Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(3):696-701. doi:10.7150/jca.32497 This issue Cite
Review
Emerging role of glucocorticoid receptor in castration resistant prostate cancer: A potential therapeutic target
Raj Kumar ![]()
Department of Biomedical Sciences, College of Medicine, University of Houston, Houston, TX, USA.
Received 2018-12-22; Accepted 2019-9-24; Published 2020-1-1
Abstract
Glucocorticoids are used as co-medication with chemotherapy for solid tumors to reduce inflammation as well as cytotoxic side effects and are effective in easing symptoms related to chemotherapy. However, emerging evidence suggests that glucocorticoids may contribute to failure of chemotherapy and tumor progression of castration resistant prostate cancer (CRPC). Thus, in recent years, glucocorticoid signaling pathway has become an important therapeutic target for CRPC. Understanding the exact mechanism of GR actions in CRPC is still work in progress. There are studies suggesting that GR expression can be upregulated following antiandrogen therapy and can contribute to resistance to hormone therapies. Therefore, attempts are being made to develop selective glucocorticoid receptor modulators that specifically antagonize GR activity in CRPC, and thereby provide clinical benefit by blocking the GR mechanism for tumor growth. However, more targeted approaches are needed to understand the role of the GR-mediated target gene expressions in the CRPC that could in near future lead to better therapeutic options for patients with CRPC. This review highlights current perspectives on the actions of glucocorticoids during tumor progression and metastasis of CRPC.
Keywords: castration resistant prostate cancer, glucocorticoid receptor, androgen receptor, therapeutic target
Introduction
Glucocorticoids, secreted from the adrenal gland, are necessary for human life and regulate various physiological processes to maintain homeostasis (1, 2). The glucocorticoids are responsible for modulating essential metabolic, cardiovascular, immune, and behavioral functions (3, 4). Because of their powerful anti-inflammatory and immunosuppressive actions, synthetic glucocorticoids are one of the most widely prescribed drugs in the world today and are used for treating inflammatory and autoimmune diseases, such as asthma, allergy, sepsis, rheumatoid arthritis, ulcerative colitis, and multiple sclerosis (5, 6). They are also commonly prescribed to prevent organ transplant rejection and to treat cancers of the lymphoid system such as leukemias, lymphomas, and myelomas (7, 8). However, patients chronically treated with synthetic glucocorticoids are prone to severe side effects including osteoporosis, diabetes, obesity, glaucoma, growth retardation in children, and hypertension among others (9-11).
In cancer therapies, glucocorticoids have not only been widely used in the treatment of lymphoid malignancies to induce cell apoptosis, but also as co-medication with chemotherapy for solid tumors to reduce inflammation as well as cytotoxic side effects (12, 13). In many types of solid tumors, co-treatment with glucocorticoids is effective in easing symptoms related to chemotherapy or cancer per se (12, 13). However, glucocorticoid therapy may also increase the risk for failure of chemotherapy (13-16). Emerging evidence suggests that glucocorticoids may contribute to failure of chemotherapy and tumor progression of many types of solid tumors including triple negative breast cancer (TNBC) and castration resistant prostate cancer (CRPC) (14-19). Interestingly, glucocorticoids appear to slow cell proliferation in estrogen receptor-positive breast cancer whereas in TNBC, glucocorticoids inhibit chemotherapy-induced cytotoxicity by preventing apoptosis, resulting into increased cell proliferation (16, 17). Similarly, in case of androgen-dependent prostate cancer, glucocorticoids appear to slow proliferation of tumor cells whereas in CRPC, glucocorticoids act quite differently, leading to tumor progression (18, 19).
It is amazing how glucocorticoids use different mechanisms depending on various cancer types and specific biological targets to promote or inhibit cancer progression and proliferation (20, 21). The role of glucocorticoids in the treatment of some cancers such leukemias and lymphomas is reasonably well understood (22, 23). However, the underlying mechanisms of the pro-tumorigenic effects of glucocorticoids in solid tumors are not well known. In recent years, though, pro-tumorigenic roles of glucocorticoids in the CRPC has emerged and glucocorticoid signaling pathway has become an important therapeutic target for these cancer types (18, 19). In this review, we are highlighting some of the latest findings and perspectives on the actions of glucocorticoids during tumor progression and metastasis of CRPC.
Structure and functions of glucocorticoid receptor
The physiological and pharmacological actions of glucocorticoids are mediated via the glucocorticoid receptor (GR) at the level of gene regulation (24, 25). The GR belongs to the superfamily of ligand- dependent intracellular transcription factors (26-28). Unliganded GR resides in the cytosol associated with various proteins including chaperones (e.g., hsp90, hsp70, and p23) and immunophilins of the FK506 family (e.g., FKBP51 and FKBP52) (29, 30). These proteins maintain the receptor in a conformation that is transcriptionally inactive but favors high affinity ligand binding (29, 30). Once ligand-bound, the GR undergoes conformational rearrangements resulting into dissociation of these proteins as well as exposing the nuclear localization signals to rapidly translocate into the nucleus where it induces or represses the transcription of its target genes by binding directly to its specific response element DNA sites and/or by physically interacting with other coregulatory proteins (Figure 1). For example, GR activation can induce apoptosis in lymphocytes (31, 32), whereas its activation results in inhibition of apoptosis in breast epithelial cells (33). The GR can also act through cross-talk with other transcription factors such as activator protein-1, signal transducers and activators of transcription-5 and nuclear factor-ĸB (24-28). In addition to the genomic mode of actions, mainly, via their transcriptional regulation of genes, increasing evidence suggests that glucocorticoids can also act through non-genomic signaling mechanism, which does not require nuclear translocation of GR and GR-mediated transcription (34, 35). These effects are thought to occur by the membrane-bound or cytoplasmic GR (34, 35). Although, the precise mechanisms of non-genomic glucocorticoids signaling are still under investigation and may provide novel therapeutic targets for related diseases in the future.
Classical action of gene regulation by the GR-mediated glucocorticoid signaling. Unliganded receptor is located in the cytosol associated with several heat shock and other chaperone proteins (shown by circular shades around GR). Ligand binding dissociates GR from these associated proteins, and ligand bound, phosphorylated GR translocates to the nucleus where it dimerizes and binds to site-specific DNA binding sequences and interacts with several other coregulatory proteins (shown by hexagonic shapes), remodels chromatin structure (shown by purple color in DNA sequences), and with certain mediators that allow cross talk between the GR and the basal transcription machinery including TATA-Box and RNA Pol II complexes (shown by arrows) affect transcription of GR-target gene. Based on (74, 75).
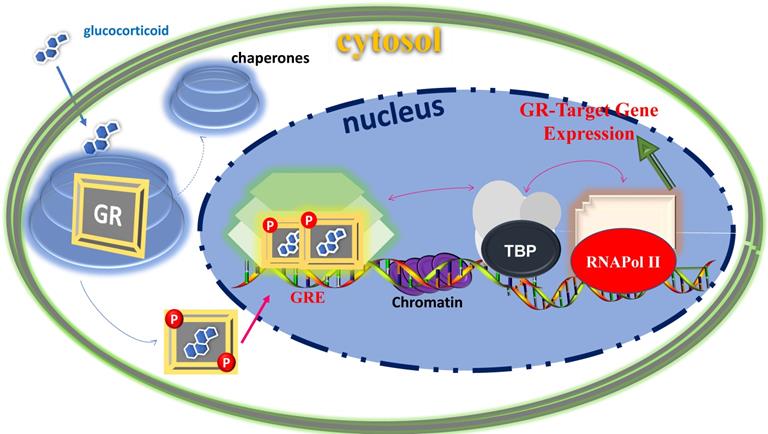
Consistent with the pleiotropic actions of glucocorticoids, GR is expressed in nearly every cell of the body and is necessary for life after birth (7). The transcriptional activity of GR varies widely between cell types, thus accounting for the diverse and sometimes opposite physiological effects of glucocorticoid in different tissues (7). Ligand bound GR also undergoes post translational modifications including phosphorylation and is tightly regulated through cell/tissue specific kinases and phosphatases (36-38). Phosphorylation affects GR stability, nuclear-cytoplasmic shuttling and its interactions with other transcriptional factors ultimately leading to different regulations of GR-responsive genes (36-39). Like other members of the steroid receptor family, GR protein consists of three major functional domains: an N-terminal domain (NTD), a central DNA-binding domain (DBD), and a C-terminal ligand binding domain (LBD) (24-28). The DBD and LBD are responsible for site-specific DNA binding and steroid/hormone binding, respectively (24-28). The NTD houses a powerful transcriptional activation function (AF1), which is constitutively active (24-28). The LBD also possesses a transcriptional activation function (AF2), which acts in a ligand-dependent manner (39). Both AF1 and AF2 are the major sites for the GR's interaction with various coregulatory proteins including the basal transcription machinery proteins (39). The complete action of the GR requires a synergistic effect of both AF1 and AF2 in a cell/tissue-dependent manner (40). Recent studies have shown that unlike DBD and LBD, which exist as globular proteins with well-defined 3-D structure (39), the NTD/AF1 exits as an intrinsically disordered protein (ID), commonly found in many transcription factors including other members of the steroid hormone receptor family (40-42). The NTD/AF1 is also the primary site for post-translational modifications, particularly all the functionally important known phosphorylation sites are located in this region (36-39). Due, in part, to the ID nature of the NTD/AF1, the full length 3-D structure determination of the GR as well as other members of the steroid hormone receptor family has been difficult so far (39). The emerging picture is that in order to access the entire GR signaling spectrum for the development of novel and potent therapeutic agents, we must determine the structure of not only individual domains but of full-length GR (39).
Glucocorticoid receptor in castration resistant prostate cancer
Prostate cancer is second leading cause of cancer-related death among men in the USA (43). Since androgen receptor (AR) plays a critical role in the development and progression of prostate cancer, androgen deprivation therapy (ADT) with lowering of serum testosterone levels to castrate levels has been the mainstay of therapy for these patients for years (44-47). Standard approaches to ADT include surgical bilateral orchiectomy or medical orchiectomy using a gonadotropin releasing hormone (GnRH) agonist alone or in combination with an anti-androgen (antagonist) (47). Some recent studies suggest that surgical orchiectomy may have lower risk for complications and side effects than medical castration with GnRH agonists in treatment of newly diagnosed patients with metastatic prostate cancer (47).
Although ADT is palliative, it can normalize serum levels of prostate-specific antigen in majority of patients, they eventually experience disease progression despite treatment (48). Because early in their development, prostate cancers need relatively high levels of androgens to grow, ADT typically works well at this stage (47, 48). Such prostate cancers are commonly called androgen-sensitive or androgen-dependent (46). Over time, however, prostate cancer tends to relapse and progresses into an incurable state and becomes refractory to ADT (49, 50). These patients ultimately progress to castration resistance, wherein prostate cancer cells become resistant to ADT and develop mechanisms to proliferate despite castrate levels of testosterone (51, 52). This state of disease which continues to grow despite the undetectable levels of androgens is known as CRPC and is highly detrimental (49, 53).
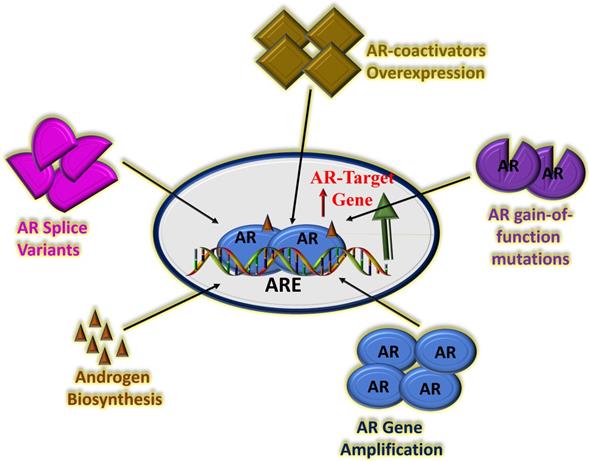
Understanding the exact mechanism of CRPC is still work in progress. In recent years, AR amplification, AR splice variant expression, AR mutation and aberrant AR co-regulators activities have been shown to be involved in CRPC (54-56). Recently, the role of AR splice variant expression in the progression of CRPC has been studied extensively including the most predominant splice variant 7 (AR-V7 or AR3), which encode protein isoforms that activate AR pathway in the absence of androgens (57). Several second-generation AR signaling inhibitors such as cytochrome P450 17α-hydroxy/17,20-lyase (CYP17) inhibitor, abiraterone acetate have been successfully tested in patients with metastatic CRPC (58). Though, the second-generation AR antagonist, enzalutamide prolongs CRPC patient survival yet prostate cancer resistance to potent AR pathway blockade is inevitable (59, 60). Multiple mechanisms of resistance have been proposed including gain-of-function mutations in AR LBD, expression of constitutively active AR splice variants, and more recently increased expression and activity of GR, which can promote CRPC progression following AR blockade (61-64). There are suggestions that GR expression can be upregulated following antiandrogen therapy including enzalutamide and that GR upregulation can bypass the AR pathway and contribute to resistance to hormone therapies (65-67). These observations are important, and therefore warrant new insights into the mechanisms of actions of the GR in the drug resistance (Figure 2). A precise understanding of the mechanisms of action will immensely help in the development of next-generation therapies for CRPC with better clinical outcomes.
Potential mechanisms of CRPC driven by continued AR transcriptional activity. There are several possible factors that can affect AR activity including amplification of the AR gene leading to overexpression of AR protein, AR gain-of-function mutations, overexpression of AR co-activator proteins, increased adrenal androgen biosynthesis and/or AR splice variants with truncated LBD. Based on (66, 67, 76, 77).
Efforts to block the effect of GR in CRPC using classical GR antagonist (RU486 or mifepristone) have been only partially successful (68). This may be due, in part, to the fact that mifepristone can also modulate AR signaling (though weakly) as well as alter the metabolism of other therapeutics through its potent effects on cytochrome P450 enzymatic activity (69, 70). Therefore, attempts are being made to develop selective glucocorticoid receptor modulators that specifically antagonize GR activity in CRPC (without significant binding to other members of the steroid hormone receptor family), and thereby provide clinical benefit by blocking the GR mechanism for tumor growth (71, 72). There are also clinical trials underway to test whether concomitant AR and GR antagonism using mifepristone and enzalutamide can increase the time to endocrine therapy resistance (Clinical Trials Entitled: “Enzalutamide and Mifepristone in Treating Patients With Metastatic Hormone Resistant Prostate Cancer”; Identifier: NCT 02012296). Despite these efforts, currently no FDA- approved therapies specific to enzalutamide-resistant CRPC are in clinical use. Therefore, GR could be a potential therapeutic target in this context. Due to the non-specific activity of mifepristone, development of highly specific GR modulators with principally GR antagonist activity have recently been tested under in vitro and in vivo studies. (73). In this study, two structurally distinct, yet highly selective GR modulators with principally GR antagonistic activity were found to selectively inhibit GR activity through the inhibition of the expression of genes associated with proliferation pathways (73).
Summary and future perspectives
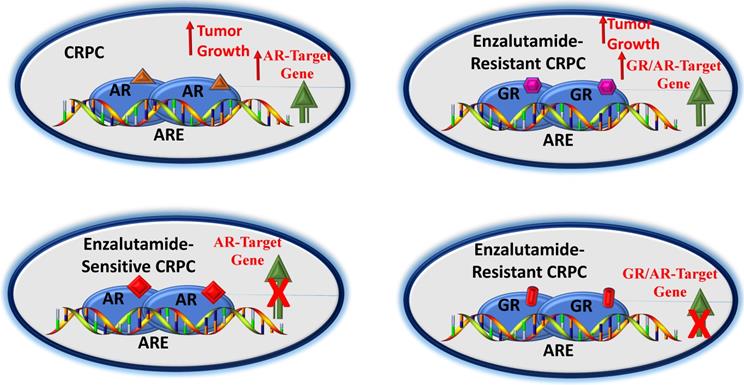
Generally, the standard treatment for men with metastatic prostate cancer involves either ADT alone or in combination with chemotherapy. A diagnosis of metastatic CRPC often means that the patient is no longer responding to ADT type of therapy. Under such circumstances, the available cancer treatment options are limited. In recent year, several pathways have been described for the progression and development of CRPC, which have led to the clinical utility of enzalutamide in the treatment of nonmetastatic CRPC. However, enzalutamide benefits are limited due to the development of drug resistance. Therefore, scientific community has been actively pursuing the mechanisms of this drug resistance. In last few years, a significant progress has been made to understanding the mechanisms of these effects that include expression of constitutively active AR- splice variants, point mutations, gene amplification and/or overexpression. More recently the role of higher expression and transcriptional activity of the GR in enzalutamide-resistant CRPC has caught an immense attention (Figure 3). Inhibition of the GR activity has been found to be effective under in vitro and in vivo conditions that has led to the development of small molecule selective GR modulators. However, more targeted approaches are needed to understand the role of the GR-mediated target gene expressions in the CRPC that could in near future lead to better therapeutic options for patients with CRPC.
A potential pathway by which GR affects CRPC. In early stages of CRPC, an agonist-bound AR can upregulate AR-target genes leading to tumor growth (A). In this androgen-sensitive stage, blockade of AR by an antagonist (e.g., enzalutamide) can block AR-target genes (B). However, enzalutamide-resistance may result into higher GR expression, leading to activation of a subset of AR- and/or as GR- target genes causing tumor growth (C). GR antagonist may overcome GR-driven resistance to antiandrogens (D). Based on (65-67, 76, 77).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev. 1996;17:245-61
2. Kumar R, Thompson EB. Gene regulation by the glucocorticoid receptor: structure:function relationship. J Steroid Biochem Mol Biol. 2005;94:383-94
3. Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Lond). 1998;94:557-72
4. Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55-89
5. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids-new mechanisms for old drugs. N Engl J Med. 2005;353:1711-23
6. Busillo JM, Cidlowski JA. The five Rs of glucocorticoid action during inflammation: ready, reinforce, repress, resolve, and restore. Trends Endocrinol Metab. 2013;24:109-19
7. Oakley RH, Cidlowski JA. The Biology of the Glucocorticoid Receptor: New Signaling Mechanisms in Health and Disease. J Allergy Clin Immunol. 2013;132:1033-44
8. Wang L, Heckmann BL, Yang X. et al. Osteoblast autophagy in glucocorticoid-induced osteoporosis. J Cell Physiol. 2019;234:3207-15
9. Ward LM, Weber DR. Growth, pubertal development, and skeletal health in boys with Duchenne Muscular Dystrophy. Curr Opin Endocrinol Diabetes Obes. 2019;26:39-48
10. Radhakutty A, Burt MG. Management of endocrine disease: Critical review of the evidence underlying management of glucocorticoid-induced hyperglycaemia. Eur J Endocrinol. 2018;179:R207-18
11. Whitcup SM, Cidlowski JA, Csaky KG. et al. Pharmacology of Corticosteroids for Diabetic Macular Edema. Invest Ophthalmol Vis Sci. 2018;59:1-12
12. Rutz HP. Effects of corticosteroid use on treatment of solid tumours. Lancet. 2002;360:1969-70
13. Schmidt S, Rainer J, Ploner C. et al. Glucocorticoid-induced apoptosis and glucocorticoid resistance: molecular mechanisms and clinical relevance. Cell Death and Differ. 2004;11:S45-55
14. Zhang C, Wenger T, Mattern J. et al. Clinical and mechanistic aspects of glucocorticoid-induced chemotherapy resistance in the majority of solid tumors. Cancer Biol Ther. 2007;6:278-87
15. Pan D, Kocherginsky M, Conzen SD. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res. 2011;71:6360-70
16. Skor MN, Wonder EL, Kocherginsky M. et al. Glucocorticoid receptor antagonism as a novel therapy for triple-negative breast cancer. Clin Cancer Res. 2013;19:6163-72
17. Chen Z, Lan X, Wu D. et al. Ligand-dependent genomic function of glucocorticoid receptor in triple-negative breast cancer. Nat Commun. 2015;6:8323
18. Montgomery B, Cheng HH, Drechsler J. et al. Glucocorticoids and prostate cancer treatment: friend or foe? Asian J Androl. 2014;16:354-8
19. Huang Y, Jiang X, Liang X. et al. Molecular and cellular mechanisms of castration resistant prostate cancer. Oncol Lett. 2018;15:6063-76
20. Ahmad N, Kumar R. Steroid hormone receptors in cancer development: A target for cancer therapeutics. Cancer Letters. 2011;300:1-9
21. Jun L, Kumar R. Crosstalk between NFkB and glucocorticoid signaling as a novel target of breast cancer therapy. Cancer Letters. 2012;322:119-26
22. Miller AL, Webb MS, Copik AJ. et al. p38 MAP kinase is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: Correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at Serine 211. Mol Endocrinol. 2005;19:1569-83
23. Thompson EB, Webb MS, Miller AL. et al. Identification of genes leading to glucocorticoid-induced leukemic cell death. Lipids. 2004;39:821-5
24. Kumar R, Thompson EB. Folding of the glucocorticoid receptor N-terminal activation domain: dynamics and regulation. Mol Cell Endo. 2012;348:450-6
25. Kumar R, Thompson EB. Gene regulation by the glucocorticoid receptor: structure and functions relationship. J Steroid Biochem Mol Biol. 2005;94:383-94
26. Kumar R, Johnson BH, Thompson EB. Overview of the structural basis for transcription regulation by nuclear hormone receptors. Essay Biochem. 2004;40:27-39
27. Kumar R, Thompson EB. Transactivation functions of the N-terminal domains of nuclear receptors: protein folding and coactivator interactions. Mol Endo. 2003;17:1-10
28. Kumar R, Thompson EB. The structure of the nuclear hormone receptors. Steroids. 1999;64:310-9
29. Grad I, Picard D. The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol. 2007;275:2-12
30. Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306-60
31. Gibson S, Tu S, Oyer R. et al. Epidermal growth factor protects epithelial cells against Fas-induced apoptosis. Requirement for Akt activation. J Biol Chem. 1999;274:17612-8
32. Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284:555-6
33. Moran TJ, Gray S, Mikosz CA. et al. The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Res. 2000;60:867-72
34. Samarasinghe RA, Witchell SF, DeFranco DB. Cooperativity and complementarity: synergies in non-classical and classical glucocorticoid signaling. Cell Cycle. 2012;11:2819-27
35. Samarasinghe RA, Di Maio R, Volonte D. et al. Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc Natl Acad Sci USA. 2011;108:16657-62
36. Khan SH, McLaughlin WA, Kumar R. Site-specific phosphorylation regulates the structure and function of an intrinsically disordered domain of the glucocorticoid receptor. Scientific Rep. 2017;7:15440
37. Garza AM, Khan SH, Kumar R. Site-specific phosphorylation induces functionally active conformation in the intrinsically disordered N-terminal activation function (AF1) domain of the glucocorticoid receptor. Mol Cell Biol. 2010;30:220-30
38. Kumar R, Calhoun WJ. Differential regulation of the transcriptional activity of the glucocorticoid receptor through site-specific phosphorylation. Biologics: Targets Therapy. 2008;2:845-54
39. Kumar R. McEwan, IJ. Allosteric modulators of steroid hormone receptors: structural dynamics and gene regulation. Endocrine Rev. 2012;33:271-99
40. Goswami D, Pascal B, Kumar R. et al. Structural dynamics and inter domain crosstalk of PR-TBP interaction probed by hydrogen/deuterium exchange Mass Spectrometry. Structure. 2014;22:961-73
41. Simons SS, Edwards DP, Kumar R. Dynamic Structures of Nuclear Hormone Receptors: New Promises and Challenges. Mol Endocrinol. 2014;28:173-82
42. Kumar R, Moure CM, Khan SH. et al. Regulation of the structurally dynamic disordered amino-terminal domain of progesterone receptor by protein induced folding. J Biol Chem. 2013;288:30285-99
43. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7-30
44. Davey RA, Grossmann M. Androgen receptor structure, function and biology: from bench to bedside. Clin Biochem Rev. 2016;37:3-15
45. Edwards J, Krishna NS, Grigor KM. et al. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br J Cancer. 2003;89:552-6
46. Crona DJ, Whang YE. Androgen receptor-dependent and -independent mechanisms involved in prostate cancer therapy resistance. Cancers. 2017;9:67
47. Tucci M, Zichi C, Buttigliero C. et al. Enzalutamide-resistant castration-resistant prostate cancer: challenges and solutions. Onco Targets Ther. 2018;11:7353-68
48. Mostaghel EA, Plymate SR, Montgomery B. Molecular pathways: targeting resistance in the androgen receptor for therapeutic benefit. Clin Cancer Res. 2014;20:791-8
49. Tucci M, Scagliotti GV, Vignani F. Metastatic castration-resistant prostate cancer: time for innovation. Future Oncol. 2015;11:91-106
50. Hussain M, Fizazi K, Saad F. et al. Enzalutamide in men with nonmetastatic, castration-resistant prostate cancer. N Engl J Med. 2018;378:2465-74
51. Mosillo C, Iacovelli R, Ciccarese C. et al. De novo metastatic castration sensitive prostate cancer: State of art and future perspectives. Cancer Treat Rev. 2018;70:67-74
52. Oudard S, Fizazi K, Sengeløv L. et al. Cabazitaxel versus docetaxel as first-line therapy for patients with metastatic castration-resistant prostate cancer: a randomized phase III trial-FIRSTANA. J Clin Oncol. 2017;35:3189-97
53. James ND, de Bono JS, Spears MR. et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med. 2017;377:338-51
54. Karantanos T, Corn PG, Thompson TC. Prostate cancer progression after androgen deprivation therapy: mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 2013;32:5501-11
55. Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253-61
56. Amaral TM, Macedo D, Fernandes I. et al. Castration-resistant prostate cancer: mechanisms, targets, and treatment. Prostate Cancer. 2012;2012:327253
57. Guo Z, Qiu Y. A new trick of an old molecule: androgen receptor splice variants taking the stage? Int J Biol Sci. 2011;7:815-22
58. Alex AB, Pal SK, Agarwal N. CYP17 inhibitors in prostate cancer: latest evidence and clinical potential. Ther Adv Med Oncol. 2016;8:267-75
59. Kregel S, Chen JL, Tom W. et al. Acquired resistance to the second-generation androgen receptor antagonist enzalutamide in castration-resistant prostate cancer. Oncotarget. 2016;7:26259-74
60. Antonarakis ES, Lu C, Wang H. et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028-38
61. Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701-11
62. Li Z, Bishop AC, Alyamani M. et al. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature. 2015;523:347-51
63. Itani OA, Liu KZ, Cornish KL. et al. Glucocorticoids stimulate human sgk1 gene expression by activation of a GRE in its 5′-flanking region. Am J Physiol Endocrinol Metab. 2002;283:E971-9
64. Bolton EC, So AY, Chaivorapol C. et al. Cell- and gene-specific regulation of primary target genes by the androgen receptor. Genes Dev. 2007;21:2005-17
65. Hirayama Y, Sadar MD. Does increased expression of glucocorticoid receptor support application of antagonists to this receptor for the treatment of castration resistant prostate cancer? AME Med J. 2018;3:66
66. Rodriguez-Vida A, Galazi M, Rudman S. et al. Sternberg Enzalutamide for the treatment of metastatic castration-resistant prostate cancer. Drug Design, Develop Therapy. 2015:9 3325-39
67. Arora VK, Schenkein E, Murali R. et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309-22
68. Taplin ME, Manola J, Oh WK. et al. A phase II study of mifepristone (RU-486) in castration-resistant prostate cancer, with a correlative assessment of androgen-related hormones. BJU Int. 2008;101:1084-9
69. Song LN, Coghlan M, Gelmann EP. Antiandrogen effects of mifepristone on coactivator and corepressor interactions with the androgen receptor. Mol Endocrinol. 2004;18:70-85
70. Clark RD. Glucocorticoid receptor antagonists. Curr Top Med Chem. 2008;8:813-38
71. Clark RD, Ray NC, Williams K. et al. 1H-Pyrazolo[3,4-g]hexahydro- isoquinolines as selective glucocorticoid receptor antagonists with high functional activity. Bioorg Med Chem Lett. 2008;18:1312-7
72. Hunt HJ, Ray NC, Hynd G. et al. Discovery of a novel non-steroidal GR antagonist with in vivo efficacy in the olanzapine-induced weight gain model in the rat. Bioorg Med Chem Lett. 2012;22:7376-80
73. Kach J, Long TM, Selman P. et al. Selective glucocorticoid receptor modulators (SGRMs) delay castrate-resistant prostate cancer growth. Mol Cancer Ther. 2017;16:1680-92
74. Kumar R, Thompson EB. Role of Phosphorylation in the Modulation of the Glucocorticoid Receptor's Intrinsically Disordered Domain. Biomolecules. 2019;9:95
75. Kumar R, McEwan IJ. Glucocorticoid Receptor Structure and Function. McEwan I, Kumar R. (eds) Nuclear Receptors: From Structure to the Clinic. Springer, Cham. 2015:41-57
76. Imamura Y, Sadar MD. Androgen receptor targeted therapies in castration-resistant prostate cancer: Bench to clinic. Int J Urol. 2016;23:654-65
77. Narayanan S, Srinivas S, Feldman D. Androgen-glucocorticoid interactions in the era of novel prostate cancer therapy. Nature Rev Urol. 2016;13:47-60
Author contact
![]() Corresponding author: Raj Kumar, Ph.D., Department of Biomedical Sciences, University of Houston - College of Medicine, Health 2 Building, 4849 Calhoun Road, Suite 8035, Houston, TX 77204. Phone: 713-743-2730; E-mail: rkumar23uh.edu
Corresponding author: Raj Kumar, Ph.D., Department of Biomedical Sciences, University of Houston - College of Medicine, Health 2 Building, 4849 Calhoun Road, Suite 8035, Houston, TX 77204. Phone: 713-743-2730; E-mail: rkumar23uh.edu