Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(6):1371-1382. doi:10.7150/jca.38603 This issue Cite
Research Paper
Practice of the New Integrated Molecular Diagnostics in Gliomas: Experiences and New Findings in a Single Chinese Center
Wan-Ming Hu1*, Fang Wang2*, Shao-Yan Xi1*, Xiao Zhang2, Jun-Peng Lai1, Hui-Yu Wu3, Li-Ling Liu1, Ke Sai4 ![]() , Jing Zeng1
, Jing Zeng1 ![]()
1. Department of Pathology, Sun Yat-sen University Cancer Center; State Key Laboratory of Oncology in South China; Collaborative Innovation Center for Cancer Medicine, Guangzhou, Guangdong, P. R. China;
2. Department of Molecular Diagnostics, Sun Yat-sen University Cancer Center;
3. Department of General, Sun Yat-sen University Cancer Center;
4. Department of Neurosurgery, Sun Yat-sen University Cancer Center;
*These authors contributed equally to this work.
Received 2019-7-21; Accepted 2019-12-2; Published 2020-1-1
Abstract
Background: The latest WHO classification of CNS tumors using the integrated phenotypic and molecular parameters (IDH, ATRX, 1p19q, TERT etc.) have reestablished the CNS tumors classification in addition to traditional histology. The establishment of glioma molecular typing can more accurately predict prognosis, better guide individualized treatment to improve survival.
Methods: The expression of IDH1, ATRX, PHH3, P53 and Ki67 was detected by IHC. Molecular status of IDH1/2 and TERT were analyzed using Sanger sequencing. MGMT was explored using methylation-specific PCR. 1p/19q codeletion status was firstly detected by FISH, then further confirmed by multiplex PCR-based next generation sequencing.
Results: The mutation frequency of IDH1 was 68.7% (79/115) in WHO II astrocytoma, and 82 cases (82/344, 23.8%) were “triple-negative glioma” in our cohort. Multivariate COX analysis revealed that only IDH, 1p/19q, TERT and MGMT were independent prognostic factors. Noteworthily, we found 7 cases of the new molecular phenotype presented as “IDH wildtype and 1p/19q codeletion”, not mentioned in the latest WHO guideline.
Conclusion: We detected the newly recommended markers in a large cohort of Chinese glioma patients. Our data demonstrated a relatively lower frequency of IDH mutations and a higher prevalence of triple-negative glioma in Chinese compared with American and European, indicating ethnic and geographical difference in some markers. In addition, the new molecular phenotype “IDH wildtype and 1p/19q codeletion” glioma deserved special focus. These findings suggest that further stratification of infiltrating gliomas is needed for different treatment strategy and precision medicine.
Keywords: IDH, ATRX, 1p/19q, TERT, MGMT, FISH, Sanger sequencing, glioma, glioblastoma
Background
Glioma is the most common malignant and highly aggressive brain tumors, possessing the characteristics of infiltrating growth and easy recurrence. The treatment is very difficult and currently considered to be incurable. The total prognosis of gliomas was dismal, especially in glioblastoma multiforme's (GBMs). GBMs account for 30% of all brain and CNS (central nervous system) tumors and 80% of all malignant brain tumors [1]. Although after several years of research, advancements in surgical techniques, and improvement of clinical care, GBMs still retains the poor prognosis of roughly 10 to 15 months. At the same time, different kinds of gliomas have a large heterogeneity. The survival ranges widely, from few months to several years. The imperfect classification of gliomas has restricted the development of new therapies for gliomas. Traditionally, the pathological diagnosis of glioma is almost entirely dependent on histomorphological features, and is classified into WHO grade I to IV. This tendency to subjective diagnosis leads to variability in outcomes between different pathologists and inability to accurately interpret prognosis [2]. In 2010, Verhaak et al [3] published a landmark paper that puts forward the idea that glioblastoma have been divided into four subtypes base on the differentiated molecular characteristics. Four subtypes were namely classical, mesenchymal, proneural and neural. More recently, intensive research on the molecular genetic changes of glioma has led to the adoption of more and more molecular markers by WHO as diagnostic, prognostic and therapeutic indicators [4]. The different subtypes of gliomas have been refurbished by the latest 2016 WHO criteria which provide the comprehensive diagnosis with phenotype and genotype feature compared with the traditional histologic classification. Using a lot of molecular parameters in addition to histology, and molecular testing of brain tumors becomes a crucial part of the diagnosis of glioma.
The purpose of this study was a retrospective analysis of 544 infiltrating glioma samples in our center. The expression of these markers in the population of gliomas in southern China was determined and compared with existing studies in the world to find out the differences and characteristics in order to further improve the diagnosis of glioma in the future, and lay the foundation for future targeted therapies.
Methods
Patients and Tissues
A cohort of 544 infiltrating glioma cases (WHO II-IV) were obtained from the Department of Pathology, Sun Yat-sen University Cancer Center, Guangzhou, China, between 1998 and 2016. Samples and data anonymization have been approved by Ethics Committee of SYSUCC. And the samples were obtained the informed consent of the patient. The malignant series consisted of 176 cases of WHO II (astrocytoma and oligodendroglioma), 159 cased of WHO III (anaplastic astrocytoma and oligodendroglioma) and 209 cases of WHO IV (glioblastoma). The ratio of male to female was 1.34:1. The median patient age at the time of primary surgery was 42 years. The median survival time was 29 months (range 0-188 months). All the tissues used for study were FFPE (formalin fixed and paraffin embedded) blocks.
Immunohistochemistry (IHC)
Many of genetic discoveries can be interrogated with cost-effective immunohistochemical stains in gliomas. The most commonly used surrogates are IDH1, ATRX and P53. Immunohistochemistry studies were performed using standard techniques as described previously [5]. IDH1-R132H (clone H09, 1:50; Dianova, Hamburg, Germany), ATRX (1:500; Sigma-Aldrich, St. Louis, MO, USA), and P53/PHH3/ Ki67(1:100; Dako, Carpinteria, CA) was detected using formalin-fixed, paraffin-embedded tumor tissue sections on an automated BenchMark Ultra (Ventana Medical systems, Roche, SW). Sections from known mutation-positive and immunoreactive tumors were used as positive controls. Negative controls consisted of sections incubated with Phosphate-buffered saline instead of the primary antibody.
Interpretation standard of IDH1, ATRX, P53, Ki67 and PHH3
Most IDH positive cases were presented as a strong diffuse cytoplasmic immunoreaction of IDH1-R312H staining. However, a strong but mottled staining could be seen in very few cases, for these cases, a threshold of 30% of positive cytoplasmic staining was used for cut-off value [6] .
Nuclear ATRX loss was scored as ATRX mutation if tumor cell nuclei were unstained while nuclei of non-neoplastic cells such as endothelia, microglia, lymphocytes and reactive astrocytes were strongly positive [7]. A threshold of 10% of strong positive tumor nuclei was used to assign immunopositivity for ATRX [8] and P53 [9], which provide the most accurate prediction of mutation.
The Ki67 index was calculated as the average percentage of positive nuclei on the total number of nuclei in high power microscopic fields, at 400× magnification. The statistical score was using a two-grade scale, with low (0) and high (1), which was representative for “< 10%” and “≥ 10%”. Score 1 was defined as high Ki-67 expression [10].
The mitotic index (PHH3) was calculated for the exact positive nuclei (the number of mitotic figures) in high power microscopic fields, at 400× magnification. The statistical score was also low (0) and high (1), which was representative for “< 5” and “≥ 5” mitotic figures per 10 HPF.
All the IHC interpretation was performed by two certified neuropathologists in all cases.
Molecular genetics
1. Fluorescent in situ hybridization (FISH) was performed to detect 1p and 19q deletion using Vysis FISH Probe Kit (Abbott Molecular, Illinois, USA). At least≥25% of counted nuclei presented one target signal and two reference signals will be considered as 1p or 19q deleted when 100 non-overlapping nuclei were counted.
2. Mutation status of IDH1/2 and TERT promoter was studied with Sanger sequencing. Hotspot codons IDH1 Arg132 (exon 4)/IDH2 Arg172 (exon 4) and the hotspot mutations of TERT promoter at positions C228T and C250T were detected on an ABI® 3130 Genetic Analyzer (Life Technologies, USA), as described in another research [11].
3. The promoter methylation status of the MGMT gene was assessed using methylation-specific PCR with the EZ DNA MethylationDirect kit (Zymo Research Corp., Orange, California, USA).
4. Multiplex PCR-Based Next generation sequencing. We used this method to confirm the true 1p/19q codeletion. Primers for several segments of chromosome 1p and 19q as well as barcoding adapter DNA oligos in the first and second enrichment separately and synthesized by Sangon Biotech (Sangon Biotech, Shanghai, China). Sequencing libraries were generated using multiplex PCR methods. Each reaction was cleaned once using Agencourt AMPure XP kit (Beckman, Indianapolis, USA) to remove unused primers, according to the manufacturer's specifications. The concentration of the barcoded PCR produced library was measured by Qubit 3.0(Thermo Fisher Scientific, MA, USA), and diluted amplicons were sequenced on the Ion Proton system (Thermo Fisher Scientific, MA, USA).
Statistical methods
Associations between categorical variables were evaluated by use of 2 × 2 contingency tables and the Chi square (χ2) test. The association between parameters was assessed using Spearman correlation coefficient. Overall survival was calculated from the time of surgery until death or the last follow-up. Univariate survival analysis was performed using Kaplan-Meier curves and the log-rank test. Multivariate analyses were done, involving a Cox proportional hazards model for which values of p<0.05 were considered significant. Analyses were carried out using SPSS16.0 (Chicago, IL, USA).
Results
The IHC results of IDH1R132H, ATRX, P53, PHH3, Ki67 and the molecular status of IDH1/2 mutation, 1p/19q chromosomal deletion, MGMT promoter methylation, TERT promoter mutation: A relatively low rate of IDH mutation and a high proportion of triple-negative gliomas in Chinese
In our cohort, the total positive rate for each marker was showed as follows: IDH1R132H (43.3%), ATRX (58.4%), P53 (58.7%), PHH3≥5/10HPF (58.4%), Ki67≥10% (66.3%), 1p/19q codeletion (19.1%), TERT promoter mutation (36.2%) and MGMT promoter mutation (43.7%). Based on these results, we have come to the following findings:
1. The traditional WHO grade was associated with the patient's age, IDH1, ATRX, P53, PHH3, Ki67, 1p/19q status and TERTp mutation(p<0.001), not related to the patient's gender and MGMTp status.
2. IDH1R132H immunoreactivity in tumor cell components occurred in most WHO II astrocytoma, and sanger sequencing revealed 68.7% (79/115) of WHO II astrocytoma occurred IDH1/2 mutated in our cohorts. Meaningfully, the incidence is lower in both our cohort and CGGA dataset than other researches focused on European and American including TCGA dataset (Table 1).
Frequency of IDH1 Mutations in WHO II astrocytoma Patients.
| Country | Total | IDH1 | IDH1 |
|---|---|---|---|
| Mutated | Mutated rate | ||
| China (present study) | 115 | 79 | 68.70% |
| China [12] | 50 | 29 | 58.00% |
| China [13] | 206 | 145 | 70.39% |
| China [14] | 125 | 86 | 68.80% |
| China [15] | 37 | 25 | 67.57% |
| China [16] | 417 | 309 | 74.10% |
| China [17] | 111 | 72 | 64.86% |
| Italy [18] | 87 | 66 | 75.86% |
| France [19] | 47 | 40 | 85.11% |
| Germany [20] | 71 | 54 | 76.06% |
| Germany [21] | 431 | 332 | 77.03% |
| America [22] | 233 | 203 | 87.12% |
| America [23] | 81 | 70 | 86.42% |
| America [24] | 75 | 64 | 85.33% |
| America [25] | 159 | 141 | 88.68% |
| CGGA [26] | 117 | 82 | 70.01% |
| TCGA | 415 | 323 | 77.83% |
Compared with Caucasians, Asians have a relatively low frequency of IDH mutations rate; CGGA=Chinese Glioma Genome Altas; TCGA=The Cancer Genome Atlas
3. Loss of nuclear ATRX expression was remarkably relevant with IDH1/2 mutations (p<0.001), on the contrary, ATRX loss was mutually exclusive with the total 1p/19q codeletion (p<0.001).
4. The frequency of ATRX loss was the highest in WHO II astrocytoma (57.1%), while PHH3 and Ki67 were both exhibited high expression rate in high grade gliomas (WHO III and IV).
5. When using the FISH method to detect the 1p/19q status, it was found that in addition to cases with 1p/19q codeletion, there were a few cases with 1p deletion alone or 19q alone, but these individual gene deletion cases had no prognostic value.
6. The TERT promoter mutation was the highest (44.9%) in the WHO IV glioblastoma, followed by WHO II oligodendroglioma with IDH mutation and 1p/19q codeletion.
7. In our study, unfortunately, the detection failure rate of MGMT promoter methylation was high. From the results obtained, it is known that the highest proportion of MGMT promoter methylation occurs in glioblastomas (51.4%). It is well known that it is associated with the efficacy of temozolomide chemotherapy (patients with MGMT mutations have a better prognosis).
8. In our cohort, 344 WHO II&III cases have full molecular information of IDH, 1p/19q and TERT, and a total of 23.8% (82/344) was triple-negative (IDH wildtype, TERT promoter wildtype and no 1p/19q codeletion). The results were similar to Ng, H K et al., for the frequency of triple-negative gliomas was higher in Chinese patients (17.4% in Chinese VS 7% in Caucasian) [27, 28].
The representative figures and detailed results were showed in Figure 1, Figure 2 and Table 2.
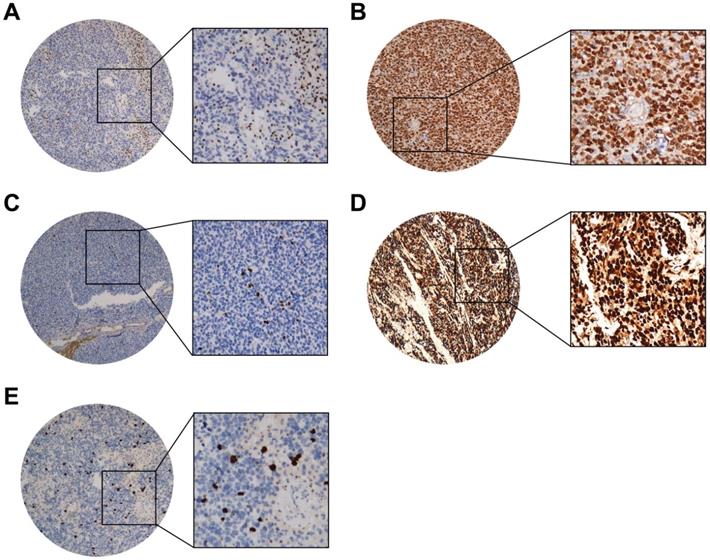
Representative IHC staining cases of (A) ATRX (nuclear loss expression while nuclei of non-neoplastic cells such as endothelia, microglia, lymphocytes and reactive astrocytes were strongly positive), (B) IDH1(strong diffuse cytoplasmic staining), (C) Ki67(Nuclear positive), (D) P53 (Nuclear positive), (E) PHH3 (Nuclear positive). (40X and 200X).
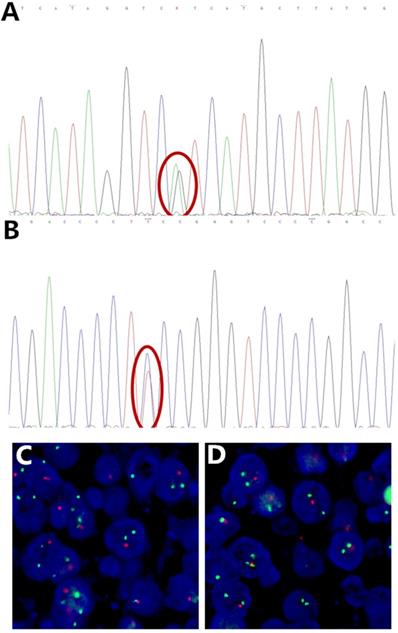
Representative Sanger sequencing images of IDH mutation, TERT promoter mutation and FISH image of 1p/19q deletion. (A) IDH1 R132H mutation (red circle), (B) TERT promoter C250T mutation (red circle), (C) 1p deletion, (D) 19q deletion, with 1 red and 2 green signals in scattered nuclei. Some signals are missing due to nuclear truncation and overlap.
The detailed results of all the markers in our cohort. (grouped by the WHO grade)
| Variables | Grade II | Grade III | Grade IV | Total | P Value | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Median age (year) | 36 | 41 | 49 | |||||||
| Age | ≥ 45 years | 135 | 76.70% | 100 | 62.89% | 88 | 42.11% | 323 | <0.001* | |
| < 45 years | 41 | 23.30% | 59 | 37.11% | 121 | 57.89% | 221 | |||
| Missing | 0 | 0 | 0 | |||||||
| Gender | Male | 102 | 57.95% | 91 | 57.23% | 119 | 56.94% | 312 | 0.983 | |
| Female | 74 | 42.05% | 68 | 42.77% | 90 | 43.06% | 232 | |||
| Missing | 0 | 0 | 0 | |||||||
| IDH1 | Positive | 116 | 67.84% | 73 | 47.40% | 36 | 17.82% | 225 | <0.001* | |
| Negative | 55 | 32.16% | 81 | 52.60% | 166 | 82.18% | 302 | |||
| Missing | 5 | 5 | 7 | 17 | ||||||
| ATRX | Positive | 73 | 42.94% | 99 | 65.13% | 120 | 60.30% | 292 | <0.001* | |
| Negative | 97 | 57.06% | 53 | 34.87% | 79 | 39.70% | 229 | |||
| Missing | 6 | 7 | 10 | 23 | ||||||
| P53 | Positive | 82 | 48.24% | 91 | 58.33% | 144 | 69.90% | 317 | <0.001* | |
| Negative | 88 | 51.76% | 65 | 41.67% | 62 | 30.10% | 215 | |||
| Missing | 6 | 3 | 3 | 12 | ||||||
| PHH3 | High (≥ 5) | 9 | 5.17% | 136 | 86.62% | 171 | 83.41% | 316 | <0.001* | |
| low(< 5) | 165 | 94.83% | 21 | 13.38% | 34 | 16.59% | 220 | |||
| Missing | 2 | 2 | 4 | 8 | ||||||
| Ki67 | High (≥ 10%) | 46 | 26.29% | 118 | 74.21% | 205 | 98.09% | 369 | <0.001* | |
| Low (< 10%) | 129 | 73.71% | 41 | 24.26% | 4 | 1.91% | 174 | |||
| Missing | 1 | 0 | 0 | 1 | ||||||
| 1p/19q | Co-deleted | 42 | 26.42% | 43 | 31.16% | 6 | 3.37% | 91 | <0.001* | |
| 1p deleted | 4 | 2.52% | 7 | 5.07% | 12 | 6.74% | 23 | |||
| 19q deleted | 6 | 3.77% | 8 | 5.80% | 15 | 8.43% | 29 | |||
| Intact | 107 | 67.30% | 80 | 57.97% | 145 | 81.46% | 332 | |||
| Missing | 17 | 21 | 31 | 69 | ||||||
| TERTp | Mutated | 39 | 24.38% | 55 | 38.73% | 84 | 44.92% | 178 | <0.001* | |
| Wildtype | 121 | 75.63% | 87 | 61.27% | 103 | 55.08% | 311 | |||
| Missing | 16 | 17 | 22 | 55 | ||||||
| MGMTp | Methylated | 35 | 37.23% | 31 | 39.24% | 62 | 51.24% | 128 | 0.058 | |
| Wildtype | 59 | 62.77% | 48 | 60.76% | 59 | 48.76% | 166 | |||
| Missing | 82 | 80 | 88 | 250 | ||||||
Prognostic factors for infiltrating gliomas: IDH, 1p/19q, TERT and MGMT were independent prognostic factors
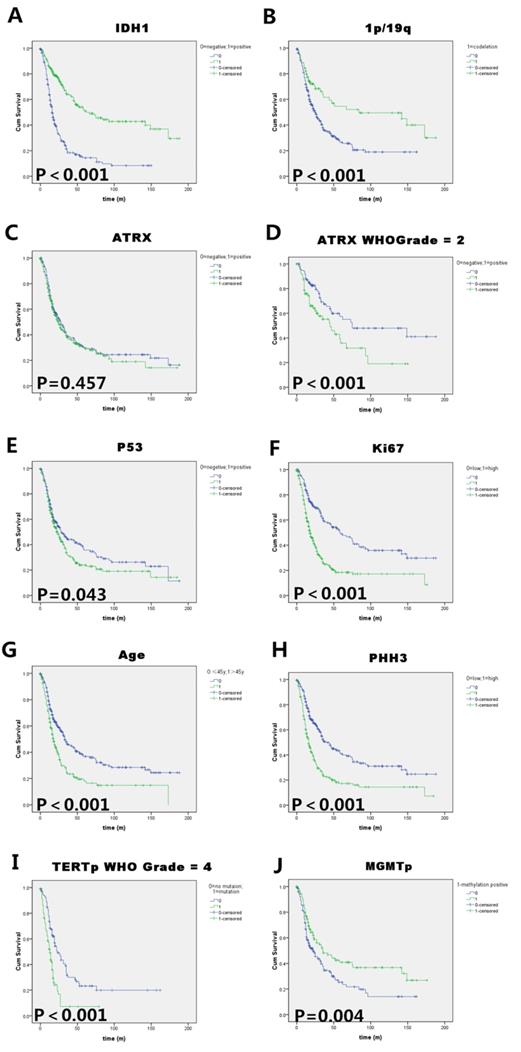
We summarize the prognostic values of each marker in univariate analysis and the results of multivariate COX analysis in Tables 3, and Figure 3 shows the significant survival curves of each useful marker in glioma patients. In Kaplan-Meier univariate analysis, we observed that patients with IDH1-R132H positive tumors had a remarkably longer survival time than those with IDH1-R132H negative tumors (Median OS of IDH1-R132H positive=67 months, Median OS of IDH1-R132H negative=17 months; p<0.001).1p/19q codeletion patients had an extremely longer survival time (Median OS=84 months; p<0.001), but 1p deletion or 19q deletion alone didn't achieve statistical significance. ATRX loss was a favorable prognostic factor only in WHO II astrocytoma patients, but in all the glioma patients, it has no statistically significant (Median OS was 28 to 25, p=0.457). P53 mutation was a relatively unfavorable prognostic factor in our cohort (Median OS was 23 to 29, p=0.043). High PHH3/Ki-67 expression was associated with shorter OS in patients (Median OS was 16 to 45 and 19 to 59, respectively; both p<0.001). For the molecular status of TERT and MGMT promoter, the patients with TERTp wild type (especially in WHO IV/GBM) and MGMTp methylated had a better survival (Median OS of TERTp wild type=33 months, Median OS of TERTp mutated=22 months; p<0.001, Median OS of MGMTp methylated=35 months, Median OS of MGMTp methylated=23 months; p=0.004). Furthermore, the multivariate analysis showed that IDH (p<0.001), 1p/19q (p<0.001) TERT (p=0.006) and MGMT (p=0.002) were independent prognostic factors, for the PR were 0.262, 0.394, 1.727 and 0.57, respectively. Notably, in our study, the traditional indicators of malignancy, PHH3 and KI67, were not independent factors of prognosis.
Univariate and multivariate analyses of different prognostic variables in infiltrating glioma patients
| Parameters | Univariate analysis | Multivariate analysis (COX) | ||||
|---|---|---|---|---|---|---|
| Median OS | p | RR | 95.0% CI for Exp(B) | P | ||
| (month) | (Log rank) | Lower | Upper | |||
| Gender | 0.278 | 1.176 | 0.826 | 1.674 | 0.368 | |
| Male | 24 | |||||
| Female | 27 | |||||
| Age | <0.001* | 0.962 | 0.668 | 1.385 | 0.835 | |
| ≤ 45 years old | 33 | |||||
| > 45 years old | 18 | |||||
| WHO Grade | <0.001* | 1.311 | 0.734 | 2.343 | 0.360 | |
| Low (II) | 62 | |||||
| High (III, IV) | 18 | |||||
| IDH1 | <0.001* | 0.262 | 0.162 | 0.425 | <0.001* | |
| Mutated | 67 | |||||
| Wildtype | 17 | |||||
| ATRX | 0.457 | 0.963 | 0.67 | 1.384 | 0.839 | |
| Mutated | 28 | |||||
| Wildtype | 25 | |||||
| P53 | 0.043* | 1.18 | 0.808 | 1.724 | 0.392 | |
| Mutated | 23 | |||||
| Wildtype | 29 | |||||
| PHH3 | <0.001* | 1.129 | 0.734 | 1.738 | 0.581 | |
| <5 | 45 | |||||
| ≥5 | 16 | |||||
| Ki67 | <0.001* | 1.064 | 0.642 | 1.765 | 0.809 | |
| <10% | 59 | |||||
| ≥10% | 19 | |||||
| 1p/19q | <0.001* | 0.394 | 0.236 | 0.581 | <0.001* | |
| Codeletion | 84 | |||||
| Intact | 25 | |||||
| TERTp | <0.001* | 1.727 | 1.172 | 2.544 | 0.006* | |
| Mutated | 22 | |||||
| Wildtype | 33 | |||||
| MGMTp | 0.004 | 0.57 | 0.397 | 0.819 | 0.002* | |
| Methylated | 35 | |||||
| Unmethylated | 23 | |||||
Kaplan-Meier survival analysis of (A) IDH1 mutation status, (B)1p/19q codeletion status, (C) ATRX immunoreactivity (negative=ATRX mutation), (D)ATRX status in WHO II astrocytoma patients, (E) P53 immunoreactivity, (F) Ki-67 labelling index (cut-off value=10%), (G) Patients' age at diagnosis (cut-off value=45 years), (H) PHH3(mitosis number/10HPF, cut-off value=5 ), (I) TERT promoter status in WHO IV glioblastoma patients and (J) MGMT promoter methylation status in all the infiltrating glioma patients. Log rank test p values are also showed for each parameter. IDH1 mutation, 1p/19q codeletion and MGMT promoter methylation were good prognostic indicators in all the infiltrating gliomas. Transitional markers such as high expression of P53/Ki67/PHH3 and old age exerted unfavorable prognosis. ATRX only have prognostic value in WHO grade II gliomas, and TERT promoter mutation only have prognostic value in WHO grade IV glioma (GBM) in our cohort.
Correlation of IDH1/2 Mutation with IDH1 R132H IHC
544 cases were evaluated for the expression of the IDH1R132H mutant protein by IHC, of which 225 (41.4%) were positive and 319 (58.6%) were negative. Then, all the cases were sent to molecular detection, and the results were effective in 482 cases (88.6%). The frequencies of IDH1 mutations detected by the Sanger sequencing were 214 cases (44.4%), with IDH1 R132H mutation accounted for the vast majority of IDH1 mutation (92.9%, 199/214), followed by IDH1 R132C (4.2%, 9/214), R132S (1.4%, 3/214) and R132G (1.4%, 3/214), but we only found one IDH2 R172K mutation WHO grade II case in our cohort. Our data showed that IDH1R132H IHC results completely matched the IDH1 mutation status, demonstrating high sensitivity and consistency of the anti-IDH1R132H antibody (clone: H09) compared with molecular detection. Details were shown in Table 4.
IDH1R132H IHC (544 cases) and sequencing analysis of IDH1/2 mutation results (482 cases)
| WHO Grade | IDH1(R132H) IHC n/N (%) | Sequencing analysis | |
|---|---|---|---|
| IDH1 mutation n/N (%) | IDH2 mutation n/N (%) | ||
| II | 116/171(67.8) | 108/158(68.4) | 1/158(0.6) |
| III | 73/152(48.0) | 70/140(50.0) | 0/140(0.0) |
| IV | 36/199(18.1) | 36/184(19.6) | 0/184(0.0) |
Conflicting molecular testing results with the phenotype presented as “IDH wild type and 1p/19q codeletion”
Among 544 infiltrating glioma cases in our cohort, we firstly found 20 cases were IDH wild type (Sanger sequencing) and 1p/19q codeletion (FISH). Although FISH is an approved and common method for 1p/19q molecular testing on routinely FFPE tissues, it was defective in some cases in the interpretation of results such as a critical value, only focal deletion, accompanied by polyploid deletion and may lead to false positive results. Based on this possibility, we sent all these 20 cases to retest the sample using Multiplex PCR-Based Next Generation Sequencing, and finally identified 7 cases of the new phenotype “IDH wild type and 1p/19q codeletion”. The age range is 25 to 75 years old. The average age is 46, mostly located in the frontal lobe. Notably, all the 7 cases were ATRX positive in IHC (Wild type), and 5/7(71.4%) cases have TERT promoter mutation (Table 5). It has no prognostic significance in our cohort due to the limited cases. To further confirm the results, we searched online CGGA database, and also found 4 cases with the same phenotype of “IDH wild type and 1p/19q codeletion”, and details was shown in Table 5 of case 11-14. Significantly, it should be brought to the forefront: although only accounted for a very small percentage, it really existed “IDH wild type and 1p/19q codeletion” oligodendroglioma and glioblastoma (GBM with oligodendrocyte components in the old version of WHO blue book) in practice.
Cases of phenotype “IDH wild type and 1p/19q codeletion”
| No. | Gender | Age | Morphological Diagnosis | Location | ATRX | TERT | OS |
|---|---|---|---|---|---|---|---|
| 1 | Male | 44 | Anaplastic oligodendroglioma | Right frontal temporal lobe | wildtype | C228T | 13m(Dead) |
| 2 | Female | 51 | Glioblastoma with oligodendrocyte components | Left frontal lobe | wildtype | C228T | 97m(Alive) |
| 3 | Female | 57 | Oligodendroglioma | Left frontal lobe | wildtype | C228T | 21m(Alive) |
| 4 | Female | 52 | Anaplastic oligodendroglioma | Right temporal occipital lobe | wildtype | Wildtype | 2m(Dead) |
| 5 | Male | 51 | Oligodendroglioma | Right frontal temporal lobe | wildtype | C228T | 16m(Alive) |
| 6 | Female | 47 | Glioblastoma | Right frontal lobe | wildtype | Wildtype | 14m(Dead) |
| 7 | Female | 34 | Oligodendroglioma | Left frontal lobe | wildtype | C250T | 12m(Alive) |
| 8* | Female | 75 | Anaplastic oligodendroglioma | Right frontal lobe | wildtype | Wildtype | 13m(Alive) |
| 9* | Male | 25 | Oligodendroglioma | Left frontal lobe | wildtype | C228T | 12m(Alive) |
| 10* | Male | 26 | Oligodendroglioma | Right frontal lobe | wildtype | C228T | 10m(Alive) |
| 11** | Male | 45 | Anaplastic oligodendroglioma | NA | wildtype | NA | 2429D(Alive) |
| 12** | Female | 56 | Glioblastoma | NA | wildtype | NA | 607D(Dead) |
| 13** | Female | 46 | Anaplastic oligoastrocytoma | NA | wildtype | NA | 1016D(Alive) |
| 14** | Male | 24 | Anaplastic astrocytoma | NA | wildtype | NA | 181D(Dead) |
*After the establishment of this database, until the publication of this article, three similar cases with “IDH wild type and 1p/19q codeletion” were found in approximately 400 cases of glioma newly treated and diagnosed in our cancer center. **4 cases (CGGA_1281, CGGA_669, CGGA_D06 and CGGA_D58) of “IDH wild type and 1p/19q codeletion” were found in CGGA datasets among 325 Gliomas using RNA sequencing. M=month, D=day
Discussion
The World Health Organization (WHO) classification of tumors of the CNS is a standard and commonly used diagnostic system for brain tumor classification. The grading system was initially employed based on the morphological appearance of tumor cells and the outcome of tumors. However, in recent years, classical histopathology classification no longer meets current clinical needs. More and more molecular markers have emerged. Crucial molecular markers such as mutations in IDH, ATRX and 1p/19q codeletion status are now central in the pathological diagnosis of glioma.
Isocitrate dehydrogenase (IDH) mutation is one of the most important molecular markers in glioma. The most common IDH1 mutation is a heterozygous missense mutation that results in the replacement of arginine by histidine in codon 132 enzyme active site [29]. IDH1 mutation status has significant favorable prognostic value, which has been reported world widely, irrespective of any other markers, and our data also showed the same result. According to reports, IDH1 exhibits high frequency mutations (70-90%) in WHO II astrocytoma [23, 29, 30]. However, our data shows that the incidence of IDH1 mutation is lower in Chinese than Caucasian, and the same results of IDH2. In our cohort, we only found one case (1/482, 0.2 %) carried IDH2 mutation (R172K). As reported in the literature, IDH2 mutations are rare, comprising only about 2.7-6.7 % of all IDH1/2 mutation cases [31-33]. Consistent with our results, other data also show that the frequency of IDH2 mutations in Chinese patients seems to be lower than in Westerner. A study from West China Hospital only found three IDH2 mutation cases (3/207, 1.4 %) in the IDH mutation Chinese patients, for two cases of IDH2 R172K and one case of R172M mutation [13], the frequency is similar to another study of IDH2(R172K) in Chinese patients (3/203, 1.5 %) [34]. Interestingly, in other diseases which carry IDH mutations such as hematological disorders, mutation rate was also lower in Chinese patients in contrast with the findings reported by North American and European populations [35]. These statistics indicate that IDH mutations may have ethnic and geographical difference, and this phenomenon is noteworthy and need to be further researched.
ATRX, alpha-thalassemia/mental retardation, X-linked, was first discovered in pancreatic neuroendocrine tumors [36]. It is able to incorporate histone variant H3.3 into heterochromatin, causing changes in telomere length and genomic instability [37, 38]. Several researches have figured out the role of ATRX in the diagnosis of diffuse glioma, with a higher rate of ATRX mutations in diffuse astrocytoma [39]. And Cai JQ et al. [14] reported that decreased expression of ATRX can cause inhibition of migration, promotion of apoptosis and reducing of proliferation in glioma cells. Some studies recommended ATRX status and IDH1-R132H initially for the top layer of “integrated decision” followed by histological classification, WHO grade, and other molecular information [40].
In 1994, Reifenberger et al. [41] first discovered the loss of heterozygosity (LOH) of chromosome arms 1p and 19q in a lot of oligodendrogliomas. Later, a lot of studies [42-45] demonstrated that the patient with 1p/19q codeletion had a higher sensitivity to chemotherapy and a better prognosis for survival. The FISH method has a higher sensitivity than the polymerase chain reaction (PCR) method when detecting the 1p/19q codeletion. Hence, FISH method has been widely used in detecting the deletion of chromosome 1p and 19q in gliomas. We firstly found 91 cases of 1p/19q codeletion in our cohort when we adopted FISH method. However, 20 cases presented as paradoxical results of IDH wildtype and 1p/19q codeletion. In consideration of conventional FISH probes hybridize to regions containing only 0.4% to 1.5 % of the corresponding chromosome arms, and other special circumstances would yield a number of “false-positive results” [46] such as a critical interpretational value, only focal deletion and polyploid deletion, we furtherly detected these 20 cases of conflicting molecular information to confirm the IDH status and 1p/19q status using the method of “Multiplex PCR-Based Next Generation Sequencing”, and finally identified 7 cases of peculiar phenotype presented as “IDH wild type and 1p/19q codeletion”. The false-positive results of 1p/19q codeletion may be due to interstitial deletions of probe hybridization or monomeric regions, rather than complete loss of 1p and 19q [46].
As to the three most important markers (IDH, ATRX and 1p19q), we found the same results as ISN-Haarlem consensus and the latest WHO CNS guildline, the loss of nuclear ATRX expression was significantly correlation with the IDH1/2 mutations status in grade II gliomas (101/124, 81.45%, P<0.001), and ATRX was seemed mutually exclusive with the total 1p/19q codeletion. Out of 91 cases of 1p/19q codeletion, only one case was ATRX negative. However, surprisingly, although we found the most 1p/19q codeletion gliomas were IDH mutated (92.31 %) in our cohort, there were still 7 cases with IDH wild type (7.7 %) after secondary detection. According to current opinion, such phenotype was not existed in glioma and many previous studies have suggested that 1p/19q codeletion tumors were all accompanied by IDH mutations [7, 47]. Under the menu of latest WHO blue book, we can only put these conflicting cases into oligodendroglioma, NOS, which lacking IDH mutation. However, articles about this situation of conflicting molecular results were published recently. Mizoguchi et al. [48] reported 8 glioblastoma cases with 1p/19q codeletion and without IDH1/IDH2 mutation by microsatellite analysis. Clark et al. [49] adopting the FISH and microsatellite analysis also detected 28 cases containing 1p/19q codeletion in total 491 glioblastoma cases, accounting for 5.7 % (28/491), and only one case harbors the IDH1-R132H mutation. All these results show IDH wild type GBM can have 1p/19q codeletion as the same as our cohort. However, besides the two cases of GBM presented as the phenotype of “IDH wildtype and 1p/19q codeletion” in our cohort, there are still 5 cases of typical oligodendroglioma morphology in our cohort. As mentioned in Fuller, G N's study [46], 6 thorny cases of 1p/19q FISH-positive and IDH wild-type did not meet the diagnostic criteria for "oligodendroglioma, IDH mutations, and 1p/19q codeletion." Ho-Keung Ng et, al [50] also found 5 in 185 cases(2.7 %) presented as “IDH wildtype and 1p/19q codeletion” and did not warrant a diagnosis of “oligodendroglioma, IDH mutant and 1p/19q co-deleted.” Our results also confirmed the presence of 1p/19q co-deleted and IDH wildtype oligodendrogliomas as described in the above studies. Although the entity of “oligodendroglioma, IDH wildtype and 1p/19q codeleted” only accounts for a very small percentage, we argue that such entity should be added to the pathological diagnosis category, and further exploration of other novel molecular markers is needed for characterization besides ATRX and TERT. Notably, other alternative methods are needed to assess the 1p/19q status when the FISH results do not meet clinical expectations or morphological criteria.
The telomerase reverse transcriptase (TERT) is a catalytic subunit with reverse transcriptase activity in telomerase. Mutations in the presence of the TERT promoter region have been reported in numerous cancers. Among gliomas, oligodendroglioma and glioblastomas (GBMs) were the most frequently affected tumors [51-55]. These mutations occurred in 2 hotspot positions (C228T and C250T). As to the prognostic significance of TERT, differences results depend on subtype of the gliomas. We found it was a poor prognostic marker in GBM, but no significance in all the infiltrating gliomas. Sanson, M et al. [56] also found that the TERT promoter mutation was a strong and independent worse prognostic marker in GBMs, and is not related to IDH status. However, Yang P et al. [57] found that the presence of TERT promoter mutation in WHO II and III gliomas means a good outcome, and it has been an independent factor relevant to a good prognosis in the IDH mutation (IDH-mut) subgroup. Because gliomas with concurrent IDH mutations and TERT promoter most possibly have a coexisting 1p/19q codeletion [22], it suggests that co-occurrence of mutations in TERT promoter and IDH plays a role in the genesis of oligodendroglioma and the favorable prognostic effect of the triple-positive tumors(IDH, 1p/19q and TERT) might be caused by 1p/19q codeletion which trump over the negative effects of TERT promoter mutation [58].
In China, there are a lot of relatively undeveloped regions which can't perform molecular testing. We hope we can establish a simple and economic testing process for the primary hospital. Based on our research, routine immunohistochemistry panel for primary hospitals include: IDH1(R132H), ATRX, P53, PHH3(mitotic figures marker) and Ki67(cell proliferation index marker). Since ATRX loss was mutually exclusive with the total 1p/19q codeletion, we can get a preliminary classification by the above package.
On the treatment of glioma according to the current classification, low grade gliomas with a poor prognosis, most cases were IDH wildtype, should receive more intensive treatment, especially in triple-negative gliomas. However, high grade gliomas with good prognosis factors, such as having 1p/19q codeletion, could appropriately use conservative management without radiation in some cases, rather than the typical treatment consisted of varying combinations of surgery, chemotherapy, and radiotherapy, which was recently, to some extent, confirmed by Sorge C et al. [59], for they found a WHO III anaplastic oligodendroglioma with IDH wildtype and 1p/19q codeletion in a children only underwent a near total resection and had a complete, durable response to temozolomide alone. Meanwhile, we also need to focus on triple-negative gliomas, for they account a relatively high proportion in our Chinese cohort. A portion of WHO Grade II or III gliomas in this group have an aggressive course and are associated with unfavorable survival and should be considered as WHO IV, which suggests the need for early adjuvant therapies.
The main limitation of the study is its retrospective nature, and there are few missing data. Its clinical significance and findings need to be demonstrated by other studies in the future. Nevertheless, it is the largest study up until now concerning the new integrated molecular diagnostics in southern China.
Conclusions
We screened recommended markers in a large cohort of Chinese glioma patients. Our data also demonstrated the subgroups of diffuse gliomas defined by the newly recommended markers possess distinctive prognostic characteristics, especially IDH mutation. IDH1R132H antibody, with good consistency with the test of gene detection, could be initially used for diagnostic and prognostic considerations in the clinical setting. Furthermore, we revealed the frequency of IDH mutation is relatively lower in Chinese compared with American and European, especially in IDH2.
Our clinicopathological study showed a higher prevalence of triple-negative gliomas (IDH wildtype, 1p/19q no-codeletion and TERT promoter wildtype WHO II&III gliomas) in Chinese glioma patients.
Importantly, we discovered the new phenotype of “infiltrating glioma, IDH wild type and 1p/19q codeletion”, which is conducive to further explore other potential molecular phenotype and further refine the classification of gliomas, and lay the foundation for future targeted therapy and precision medicine.
Abbreviations
WHO: World Health Organization; IDH: isocitrate dehydrogenase; ATRX: alpha-thalassemia/ mental retardation, X-linked; TERT: telomerase reverse transcriptase; MGMT: O6-methylguanine- DNA-methyltransferase; CGGA: Chinese Glioma Genome Atlas; TCGA: The Cancer Genome Atlas.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the National Nature Science Foundation of China (No.81772675) and the Neuro-oncology Project from the Chinese Society of Neuro-oncology (No. CSNO-2016-MSD02). KS was the project director and participated in writing the manuscript.
Author contributions
Conceptualization, WH and JZ; Data curation, FW, SX and XZ; Formal analysis, SX; Funding acquisition, KS; Methodology, FW, XZ and LL; Resources, JL and HW; Software, XZ; Validation, JL and LL; Writing - original draft, WH; Writing - review & editing, WH, KS and JZ. All authors have read and approved the manuscript.
Availability of data and materials
The TCGA and CGGA datasets generated and analyzed during this study is available in (http://www.cgga.org.cn) and (https://tcga-data.nci.nih.gov/tcga/tcgaDownload.jsp). All additional data used and/or analyzed during the current study are included in this published article and available from the corresponding author on reasonable request.
Ethics approval and consent to participate
The study was approved by the Institutional Review Board of Sun Yat-sen University Cancer Center, and all the patients provided written informed consent for the collection and publication of their medical information during the first visit to our center. SYSUCC ethics committee ruled that no formal ethics approval was required in this particular case since it only contained anonymized results of diagnostics on tissue.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Goodenberger ML, Jenkins RB. Genetics of adult glioma. Cancer Genet. 2012;205(12):613-621
2. van den Bent MJ. Interobserver variation of the histopathological diagnosis in clinical trials on glioma: a clinician's perspective. Acta Neuropathol. 2010;120(3):297-304
3. Verhaak RG, Hoadley KA, Purdom E. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98-110
4. Appin CL, Brat DJ. Molecular pathways in gliomagenesis and their relevance to neuropathologic diagnosis. Adv Anat Pathol. 2015;22(1):50-58
5. Hu W, Yang Y, Xi S. et al. Expression of CPEB4 in Human Glioma and Its Correlations With Prognosis. Medicine (Baltimore). 2015;94(27):e979
6. Capper D, Weissert S, Balss J. et al. Characterization of R132H mutation-specific IDH1 antibody binding in brain tumors. Brain Pathol. 2010;20(1):245-254
7. Reuss DE, Sahm F, Schrimpf D. et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an "integrated" diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol. 2015;129(1):133-146
8. Wiestler B, Capper D, Holland-Letz T. et al. ATRX loss refines the classification of anaplastic gliomas and identifies a subgroup of IDH mutant astrocytic tumors with better prognosis. Acta Neuropathol. 2013;126(3):443-451
9. Takami H, Yoshida A, Fukushima S. et al. Revisiting TP53 Mutations and Immunohistochemistry-A Comparative Study in 157 Diffuse Gliomas. Brain Pathol. 2015;25(3):256-265
10. Cai J, Yang P, Zhang C. et al. ATRX mRNA expression combined with IDH1/2 mutational status and Ki-67 expression refines the molecular classification of astrocytic tumors: evidence from the whole transcriptome sequencing of 169 samples samples. Oncotarget. 2014;5(9):2551-2561
11. Koelsche C, Sahm F, Capper D. et al. Distribution of TERT promoter mutations in pediatric and adult tumors of the nervous system. Acta Neuropathol. 2013;126(6):907-915
12. Cai J, Zhang C, Zhang W. et al. ATRX, IDH1-R132H and Ki-67 immunohistochemistry as a classification scheme for astrocytic tumors. Oncoscience. 2016;3(7-8):258-265
13. Chen N, Yu T, Gong J. et al. IDH1/2 gene hotspot mutations in central nervous system tumours: analysis of 922 Chinese patients. Pathology. 2016;48(7):675-683
14. Cai J, Zhu P, Zhang C. et al. Detection of ATRX and IDH1-R132H immunohistochemistry in the progression of 211 paired gliomas. Oncotarget. 2016;7(13):16384-16395
15. Zhou YX, Wang JX, Feng M. et al. Analysis of isocitrate dehydrogenase 1 mutation in 97 patients with glioma. J Mol Neurosci. 2012;47(3):442-447
16. Li MY, Wang YY, Cai JQ. et al. Isocitrate dehydrogenase 1 Gene Mutation Is Associated with Prognosis in Clinical Low-Grade Gliomas. PLOS ONE. 2015;10(6):e130872
17. Qi S, Yu L, Li H. et al. Isocitrate dehydrogenase mutation is associated with tumor location and magnetic resonance imaging characteristics in astrocytic neoplasms. ONCOL LETT. 2014;7(6):1895-1902
18. Mellai M, Annovazzi L, Senetta R. et al. Diagnostic revision of 206 adult gliomas (including 40 oligoastrocytomas) based on ATRX, IDH1/2 and 1p/19q status. J Neurooncol. 2017;131(2):213-222
19. Metellus P, Coulibaly B, Colin C. et al. Absence of IDH mutation identifies a novel radiologic and molecular subtype of WHO grade II gliomas with dismal prognosis. Acta Neuropathol. 2010;120(6):719-729
20. Juratli TA, Kirsch M, Robel K. et al. IDH mutations as an early and consistent marker in low-grade astrocytomas WHO grade II and their consecutive secondary high-grade gliomas. J Neurooncol. 2012;108(3):403-410
21. Hartmann C, Meyer J, Balss J. et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta Neuropathol. 2009;118(4):469-474
22. Killela PJ, Pirozzi CJ, Healy P. et al. Mutations in IDH1, IDH2, and in the TERT promoter define clinically distinct subgroups of adult malignant gliomas. Oncotarget. 2014;5(6):1515-1525
23. Yan H, Parsons DW, Jin G. et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765-773
24. Sabha N, Knobbe CB, Maganti M. et al. Analysis of IDH mutation, 1p/19q deletion, and PTEN loss delineates prognosis in clinical low-grade diffuse gliomas. Neuro Oncol. 2014;16(7):914-923
25. Leeper HE, Caron AA, Decker PA. et al. IDH mutation, 1p19q codeletion and ATRX loss in WHO grade II gliomas. Oncotarget. 2015;6(30):30295-30305
26. Yan W, Zhang W, You G. et al. Molecular classification of gliomas based on whole genome gene expression: a systematic report of 225 samples from the Chinese Glioma Cooperative Group. Neuro Oncol. 2012;14(12):1432-1440
27. Eckel-Passow JE, Lachance DH, Molinaro AM. et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med. 2015;372(26):2499-2508
28. Chan AK, Mao Y, Ng HK. TP53 and Histone H3.3 Mutations in Triple-Negative Lower-Grade Gliomas. N Engl J Med. 2016;375(22):2206-2208
29. Balss J, Meyer J, Mueller W. et al. Analysis of the IDH1 codon 132 mutation in brain tumors. ACTA NEUROPATHOL. 2008;116(6):597-602
30. Parsons DW, Jones S, Zhang X. et al. An integrated genomic analysis of human glioblastoma multiforme. SCIENCE. 2008;321(5897):1807-1812
31. Hartmann C, Meyer J, Balss J. et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. ACTA NEUROPATHOL. 2009;118(4):469-474
32. Horbinski C, Kelly L, Nikiforov YE. et al. Detection of IDH1 and IDH2 mutations by fluorescence melting curve analysis as a diagnostic tool for brain biopsies. J MOL DIAGN. 2010;12(4):487-492
33. Horbinski C, Kofler J, Kelly LM. et al. Diagnostic use of IDH1/2 mutation analysis in routine clinical testing of formalin-fixed, paraffin-embedded glioma tissues. J Neuropathol Exp Neurol. 2009;68(12):1319-1325
34. Zhang CB, Bao ZS, Wang HJ. et al. Correlation of IDH1/2 mutation with clinicopathologic factors and prognosis in anaplastic gliomas: a report of 203 patients from China. J Cancer Res Clin Oncol. 2014;140(1):45-51
35. Zou Y, Zeng Y, Zhang DF. et al. IDH1 and IDH2 mutations are frequent in Chinese patients with acute myeloid leukemia but rare in other types of hematological disorders. Biochem Biophys Res Commun. 2010;402(2):378-383
36. Jiao Y, Shi C, Edil BH. et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. SCIENCE. 2011;331(6021):1199-1203
37. Schwartzentruber J, Korshunov A, Liu XY. et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. NATURE. 2012;482(7384):226-231
38. Lovejoy CA, Li W, Reisenweber S. et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLOS GENET. 2012;8(7):e1002772
39. Kannan K, Inagaki A, Silber J. et al. Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget. 2012;3(10):1194-1203
40. Reuss DE, Sahm F, Schrimpf D. et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an "integrated" diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol. 2015;129(1):133-146
41. Reifenberger J, Reifenberger G, Liu L. et al. Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol. 1994;145(5):1175-1190
42. Cairncross G, Berkey B, Shaw E. et al. Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol. 2006;24(18):2707-2714
43. Giannini C, Burger PC, Berkey BA. et al. Anaplastic oligodendroglial tumors: refining the correlation among histopathology, 1p 19q deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. BRAIN PATHOL. 2008;18(3):360-369
44. McLendon RE, Herndon JN, West B. et al. Survival analysis of presumptive prognostic markers among oligodendrogliomas. Cancer-Am Cancer Soc. 2005;104(8):1693-1699
45. Smith JS, Perry A, Borell TJ. et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol. 2000;18(3):636-645
46. Ballester LY, Huse JT, Tang G. et al. Molecular classification of adult diffuse gliomas: conflicting IDH1/IDH2, ATRX, and 1p/19q results. Hum Pathol. 2017;69:15-22
47. Wang XW, Ciccarino P, Rossetto M. et al. IDH mutations: genotype-phenotype correlation and prognostic impact. Biomed Res Int. 2014;2014:540236
48. Mizoguchi M, Yoshimoto K, Ma X. et al. Molecular characteristics of glioblastoma with 1p/19q co-deletion. Brain Tumor Pathol. 2012;29(3):148-153
49. Clark KH, Villano JL, Nikiforova MN. et al. 1p/19q testing has no significance in the workup of glioblastomas. Neuropathol Appl Neurobiol. 2013;39(6):706-717
50. Li YX, Shi Z, Aibaidula A. et al. Not all 1p/19q non-codeleted oligodendroglial tumors are astrocytic. Oncotarget. 2016;7(40):64615-64630
51. Liu X, Wu G, Shan Y. et al. Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell Cycle. 2013;12(10):1637-1638
52. Arita H, Narita Y, Fukushima S. et al. Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol. 2013;126(2):267-276
53. Killela PJ, Reitman ZJ, Jiao Y. et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A. 2013;110(15):6021-6026
54. Chan AK, Yao Y, Zhang Z. et al. TERT promoter mutations contribute to subset prognostication of lower-grade gliomas. Mod Pathol. 2015;28(2):177-186
55. Zhang ZY, Chan AK, Ding XJ. et al. TERT promoter mutations contribute to IDH mutations in predicting differential responses to adjuvant therapies in WHO grade II and III diffuse gliomas. Oncotarget. 2015;6(28):24871-24883
56. Labussiere M, Boisselier B, Mokhtari K. et al. Combined analysis of TERT, EGFR, and IDH status defines distinct prognostic glioblastoma classes. NEUROLOGY. 2014;83(13):1200-1206
57. Yang P, Cai J, Yan W. et al. Classification based on mutations of TERT promoter and IDH characterizes subtypes in grade II/III gliomas. Neuro Oncol. 2016;18(8):1099-1108
58. Vuong HG, Altibi A, Duong U. et al. TERT promoter mutation and its interaction with IDH mutations in glioma: Combined TERT promoter and IDH mutations stratifies lower-grade glioma into distinct survival subgroups-A meta-analysis of aggregate data. Crit Rev Oncol Hematol. 2017;120:1-9
59. Sorge C, Li R, Singh S. et al. Complete durable response of a pediatric anaplastic oligodendroglioma to temozolomide alone: Case report and review of literature. Pediatr Blood Cancer. 2017
Author contact
![]() Corresponding authors: Sai Ke; saikeorg.cn and Zeng Jing; zengjing1320com
Corresponding authors: Sai Ke; saikeorg.cn and Zeng Jing; zengjing1320com