC and Propidium Iodide for 5-10 min in the dark condition. Flow cytometry analysis (Becton Dickinson FACS Calibur; Becton-Dickinson, USA) was conducted to detect apoptosis.
Western blotting assay
Cells were treated with indicated drugs. Then the total protein was collected from cells. The concentration of protein in the supernatants was detected according to BCA protein assay kit instructions (Beyotime, P0010). Then, 60 μg protein was separated by 8%-12% SDS-PAGE and analyzed with antibodies. The final detection was performed by ECL reagents (KeyGen, KGP1121).
Immunofluorescence assay
Cells (1×105 cells/mL) were cultured on dish and treated with fluorescent-labeled quinazoline skeleton of gefitinib (10 µM) alone or in combination with SFI (1:10) for 3 h. Cells were washed with PBS, then incubated within DAPI (Beyotime, C1006) for further 20 min. Finally, the images were gained by a ZEISS.
Results
Gefitinib induced cytotoxicity in NSCLC cells with high constitutive levels of MAPK and SREBP1
We chose gefitinib sensitive PC-9 cells harboring EGFR exon 19 deletion and gefitinib resistant PC-9/GR cells for experiment. To quantify gefitinib cytotoxicity, MTT assay was performed. The cells were incubated for 24, 48 or 72 h at indicated doses of gefitinib (1, 3, 9, 27, 81, 243, 729 and 2187 nmol/L). As shown in Fig. 1A, the results indicated that gefitinib significantly inhibited the proliferation of PC-9 cells in a time and dose-dependent manner, and slightly inhibited the proliferation of PC-9/GR cells.
Gefitinib suppresses PC-9 and PC-9/GR cell proliferation. (A) Growth inhibition assay of gefitinib was determined by MTT under indicated concentration of gefitinib. (B) Western blotting for p-EGFR, EGFR, p-MEK, MEK, p-ERK, ERK, flSREBP1 and mSREBP1 protein expression in PC-9 and PC-9/GR cells treated with EGF. *p<0.05, **p<0.01 or ***p<0.001 compared to PC-9 cells.
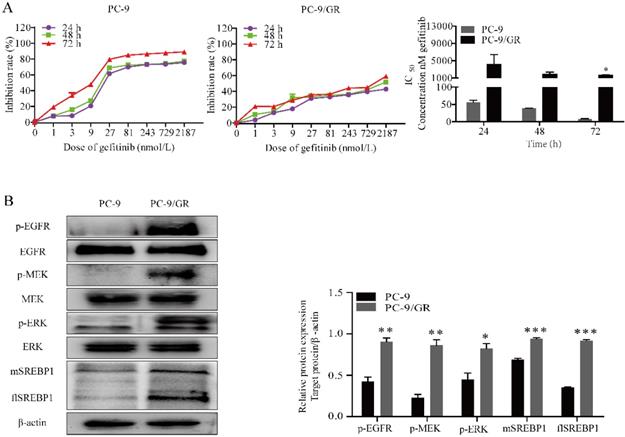
To test whether MAPK signaling cascades phosphorylated and activated in cells with resistance to gefitinib, western blotting was conducted. As expected, phosphorylated MEK and ERK were elevated in PC-9/GR compared with PC-9 cells (Fig. 1B). Since it has been reported that MAPK pathway affected the transcriptional activity of SREBP1, directly [21]. Here, we compared the expression of SREBP1 in different cells. Notably, we found a higher expression of both flSREBP1and mSREBP1 in PC-9/GR cells.
Inhibition of SREBP1 reversed Gefitinib resistance of NSCLC cells
U0126 is a selective inhibitor of MEK kinases [22], which has been widely used as an inhibitor for the MAPK pathway in diverse fields [23]. To confirm the effects of MAPK pathway on SREBP1 expression, PC-9/GR cells were treated with 10 μM U0126. We found that both flSREBP1 and mSREBP1 expression were inhibited by U0126.These data indicated that overexpression of SREBP1 was due to activated MAPK pathway in PC-9/GR cells (Fig. 2A).
Effects of MPAK pathway inhibition on SREBP1 expression and SREBP1 inhibition on migration and proliferation in PC-9/GR cells. (A) Western blotting for p-MEK, MEK, flSREBP1 and mSREBP1 protein expression in PC-9/GR cells after being treated with 10 μM U0126. **p<0.01 or ***p<0.001 compared to control group. (B) Cells were treated with gradient concentration of betulin (5 and 10 μM). Closed wound area of 5 and 10 μM betulin group at 12 h were significantly smaller compared to Control group. (C) PC-9/GR cells were treated with betulin 5 μM alone or combined with gefitinib 30 μM for 48 h. Relative cell viability was measured by MTT assay. **p<0.01 or ***p<0.001 compared to combination group.
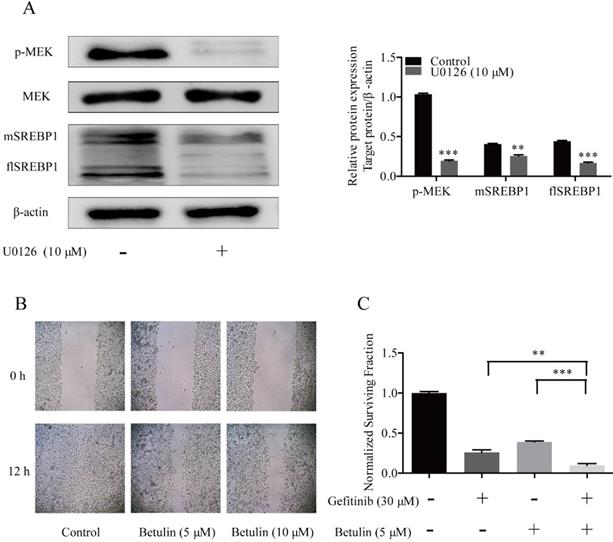
Betulin was previously identified as an inhibitor of SREBP pathway [24]. Treating cells with gradient concentration of betulin, the migration of PC-9/GR cells was inhibited (Fig. 2B). To examine whether inhibit SREBP1 could reverse cells resistance to gefitinib, 5 μM betulin was then combined with 30 μM gefitinib. The combined treatment exhibited a greater anti-proliferation effect on PC-9/GR cells than a single drug alone (Fig. 2C). These data confirmed that the MAPK/SREBP1 pathway mediated resistance to gefitinib in NSCLC cells.
SFI synergizes with gefitinib to inhibit cell proliferation and clonogenicity in PC-9/GR, H1975 and H1650 cells
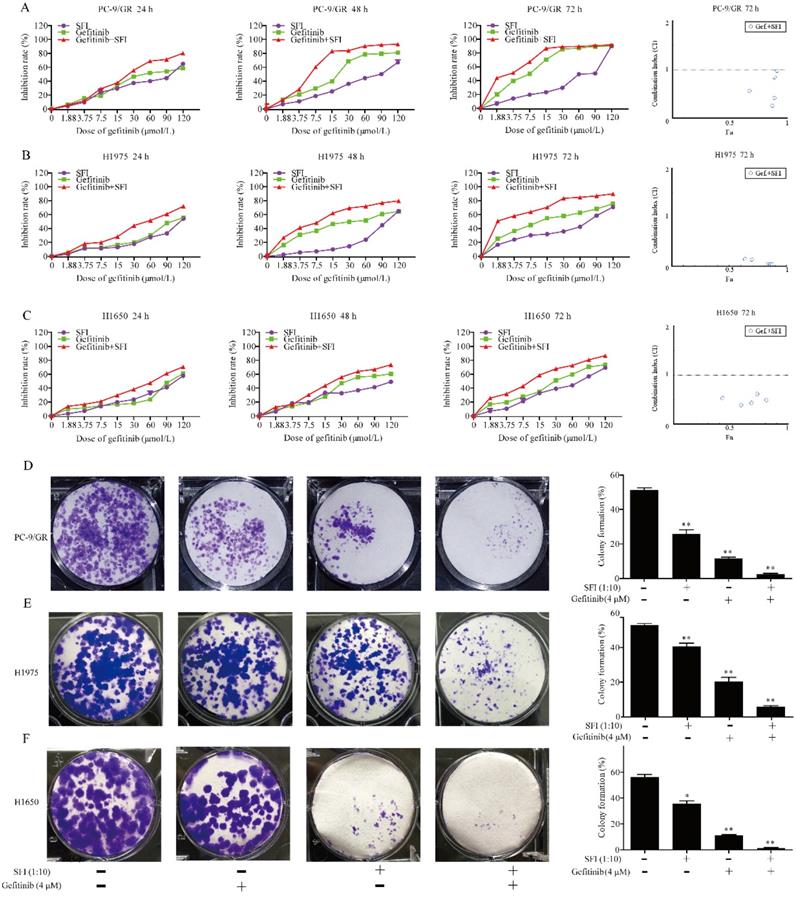
Shenqi fuzheng injection (SFI) was extracted from astragali and codonopsis, which was generally used to improve the immune system of patients with NSCLC [25]. Previous study has reported that astragaloside IV inhibited accumulation and nuclear translocation of SREBP1 [26]. We presumed that SFI might show synergistic antitumor effects with gefitinib by inhibiting SREBP1 expression in NSCLC cells. To investigate the synergistic effects on NSCLC cell motility, PC-9/GR, H1975 and H1650 cells were selected for further study. Cells were treated with different concentrations of gefitinib (1.875, 3.75, 7.5, 15, 30, 60, 90 and 120 μmol/L), SFI (1:1, 1:2, 1:4, 1:8, 1:16, 1:32, 1:64, 1:128) alone or in combination for 24, 48 or 72 h. The combined treatment had significant inhibition on PC-9/GR, H1975 and H1650 cells when compared with a single drug alone. (Fig. 3A, B and C). The results of the CI-isobologram analysis showed that SFI and gefitinib had synergistic effects on the PC-9/GR (CI: 0.179-0.982), H1975 (CI: 0.032-0.582), and H1650 (CI: 0.360-0.834) cells at 72 h. The combination of SFI and gefitinib rendered the PC-9/GR, H1975 and H1650 cells more sensitive to gefitinib. To further evaluate the synergistic anti-cancer effects of gefitinib and SFI, clonogenic assay was performed. We found that SFI, gefitinib alone or in combination inhibited cell colony formation in all three kinds of cells. Furthermore, combined treatment could significantly reduce the number of clones compared to each drug alone (Fig. 3D, E and F).
The synergistic effects of SFI and gefitinib on cell proliferation and clonogenicity. (A, B and C) Cells were treated with gefitinib alone or in combination with SFI for 24, 48 or 72 h. MTT was used to detect the cell viability. The Fa-CI (Combination Index) Plot was generated by CompuSyn software. Points that fall below the line indicate synergic relationship between the two drugs. The CI was calculated using the data and CompuSyn software. (D, E and F) Clonogenic assays were performed in PC-9/GR, H1975 and H1650 cells with no drug solution, SFI (1:10), Gefitinib 4 μM alone or in combination. *p<0.05 or **p<0.01 compared to control group.
SFI enhances the effect of gefitinib on inducing apoptosis in NSCLC cells
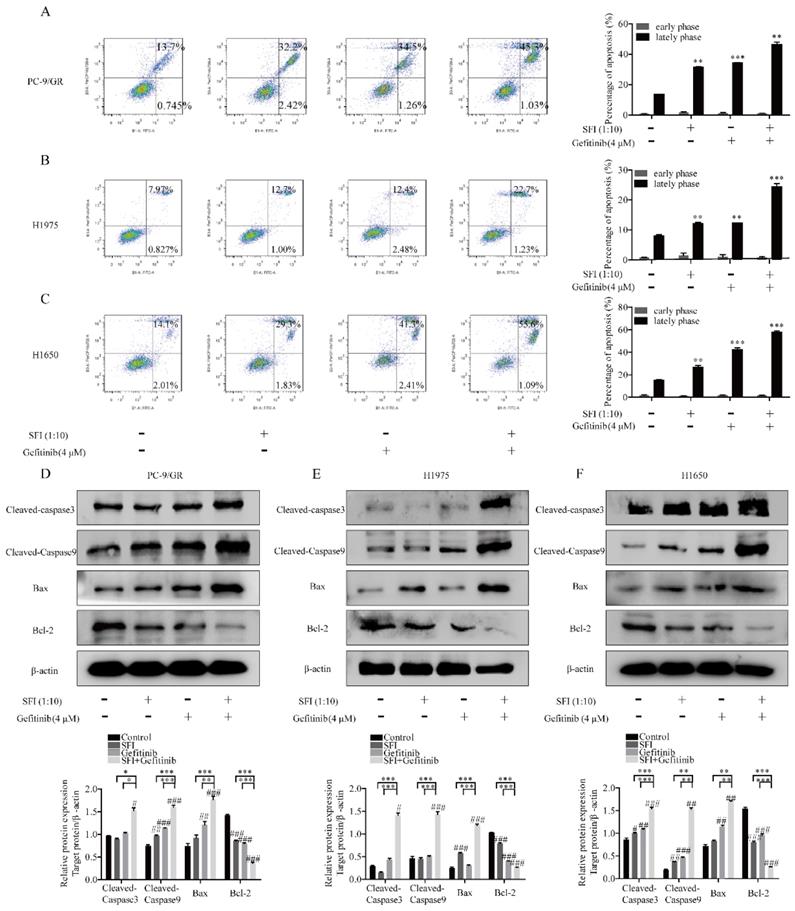
To test the regulation of SFI on cell apoptosis induced by gefitinib, cells were co-treated with SFI and gefitinib. As shown in Fig. 4A-C, SFI synergized with gefitinib in inducing cell apoptosis. The combined treatment increased cleaved-caspase 3, cleaved-caspase 9 and pro-apoptotic proteins Bax expression, as well as decreased the expression of anti-apoptotic protein Bcl-2. (Fig. 4D, E and F).
SFI synergizes with Gefitinib to induce apoptosis. (A, B and C) Apoptosis assay was conducted with SFI (1:10) and Gefitinib 4 μM alone or in combination for 24 h by Flow cytometry. **p<0.01 or ***p<0.001 compared to control group. (D, E and F) Western blotting was used to detect cleaved-caspase 3, cleaved-caspase 9, bax, and Bcl-2 protein expression after being treated with SFI (1:10) and Gefitinib 4 μM alone or in combination for 24 h. #p<0.05, ##p<0.01 or ###p<0.001 compared to control group. *p<0.05, **p<0.01 or ***p<0.001 compared to combination group.
The synergistic efficacy of SFI and gefitinib is dependent on inhibition of MAPK/SREBP1 pathway
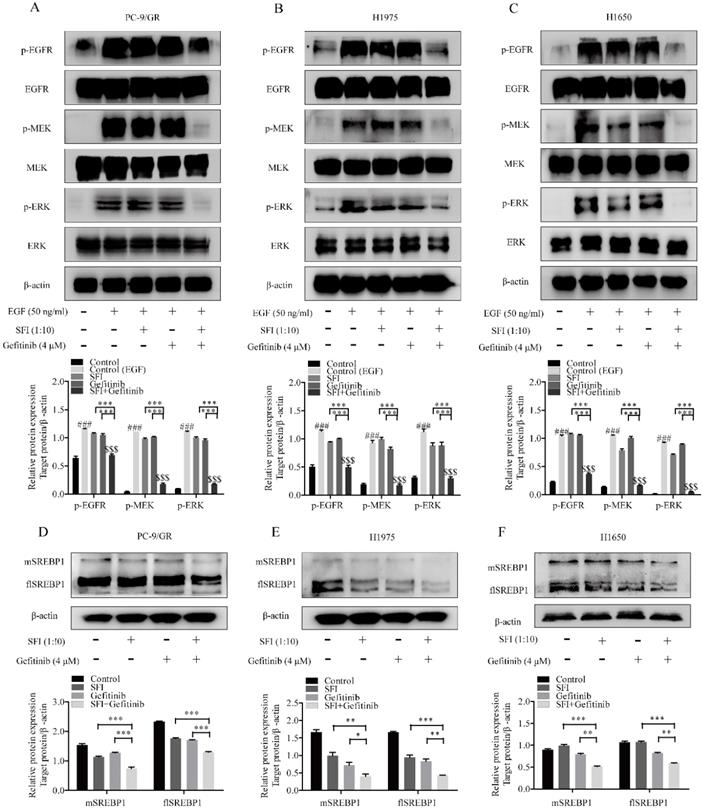
To elucidate whether a possible mechanism of synergy is due to the involvement of regulating MAPK/SREBP1 pathway, cells were treated with SFI and gefitinib respectively, or in combination for 24h, and then analyzed EGFR-related proteins levels by western blotting. As shown in Fig. 5A-C, gefitinib and SFI alone had no regulatory effects on phosphorylated EGFR, MEK, ERK proteins levels. Nevertheless, combined treatment showed a significantly inhibition on p-EGFR, p-MEK and p-ERK expression. We then investigated the expression of flSREBP1 and mSREBP1, a similar tendency was observed (Fig. 5D, E and F). To sum up, the combination of SFI and gefitinib could be a potential therapeutic strategy for gefitinib resistant treatment in NSCLC cells via regulating MAPK/SREBP1 pathway.
The mechanism of SFI showing synergistic efficacy on cells with Gefitinib is inhibiting MAPK/SREBP1 pathway. (A, B and C) Western blotting was performed to determined p-EGFR, EGFR, p-MEK, MEK, p-ERK, ERK protein expression after being treated with SFI (1:10) and Gefitinib 4 μM alone or in combination in PC-9/GR, H1975 and H1650 cells. ###p<0.001 compared to control group. $$$p<0.001 compared to control (EGF) group. ***p<0.001 compared to combination group. (D, E and F) Western blotting was conducted to detect flSREBP1 and mSREBP1 protein expression after being treated with SFI (1:10) and Gefitinib 4 μM alone or in combination in PC-9/GR, H1975 and H1650 cells. *p<0.05, **p<0.01 or ***p<0.001 compared to combination group.
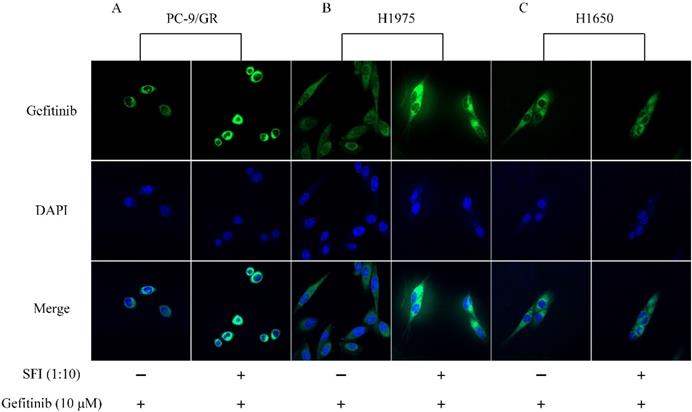
SFI enhances gefitinib binding to EGFR resulting in restoration of sensitivity to gefitinib in PC-9/GR and H1975 cells
SREBP1 is a transcription factor that maintain cellular lipid homeostasis by regulating the expression of many enzymes needed for the formation of cholesterol and fatty acid. Cholesterol and fatty acid are main components of mammalian cell membrane. EGFR is known to be a plasma membrane-resident protein, whose function is modulated by its surrounding lipid environment [27]. To determine whether SFI can cause changing in gefitinib affinity to EGFR, cells were treated with gefitinib alone or in combination with SFI. The fluorescence intensity was represented for the binding capacity of gefitinib to EGFR. Enhanced fluorescence intensity was observed by Confocal imaging (Fig. 6A, B and C) when cells were co-treated with SFI and gefitinib in PC-9/GR and H1975 cells. These results revealed that SFI increased gefitinib affinity in acquired resistant PC-9/GR and H1975 cells, but not in primary resistant H1650 cells.
SFI enhances Gefitinib binding to EGFR in PC-9/GR, H1975 cells. (A, B and C) Cells were exposed to fluorescent labeled gefitinib quinazoline skeleton (10 µM) alone or in combination with SFI (1:10) for 3 h. Immunofluorescence assay was conducted to detect the affinity of gefitinib to EGFR tyrosine kinase domain (green fluorescence).
Discussion
Gefitinib is the first EGFR-TKI that was approved for the therapy of patients with NSCLC [28]. By competitively interacting with the ATP-binding site, gefitinib can inhibit EGFR kinase activity, prevent auto-phosphorylation and suppress downstream signaling. NSCLC patients harboring EGFR mutation demonstrate good responses to gefitinib. Unfortunately, the clinical application of gefitinib is limited by drug resistance due to many mechanisms including the secondary T790M mutation, a most common mechanism for gefitinib resistance manifested in approximately 60% of patients. The third generation EGFR-TKIs, such as osimertinib, is designed to overcome T790M mutation. This new agent significantly increases the overall response rates of patients. However, similar to gefitinib, the application of osimertinib has been accompanied by the drug resistance. Several mechanisms of resistance have been identified including EGFR C797S mutation, MET amplification and epithelial-mesenchymal transition (EMT) [29]. Even with the fourth generation EGFR-TKIs on the clinical research, the complex mechanisms of drug resistance have not been fully revealed. Thus, there is a need to understand the underlying mechanism and identify the key molecule target so as to develop new strategies to overcome EGFR-TKIs resistance.
The study is based on our previous work which showed that high levels of cholesterol in lipid rafts are responsible for gefitinib resistance in NSCLC cells and the depletion of cholesterol can restore the sensitivity of gefitinib. We presumed that the key molecules involved in the regulation of cellular cholesterol level could be targets to overcome EGFR-TKIs resistance. SREBP1 is a key transcription factor for cholesterol homeostasis by regulating the transcriptional activation of target genes, such as 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR) and low-density lipoprotein receptor (LDLR) [30]. In the present study, we found a higher expression of SREBP1 in PC-9/GR cells compared to PC-9 cells (p<0.001). As it was documented before, SREBP1 could promote proliferation, metastasis and EMT in cancer cells by providing the membrane building materials [31]. We acquired similar results where the suppression of SREBP1 by betulin inhibited the migration of PC-9/GR cells. Further study was conducted to investigate the role of SREBP1 playing in gefitinib resistance by combining betulin and gefitinib to treat cells. Results showed that inhibition of SREBP1 enhanced cell sensitivity to gefitinib in NSCLC cells.
The Ras-Raf-MEK-ERK mitogen-activated protein kinase (MAPK) pathway governs fundamental physiological processes, such as cell proliferation, metabolism, cell death and survival in NSCLC [32]. It is activated by extracellular ligands, such as epidermal growth factor (EGF), and motivates cell survival by regulating a range of targets including caspase 3, caspase 9, Bcl-xl and Bad transcription factors [33]. We found MAPK signaling cascades phosphorylated and activated in PC-9/GR cells. Previous studies have identified SREBP as a downstream effector of MAPK cascades in prostate cancer or melanoma, but not in NSCLC. To determine the targeted relationship between MAPK pathway and SREBP1 in NSCLC gefitinib resistant cells, we pharmacologically inhibited MAPK pathway by U0126. We discovered that in PC-9/GR cells, the expression of SREBP1 (both flSREBP1 and mSREBP1) was regulated by MAPK pathway. Our results demonstrated that MAPK/SREBP1 pathway was responsible for gefitinib resistance in NSCLC cells.
Regarding SFI are mainly composed of codonopsis and astragali. Previous studies have reported that inhibitory impacts of astragaloside on cancer cells were probably related to its regulating MAPK pathway [34, 35]. Moreover, astragaloside IV inhibited the accumulation and nuclear translocation of mature SREBP1 [36]. Above all, we presumed that SFI might show synergistic antitumor effects of gefitinib by regulating MAPK/SREBP1 pathway in NSCLC gefitinib resistant cells. Here, we selected PC-9/GR, H1975 and H1650 cells to detect synergistic effects of SFI and gefitinib. Results suggested that SFI combined with gefitinib to inhibit cell proliferation, clonogenicity and induce apoptosis, which were consistant with the study conducted by Xiong Y, et al. They documented that SFI increased chemotherapy sensitivity in cisplatin resistance of NSCLC cells through regulating cell cycle and initiating mitochondrial apoptosis [37]. We further validated the underlying mechanism of SFI reversing gefitinib resistance was the inhibition of MAPK/SREBP1 pathway.
The secondary T790M mutation within the ATP site of EGFR is the most common mechanism of resistance to the first generation EGFR-TKIs in lung cancers [38], which reduces the binding efficacy of EGFR-TKIs to EGFR kinase domain [39]. The EGFR is a membrane-bound receptor, which consists of an extracellular module and an intracellular kinase domain. The activation and function of EGFR is related to membrane lipids [40]. Main components of membrane such as phospholipids, fatty acids and cholesterol have been reported to regulate drug sensitivity of gefitinib in NSCLC [41, 42]. SREBP1 is the key factor transcription factor playing a central role in lipid metabolism. Based on the result that SFI combined with gefitinib can reduce the SREBP1 protein expression. Here, we speculated that SFI might enhance gefitinib binding to EGFR by inhibiting SREBP1 and then detected the binding of gefitinib to EGFR by Immunofluorescence. We found that SFI enhanced gefitinib binding to EGFR in acquired resistant NSCLC PC-9/GR and H1975 cells, but not in primary resistant H1650 cells. The resistant mechanism of H1650 cells is associated with PTEN deletion without affecting gefitinib binding to EGFR [43]. Therefore, it is not surprising that SFI did not exert any impacts on binging gefitinib to EGFR in H1650 cells.
In summary, our data initially exhibit the MAPK/SREBP1 pathway were responsible for gefitinib resistance in NSCLC cells and draw a conclusion that the combined treatment of SFI augmented gefitinib's anti-proliferation and pro-apoptosis potential in NSCLC gefitinib resistant cells through regulating MAPK/SREBP1 pathway. Moreover, the combined treatment enhanced gefitinib binding to EGFR resulted in restoration of sensitivity to gefitinib in acquired resistant NSCLC cells. Thus it can be seen that inhibition of MAPK/SREBP1 pathway is a prospective strategy to conquer gefitinib resistance in NSCLC cells and adjuvant therapy of SFI could be a potential therapeutic strategy for gefitinib resistant treatment.
Abbreviations
AKT: protein kinase B; ALK: anaplastic lymphoma kinase; ATP: adenosine triphosphate; BCA: bicinchoninic acid; CI: combination index; DAPI: 4′:6-diamidino-2-phenylindole; DMEM: dulbecco's modified eagle medium; DMSO: dimethyl sulfoxide; EGF: epidermal growth factor; EGFR: epidermal growth factor receptor; EGFR-TKIs: EGFR tyrosine kinase inhibitors; EMT: epithelial-mesenchymal transition; flSREBP1: full length sterol regulatory element binding protein 1; HMGCR: 3-hydroxy-3-methylglutaryl-CoA reductase; KRAS: kirsten rat sarcoma 2 viral oncogene homolog; LDLR: low-density lipoprotein receptor; MAPK: mitogen-activated protein kinase; MET: mesenchymal-epithelial transition; mSREBP1: mature sterol regulatory element binding protein 1; MTT: 3-(4:5-dimethylthiazol-2-yl)-2:5-diphenyltetrazolium bromide; NSCLC: non-small cell lung cancer; PBS: phosphate buffer solution; PI3K: phosphatidylinositol-4:5-bisphosphate 3-kinase; PTEN: phosphatase and tensin homolog; SFI: shenqi fuzheng injection; SREBP1: sterol regulatory element binding protein 1; TCM: traditional Chinese medicine.
Acknowledgements
The study was supported by grants from Postgraduate Innovative Project of Jiangsu Province (KYCX18_0817).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
2. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359-86
3. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008;83:584-94
4. Kris MG, Johnson BE, Berry LD, Kwiatkowski DJ, Iafrate AJ, Wistuba II. et al. Using Multiplexed Assays of Oncogenic Drivers in Lung Cancers to Select Targeted Drugs. JAMA. 2014;311:1998-2006
5. Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C. et al. Screening for Epidermal Growth Factor Receptor Mutations in Lung Cancer. N Engl J Med. 2009;361:958-67
6. Pirazzoli V, Ayeni D, Meador CB, Sanganahalli BG, Hyder F, de Stanchina E. et al. Afatinib plus Cetuximab Delays Resistance Compared to Single-Agent Erlotinib or Afatinib in Mouse Models of TKI-Naive EGFR L858R-Induced Lung Adenocarcinoma. Clin Cancer Res. 2016;22:426-35
7. Rotow J, Bivona TG. Understanding and targeting resistance mechanisms in NSCLC. Nat Rev Cancer. 2017;17:637-58
8. Niederst MJ, Engelman JA. Bypass Mechanisms of Resistance to Receptor Tyrosine Kinase Inhibition in Lung Cancer. Sci Signal. 2013;6:re6
9. Chen QF, Pan ZZ, Zhao M, Wang Q, Qiao C, Miao LY. et al. High cholesterol in lipid rafts reduces the sensitivity to EGFR-TKI therapy in non-small cell lung cancer. J Cell Physiol. 2018;233:6722-32
10. Calvisi DF, Wang CM, Ho C, Ladu S, Lee SA, Mattu S. et al. Increased Lipogenesis, Induced by AKT-mTORC1-RPS6 Signaling, Promotes Development of Human Hepatocellular Carcinoma. Gastroenterology. 2011;140:1071-83
11. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology-divergent pathophysiology. Nat Rev Endocrinol. 2017;13:710-30
12. Wen YA, Xiong XP, Zaytseva YY, Napier DL, Vallee E, Li AT. et al. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018;9:265
13. Chen M, Zhang JW, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E. et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet. 2018;50:206-18
14. Talebi A, Dehairs J, Rambow F, Rogiers A, Nittner D, Derua R. et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat Commun. 2018;9:2500
15. Wu ZM, Zhu QY, Yin YY, Kang D, Cao RY, Tian Q. et al. Traditional Chinese Medicine CFF-1 induced cell growth inhibition, autophagy, and apoptosis via inhibiting EGFR-related pathways in prostate cancer. Cancer Med. 2018;7:1546-59
16. Bing ZT, Cheng ZY, Shi DF, Liu XK, Tian JH, Yao XJ. et al. Investigate the mechanisms of Chinese medicine Fuzhengkangai towards EGFR mutation-positive lung adenocarcinomas by network pharmacology. BMC Complement Altern Med. 2018;18:293
17. Dong J, Su SY, Wang MY, Zhan Z. Shenqi fuzheng, an injection concocted from chinese medicinal herbs, combined with platinum-based chemotherapy for advanced non-small cell lung cancer: a systematic review. J Exp Clin Cancer Res. 2010;29:137
18. Cao DD, Xu HL, He AB, Xu XM, Ge W. The Effect of ShenQi FuZheng Injection in Combination with Chemotherapy versus Chemotherapy Alone on the Improvement of Efficacy and Immune Function in Patients with Advanced Non-Small Cell Lung Cancer: A Meta-Analysis. PLoS One. 2016;11:e0152270
19. Hao TT, Xie YM, Liao X, Wang J. Systematic review and Meta-analysis of Shenqi Fuzheng injection combined with first-line chemotherapy for non-small cell lung cancer. Zhongguo Zhong Yao Za Zhi. 2015;40:4094-107
20. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27-55
21. Kotzka J, Muller-Wieland D, Roth G, Kremer L, Munck M, Schurmann S. et al. Sterol regulatory element binding proteins (SREBP)-1a and SREBP-2 are linked to the MAP-kinase cascade. J Lipid Res. 2000;41:99-108
22. Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS. et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623-32
23. Hawkins TA, Cavodeassi F, Erdelyi F, Szabo G, Lele Z. The small molecule Mek1/2 inhibitor U0126 disrupts the chordamesoderm to notochord transition in zebrafish. BMC Dev Biol. 2008;8:42
24. Tang JJ, Li JG, Qi W, Qiu WW, Li PS, Li BL. et al. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab. 2011;13:44-56
25. Xu R, Lin L, Li Y, Li Y. ShenQi FuZheng Injection combined with chemotherapy in the treatment of colorectal cancer: A meta-analysis. PLoS One. 2017;12:e0185254
26. Zhou B, Zhou DL, Wei XH, Zhong RY, Xu J, Sun L. Astragaloside IV attenuates free fatty acid-induced ER stress and lipid accumulation in hepatocytes via AMPK activation. Acta Pharmacol Sin. 2017;38:998-1008
27. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497-510
28. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446-54
29. Tan CS, Kumarakulasinghe NB, Huang YQ, Ang YLE, Choo JR, Goh BC. et al. Third generation EGFR TKIs: current data and future directions. Mol Cancer. 2018;17:29
30. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331-40
31. Zhai D, Cui C, Xie L, Cai L, Yu J. Sterol regulatory element-binding protein 1 cooperates with c-Myc to promote epithelial-mesenchymal transition in colorectal cancer. Oncol Lett. 2018;15:5959-65
32. Ciuffreda L, Incani UC, Steelman LS, Abrams SL, Falcone I, Del Curatolo A. et al. Signaling Intermediates (MAPK and PI3K) as Therapeutic Targets in NSCLC. Curr Pharm Design. 2014;20:3944-57
33. Grant S, Qiao L, Dent P. Roles of ERBB family receptor tyrosine kinases, and downstream signaling pathways, in the control of cell growth and survival. Front Biosci. 2002;7:d376-89
34. Jiang K, Lu Q, Li Q, Ji Y, Chen W, Xue X. Astragaloside IV inhibits breast cancer cell invasion by suppressing Vav3 mediated Rac1/MAPK signaling. Int Immunopharmacol. 2017;42:195-202
35. Li B, Wang F, Liu N, Shen W, Huang T. Astragaloside IV inhibits progression of glioma via blocking MAPK/ERK signaling pathway. Biochem Biophys Res Commun. 2017;491:98-103
36. Wang C, Li Y, Hao M, Li W. Astragaloside IV Inhibits Triglyceride Accumulation in Insulin-Resistant HepG2 Cells via AMPK-Induced SREBP-1c Phosphorylation. Front Pharmacol. 2018;9:345
37. Xiong Y, Zhao QY, Gu LY, Liu CY, Wang C. Shenqi Fuzheng Injection Reverses Cisplatin Resistance through Mitofusin-2-Mediated Cell Cycle Arrest and Apoptosis in A549/DDP Cells. Evid Based Complement Alternat Med. 2018;2018:8258246
38. Jia Y, Yun CH, Park E, Rcan DE, Manuia M, Juarez J. et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534:129-32
39. Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK. et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070-5
40. Arkhipov A, Shan YB, Das R, Endres NF, Eastwood MP, Wemmer DE. et al. Architecture and Membrane Interactions of the EGF Receptor. Cell. 2013;152:557-69
41. Jung JH, Lee MY, Choi DY, Lee JW, You SY, Lee KY. et al. Phospholipids of tumor extracellular vesicles stratify gefitinib-resistant nonsmall cell lung cancer cells from gefitinib-sensitive cells. Proteomics. 2015;15:824-35
42. Bach DH, Luu TTT, Kim D, An YJ, Park S, Park HJ. et al. BMP4 Upregulation Is Associated with Acquired Drug Resistance and Fatty Acid Metabolism in EGFR-Mutant Non-Small-Cell Lung Cancer Cells. Mol Ther Nucleic Acids. 2018;12:817-28
43. Zhong J, Li L, Wang ZJ, Bai H, Gai F, Duan JC. et al. Potential Resistance Mechanisms Revealed by Targeted Sequencing from Lung Adenocarcinoma Patients with Primary Resistance to Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Inhibitors (TKIs). J Thorac Oncol. 2017;12:1766-78
Author contact
![]() Corresponding author: Xuansheng Ding, Tel: +86 13357823738 (Ding); E-mail: dxs0162com. School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, No.639 Longmian Avenue, Jiangning District, Nanjing, Jiangsu, China.
Corresponding author: Xuansheng Ding, Tel: +86 13357823738 (Ding); E-mail: dxs0162com. School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, No.639 Longmian Avenue, Jiangning District, Nanjing, Jiangsu, China.