Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(8):2139-2149. doi:10.7150/jca.38796 This issue Cite
Research Paper
A Combined four-mRNA Signature Associated with Lymphatic Metastasis for Prognosis of Colorectal Cancer
Xueping Li1,2*, Qiang Zhang1,2*, Lan Zhao1,2, Longyang Jiang1,2, Aoshuang Qi1,2, Qian Wei1,2, Xinyue Song1,2, Lin Wang1,2, Liwen Zhang1,2, Yanyun Zhao1,2, Xuemei Lv1,2, Minjie Wei1,2 ![]() , Lin Zhao1,2
, Lin Zhao1,2 ![]()
1. Department of Pharmacology, School of Pharmacy, China Medical University, No.77 Puhe Road, Shenyang North New Area, Shenyang City, 110122, Liaoning, China.
2. Liaoning Engineering Technology Research Center, China Medical University, No.77 Puhe Road, Shenyang North New Area, Shenyang City, 110122, Liaoning, China.
*Xueping Li and Qiang Zhang have contributed equally to this work.
Received 2019-7-28; Accepted 2020-1-4; Published 2020-2-3
Abstract
Background: Colorectal cancer (CRC) is one of the most common malignant tumors in the world. Lymph node metastasis (LNM) is a common mode of metastasis of CRC. However, the combined mRNA biomarkers associated with LNM of CRC that can effectively predict CRC prognosis have not been reported yet.
Methods: To identify biomarkers that are associated with LNM, we collected data from the The Cancer Genome Atlas (TCGA) database. The edgeR package was searched to seek LNM-related genes by comparisons between cancer samples and normal colorectal tissues and between LNM and non-LNM (NLNM) of CRC. Univariate and multivariate regression analysis of genes in the intersection to build gene signature associated with independent prognosis of CRC, and then verified by Kaplan-Meier curve and log-rank test, receiver operating characteristic (ROC) curve was used to determine the efficiency of survival prediction of our four-mRNA signature. Finally, the potential molecular mechanisms and properties of these gene signature were also explored with functional and pathway enrichment analysis.
Results: 329 mRNAs were up-regulated in CRC tissues with LNM, and 8461 mRNAs were up-regulated in CRC tissues, the intersection is 100 mRNAs. After univariate and multivariate Cox regression analysis of 100 mRNAs, a novel four LNM related mRNAs (EPHA8, KRT85, GABRA3, and CLPSL1) were screened as independent prognostic indicators of CRC. Surprisingly, the four-mRNA signature can predict the prognosis of CRC patients independently of clinical factors andthe area under the curve (AUC) of the ROC is 0.730. The novel four-mRNA signature was used to identify high and low-risk groups. Stratified analysis indicated the risk score based on four-mRNA signature was an independent prognostic indicator for female, T3+T4, N1+N2 ,stage III+IV and patients with no new tumor event. Functional annotation of this risk model in high-risk patients revealed that pathways associated with neuroactive ligand-receptor interaction, estrogen signaling pathway, and steroid hormone biosynthesis.
Conclusions: By conducting TCGA data mining, our study demonstrated that a four-mRNA signature associated with LNM can be used as a combined biomarker for independent prognosis of CRC.
Keywords: biomarker, lymph node metastasis, colorectal cancer, mRNA, prognosis
Background
Colorectal cancer (CRC) is one of the common malignant tumors of the digestive tract [1], which seriously threatens the life and quality of life of patients. In both sexes combined, the incidence of CRC is the third (10.2% of the total cases), and the mortality rate is the second (9.2% of the total cancer deaths) [2]. At present, the effective treatment of CRC is surgical resection [3], but it is easy to relapse and metastasis after surgery. The most common metastatic sites of CRC are liver, abdominal lymph nodes and lung metastases. According to reports, 5-year survival rate in the United States of CRC is close to 70%, but due to the presence of lymph node metastasis (LNM) or distant metastases, 5-year survival rate under the same conditions fell to 13% [4].The LNM severely affects the survival of CRC patients[5], so the exploration of biomarkers with high sensitivity and specificity for diagnosis of CRC associate with LNM has become a key problem in the medical field.
Currently researches on CRC-related biomarkers continue to grow [6], for example, related studies have shown that vascular endothelial growth factor (VEGF) is expressed in about 50% of CRC, which is very rarely expressed in normal colon mucosa and adenoma, and VEGF-1 can effectively predict the prognosis of CRC patients [7]. PTTG1 (Pituitary tumor transforming gene-1) is an independent prognostic factor that affects the proliferation, invasion and migration of CRC cells [8]. Moreover, Zhang JS et al. found patients with high expression of RABEX-5 mRNA had a poor prognosis, RABEX-5 mRNA may be a potential biomarker for evaluating the prognosis of CRC [9]. miRNAs are involved in the development and progression of tumors [10], in recent years, miRNAs have also been found as biomarkers for invasion, metastasis and prognosis of CRC [11]. Compared with a single biomarker, the advantage of combining biomarkers is that it can increase the sensitivity of detection. Therefore, in order to improve the sensitivity of clinical diagnosis of tumor biomarkers, we can screen combined biomarkers of CRC. Andrea Angius et al. found an integrated signature of 20 deregulated miRNAs that could be evaluated as potential prognostic biomarkers [12], Chuanpeng Dong et al. identified that an eight-gene signature in cancer stem cell signaling was associated with the overall survival patients with CRC [13]. Furthermore, the researcher identified a 6-gene signature predicting prognosis for CRC [14]. Moreover, LNM is a common mode of metastasis and an important factor affecting the prognosis of CRC [15], there are very few reports on mRNA combination biomarkers for LNM of CRC, so differentially expressed mRNA associated with LNM should be the key to the progression of CRC, we can screen for mRNA combined biomarkers of CRC from this perspective.
In this work, we analyzed 614 patients with CRC in The Cancer Genome Atlas (TCGA) database and found that 100 mRNAs were up-regulated both in CRC patients and CRC patients with LNM. By further analysis we found that based on four-mRNA signature patients with high risk scores have a poorer prognosis and four-mRNA signature associated with LNM can effectively predict the prognosis of CRC patients.
Methods and Materials
Patient and mRNA expression data procession
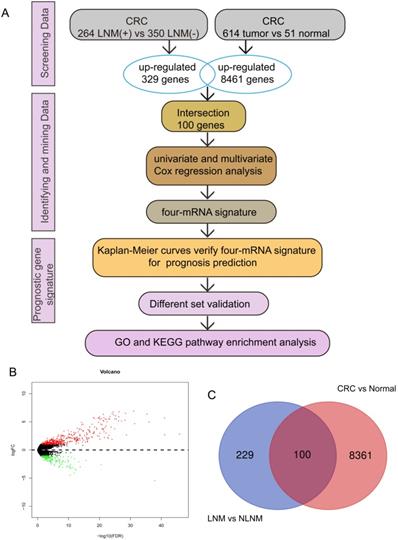
The expression of CRC mRNA and the corresponding clinical data were downloaded from the TCGA database. According to the inclusion criteria: (a) complete gene expression and survival information (b) the CRC patients with LNM were filtered by the criteria that N stage of patients was I-IV, and the exclusive criteria were as follows: (a) survival information or gene expression is incomplete (b) N stage in clinical pathological parameters is not available, we finally obtained a total of 614 CRC tissues and 51 normal colorectal tissues of mRNA expression profiles for further research, among them, 614 tissues included 264 tissues with LNM and 350 tissues with non-LNM (NLNM) (Figure 1A).
DEGs associated with LNM. (A) study design. (B) The volcano map of DEGs in CRC tissues with LNM vs NLNM, the red represents the up-regulated genes. (C) the intersection of the up-regulated genes in LNM vs NLNM tissues and CRC vs non-cancer tissues.
Differential expression of CRC mRNA data mining
Screening for differentially expressed mRNA of 614 CRC tissues and 51 normal colorectal tissues in TCGA using edgeR package, the threshold was set to |logFC| > 1and adjusted p value < 0.05, 264 CRC tissues with LNM and 350 CRC tissues with NLNM for differential mRNA mining under the same conditions.
Using Venn digram web-tool (http://bioinformatics.psb.ugent.be/webtools/Venn/) to find the intersection of up-regulated genes in CRC tissues and CRC tissues with LNM.
Construction of independent prognostic indicators based on mRNA
In our work, 614 CRC patients were randomly divided into two groups (test set N=322, validation set N=292) (Table 1). Then the mRNA expression profile was subjected to log2 transformation for further statistical analysis, and univariate Cox was used to screen mRNAs affecting OS of patients (p < 0.05), followed by multivariate Cox regression analysis to identify mRNAs as independent prognostic indicators. Subsequently, based on the expression level of each mRNA and the regression coefficient obtained from multivariate Cox conduct a risk score, risk score =
Clinical characteristic of patients with CRC in each set.
| Clinical feature | Test set(%) | Validation set(%) | Entire set (%) |
|---|---|---|---|
| Gender | |||
| Male | 178(55.3) | 148(50.7) | 326(53.1) |
| Female | 144(44.7) | 144(49.3) | 288(46.9) |
| Age | |||
| < = 68 | 166(51.6) | 162(55.5) | 328(53.4) |
| > 68 | 156(48.4) | 130(44.5) | 286(46.6) |
| T stage | |||
| T1+T2 | 61(19.0) | 64(21.9) | 125(20.4) |
| T3+T4 | 260(80.7) | 228(78.1) | 488(79.5) |
| unknown | 1(0.3) | 0(0.0) | 1(0.1) |
| N stage | |||
| N0 | 194(60.2) | 156(53.4) | 350(57.0) |
| N1+N2 | 128(39.8) | 136(46.6) | 264(43.0) |
| M stage | |||
| M0 | 235(73.0) | 222(76.0) | 457(74.4) |
| M1 | 46(14.3) | 41(14.0) | 87(14.2) |
| unknown | 41(12.7) | 29(10.0) | 70(11.4) |
| Stage | |||
| I+II | 181(56.2) | 150(51.4) | 331(53.9) |
| III+IV | 131(40.7) | 136(46.6) | 267(43.5) |
| unknown | 10(3.1) | 6(2.0) | 16(2.6) |
| Neoplasm cancer status (with tumor/tumor free) | |||
| Tumor free | 135(41.9) | 120(41.1) | 255(41.5) |
| With tumor | 152(47.2) | 137(46.9) | 289(47.1) |
| unknown | 35(10.9) | 35(12.0) | 70(11.4) |
| New tumor event after initial treatment(yes/no) | |||
| No | 222(68.9) | 187(64.0) | 409(66.6) |
| Yes | 52(16.1) | 47(16.1) | 99(16.1) |
| unknown | 48(15.0) | 58(19.9) | 106(17.3) |
| Lymphatic invasion(yes/no) | |||
| No | 171(53.1) | 157(53.8) | 328(53.4) |
| Yes | 115(35.7) | 112(38.4) | 227(37.0) |
| unknown | 36(11.2) | 23(7.8) | 59(9.6) |
| Residual tumor(yes/no) | |||
| No | 230(71.4) | 219(75.0) | 449(73.2) |
| Yes | 22(6.8) | 20(6.8) | 42(6.8) |
| unknown | 70(31.8) | 53(18.2) | 123(20.0) |
ExpmRNA1×βmRNA1+ExprnRNA2×βmRNA2+⋯+ExpmRNAn×βmRNAn
(Exp represents the expression level of each mRNA and β represents the regression coefficient of each mRNA).
Functional enrichment analysis
In this study, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis was performed for these up-regulated mRNAs in patients with high risk score by using the Database for Annotation, Visualization, and Integrated Discovery online tool (https://david.ncifcrf.gov/). GO terms and KEGG pathways with a false discovery rate (FDR) < 0.05 were statistically significant.
Statistical Analysis
According to the median value of risk scores, 614 CRC patients were divided into high-risk group and low-risk group. Kaplan-Meier curve and log-rank test were used for plotting survival curves. The area under the curve (AUC) of the receiver operating characteristic (ROC) curves used to determine the predicted power of the prognostic gene signature. Moreover, we applied univariate and multivariate Cox analysis to evaluate whether or not the risk score was an independent factor of the other clinical variables including age, T, N, M, stage, residual tumor and neoplasm cancer status in patients with CRC. We used Pearson test or Fisher's exact test to analyze the correlation between LNM and clinical pathological parameters. All statistical analysis was using SPSS 16.0 and GraphPad Prism7.
Results
The intersection of differentially expressed genes (DEGs) in CRC
We screened differentially expressed genes (|logFC| > 1, p < 0.05) in 614 CRC tissues and 51 normal colorectal tissues and found that 8461 genes were up-regulated in CRC tissues, while 264 CRC tissues with LNM and 350 CRC tissues with NLNM for differential mRNA mining under the same conditions (Figure 1B), then a total of 329 genes were obtained from up-regulated genes in CRC tissues with LNM. In addition, we used the Venn diagram web-tool to cross the two sets of up-regulated genes, as shown in Figure 1C, 100 genes were in the intersection (Supplementary Table S1).
Identification of four mRNAs associated with prognosis in CRC
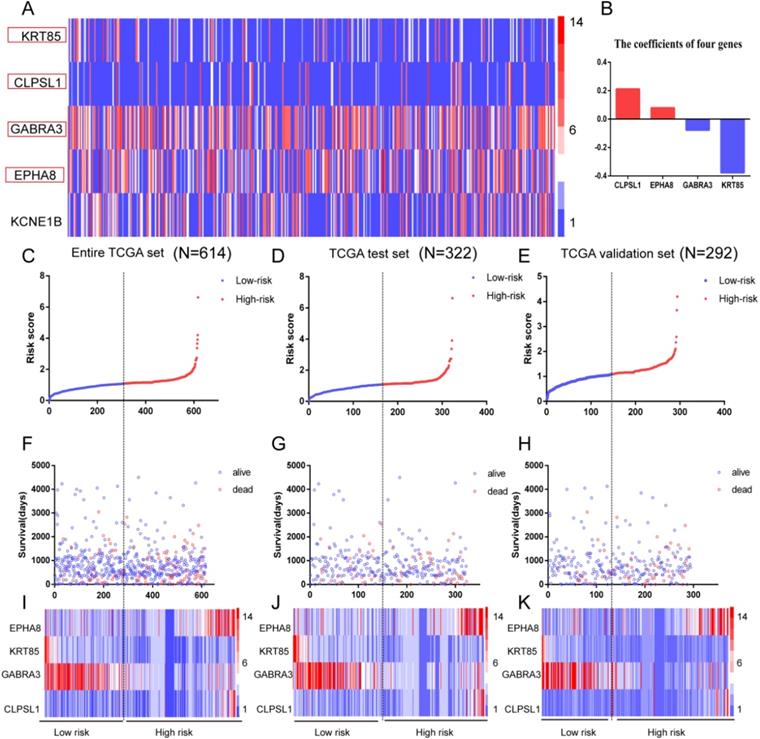
We first used univariate Cox regression analysis to identify 100 intersection genes associated with prognosis, and got five genes with p values < 0.05 (Figure 2A). Then multivariate Cox regression analysis was performed and four mRNAs (EPHA8, KRT85, GABRA3, and CLPSL1) were finally screened as prognostic signature models (as shown in Table 2). Among them, EPHA8 and CLPSL1 showed positive coefficients, indicating they are risk factors since their high expression is accompanied by a shorter survival. Instead, we found that KRT85 and GABRA3 are negative coefficients, which means they can be considered protective mRNA and high expression of these mRNAs suggests that patients have longer survival (Figure 2B).
Risk score analysis of the four-mRNAs signature of CRC. (A) The heat map of five genes in CRC patients. Each column represents a patient and each row represents a gene. The expression levels of genes are displayed in different colors. From blue to red, the expression is gradually increasing. (B) The coefficients of the four genes, red for positive numbers and blue for negative numbers. (C-E)The distribution of high and low risk scores of four mRNAs in entire TCGA set (N=614), TCGA test set (N=322) and validation set (N=292). (F-H) Survival time and status of patients based on the high and low risk scores of four mRNAs in entire TCGA set (N=614)、TCGA test set (N=322) and validation set (N=292). (I-K) The heat map of four genes in entire TCGA set (N=614), TCGA test set (N=322) and validation set (N=292).
Details of four prognostic mRNAs significantly associated with OS in CRC.
| mRNA | Ensemble ID | Location | β | HR | p |
|---|---|---|---|---|---|
| EPHA8 | ENSG00000070886 | chr1: 22563489-22603595 | 0.079 | 1.083 | 0.045 |
| KRT85 | ENSG00000135443 | chr12:52360006-52367481 | -0.377 | 0.686 | 0.014 |
| GABRA3 | ENSG00000011677 | chrX:152166234-152451359 | -0.077 | 0.926 | 0.036 |
| CLPSL1 | ENSG00000204140 | chr6:35781017-35794039 | 0.212 | 1.236 | 0.016 |
Next, the risk score for predicting OS was established using the formula of the four mRNAs based on the multivariate Cox regression analysis results above: risk score = 0.079 × expression of EPHA8 + 0.212 × expression of CLPSL1-0.377 × expression of KRT85-0.077 × expression of GABRA3. Entire TCGA set (N=614), test set (N=322) and validation set (N=292) respectively were divided into low-risk and high-risk groups according to the median of the prognosis risk score (Figure 2C-E). Next, we analyzed the survival and status of patients in the high and low risk score group, and the results showed that the mortality rate of the high-risk score group is higher than that of the low-risk score group (Figure 2F-H). The heat map results showed that the risk mRNA (EPHA8 and CLPSL1) was up-regulated with increasing risk score and the protective mRNA (KRT85 and GABRA3) expression was down-regulated (Figure 2I-K).
The four-mRNA signature as a prognostic indicator independent of clinical characteristics
First of all, we looked at the distribution of different clinical parameters in patients with low to high risk scores (Figure 3A). Next, the risk score and the clinic pathological parameters including age, T stage, N stage, M stage, stage, residual tumor and neoplasm cancer status were used as explanatory variables, and the OS rate was used as a dependent variable for univariate and multivariate Cox regression analysis. As shown in Figure 3B, univariate Cox regression analysis showed that the four-mRNA risk score and the above mentioned conventional clinic pathological factors can effectively predict the prognosis of patients with CRC.
Univariate and multivariate Cox regression analysis of OS. (A) Distribution of the clinic pathological parameters including age, T, N, M, stage, residual tumor and neoplasm cancer status in CRC patients with low-risk score to high-risk score. (B) univariate Cox regression analysis of OS. (C) multivariate Cox regression analysis of OS.
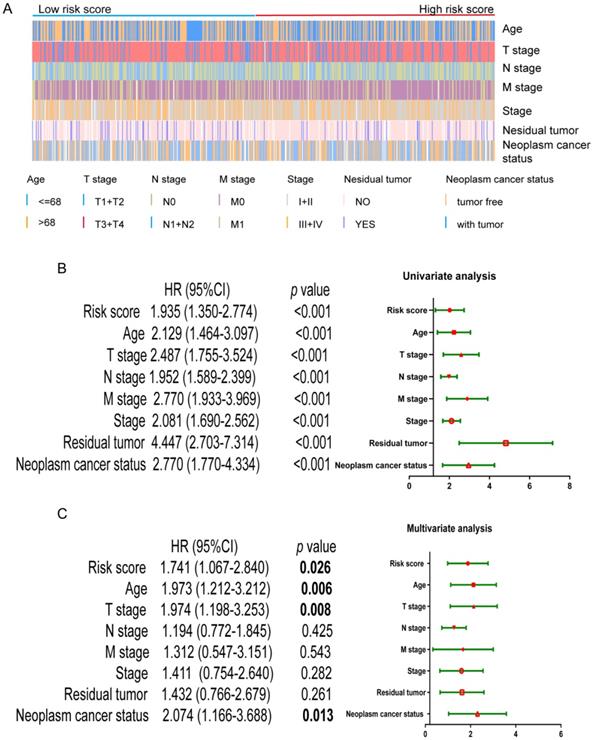
Among them, "Residual tumor" is the most obvious clinical and pathological parameters predicting the prognosis of patients with CRC, because the probability of death in patients with residual tumors is 4.472 times that of patients without residual tumors. In addition, risk score, age, T stage, and neoplasm cancer status were also significantly different in multivariate analysis (p < 0.05), indicating that they can be used as independent prognostic indicators for CRC patients (Figure 3C).
Kaplan-Meier curves verify four-mRNA signature for survival prediction
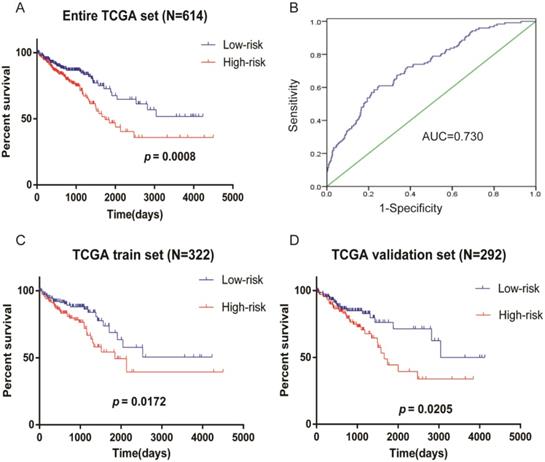
The Kaplan-Meier curves showed that the prognosis of patients with high-risk scores was poorer (Figure 4A), the AUC of the ROC curves used to determine the predicted power of the prognostic gene signature, the AUC of the four-mRNA signature was 0.730 (Figure 4B). It is also confirmed in test set (N=322) (Figure 4C) and validation set (N=292) (Figure 4D) that patients with high-risk scores had a worse prognosis. These results indicate the four-mRNA signature can effectively predict the prognosis of patients with CRC. Previous univariate Cox regression analysis of OS showed that age, T stage, N stage, M stage, stage, residual tumor and neoplasm cancer status in clinical pathological parameters could effectively predict survival in patients with CRC. Next, we use the Kaplan-Meier method to verify the above conclusion, the results indicate that patients who were older than 68 years, who were in T3+T4, N1+N2, M1, and stage III+IV, who had residual tumors and who had neoplasm cancer have a poorer prognosis(Supplementary Figure S1A). This result further confirms the accuracy of our previous analysis.
Kaplan-Meier survival analysis of CRC patients in TCGA data. (A) Risk score predicts survival of patients with CRC in entire TCGA set (N=614). (B) ROC curves of the four-mRNA signature in CRC (AUC = 0.730). (C) Risk score predicts survival of patients with CRC in TCGA test set (N=322). (D) Risk score predicts survival of patients with CRC in TCGA validation set (N=292).
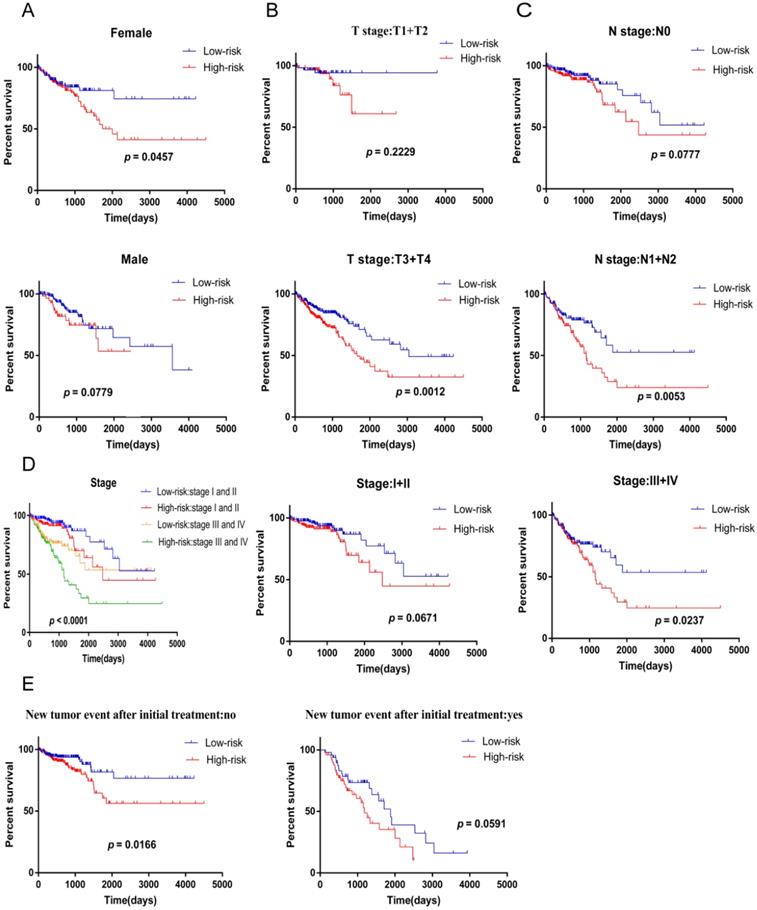
Next, we used stratified analysis for further data mining and we found that four-mRNA signature is a prognostic marker for female patients with CRC (Figure 5A). After stratification of T stage, N stage, and stage, respectively, the risk score based on four-mRNA signature was an independent prognostic indicator for T3+T4, N1+N2 and stage III+IV, and patients with high risk scores had a poorer prognosis (Figure 5B-D). However, according to the new tumor event after initial treatment, four-mRNA signature was found to be a prognostic marker for patients with no new tumor event, and the high-risk subgroup survived for a shorter period of time (Figure 5E).
Kaplan-Meier curves predicts patient survival with clinical features for the patients divided into high and low risk scores. (A) gender. (B) T stage. (C) N stage. (D) stage.( E) new tumor event after initial treatment.
Identification of related potential functions of the four-mRNA signature
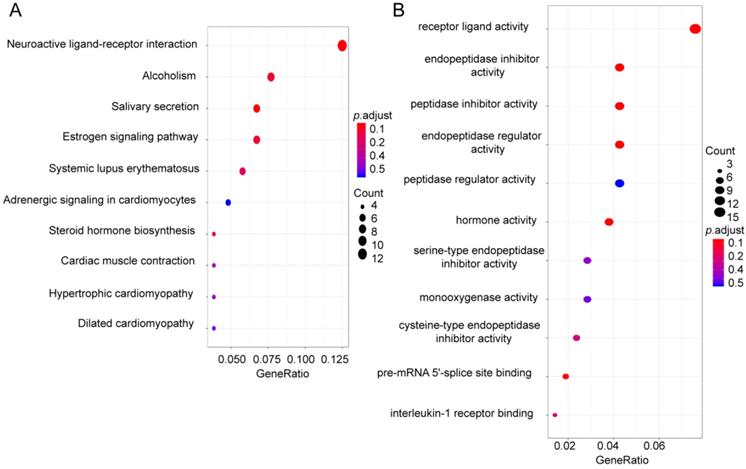
To identify pathways and biological processes in which four-mRNA signatures work, we divided 614 CRC patients into low risk group (N=307) and high risk group (N=307) according to the median risk score and screened for differential genes (|logFC| > 1, p < 0.05), among them, 112 genes were up-regulated in high risk group (Supplementary Table S2). Next, the GO and KEGG enrichment analysis was performed on these 112 genes. The results showed that these genes were together enriched in pathways in neuroactive ligand-receptor interaction, estrogen signaling pathway, and steroid hormone biosynthesis (Figure 6A) and the results suggested that the top GO biological process were receptor ligand activity, endopeptidase inhibitor activity and peptidase inhibitor activity (Figure 6B).
Functional enrichment analysis. (A) KEGG analysis of up-regulated genes in high risk score group. (B) GO analysis of up-regulated genes in high risk score group.
Discussion
CRC is a heterogeneous malignant tumor, which makes some clinical parameters such as gender, age, residual tumors, and stage unable to accurately predict the prognosis of CRC patients [16]. With the development of high-throughput sequencing, microarray technology and bioinformatics, more and more biomarkers have been discovered to effectively predict the prognosis of patients with CRC [17]. For example, Ai-Jun Sun et al. started bioinformatics data mining from the DNA methylation information in the tcga database and found that MSX1 and DCLK1, which are involved in DNA methylation, may be used as biomarkers for CRC [18]. Zong Z et al found that MSI-2 is highly expressed in CRC tissues. Surprisingly, logistic regression analysis showed that MSI-2 is associated with liver metastasis of CRC, and MSI-2 is expected to be a biomarker for liver metastasis of CRC [19]. Moreover, recent studies have found that TIMP1 is an independent prognostic indicator for disease-free survival and OS in colon cancer patients through the Cox proportional hazards model [20]. These findings make mRNAs a promising biomarker for predicting CRC survival. However, in the clinically, a single biomarker is susceptible to various factors, making the combination marker a research hotspot. In recent years, research on combined biomarkers has also been emerging. Zhang Z et al. [21] indicated a set of circRNAs that may serve as a candidate diagnostic biomarker, Dai W et al. [22] revealed an integrated mRNA-lncRNA signature with predictive value of early relapse in colon cancer, Dai W et al. [23] discovered a gene signature for the prediction of early relapse in stage I-III colon cancer and researcher identified a 14-lncRNA prognostic signature for patients with colon adenocarcinoma [24].
It is well known that the treatment of tumors has made great progress in recent years, but metastatic malignant tumors are often incurable, and metastasis is considered to be the main cause of tumor treatment failure [25]. Metastasis is a multistep and complex process [26], and the specific mechanism behind it is still not very clear. Tumor metastasis refers to the process in which tumor cells migrate from the primary site through lymphatic vessels or blood vessels to other parts to continue to grow [27]. Biomarkers associated with LNM are critical for diagnosis and prognosis of cancer. For example, studies have found that 14-3-3β and profilin-1 can be used to predict LNM of gastric cancer and 14-3-3β may become an independent prognostic marker for gastric cancer [28]. Metastasis is an important factor affecting the prognosis and survival of CRC patients [29]. Moreover, LNM is a major shift in the form of CRC, the effective removal of LNM of CRC surgery is the focus of a direct impact on the prognosis of patients. Compared with CRC patients without LNM, the OS of patients with LNM was shorter [30]. Tsuyoshi Ozawa et al. discovered a 5-miRNA signature which associated with LNM of CRC by tcga RNA sequence mining [31]. A recent study showed that SATB1 is highly expressed in the CRC of LNM and may be used as a biomarker for CRC [15]. In this study, we analyzed the correlation of LNM and clinic parameters, and the results showed that LNM was associated with T stage, M stage, stage, new tumor event after initial treatment, lymphatic invasion and residual tumor (p < 0.05, Supplementary Table 3).Therefore, the identification of genes associated with LNM can be indispensable for the diagnosis and treatment of CRC.
In this research, we analyzed 614 CRC patients and their clinical information in the TCGA database. We used the up-regulated genes screened in CRC patients with LNM to intersect with genes up-regulated in CRC tissues. Next, through COX regression analysis, it was found that the four-mRNA signature (EPHA8, CLPSL1, KRT85, and GABRA3) was closely related to the prognosis of patients with CRC. We further compared the four mRNAs expression between patients with LNM and with NLNM. The results showed that EPHA8, KRT85, and GABRA3 and CLPSL1 were highly expressed in patients with LNM (p < 0.05, Supplementary Figure S2A-D). Patients were divided into low risk group and high risk group according to the risk score of four-mRNA signature. Surprisingly, patients in high-risk group had worse prognosis, then the training set and validation set are well validated. By further stratification analysis, the risk score based on four-mRNA signature was an independent prognostic indicator for female, T3+T4, N1+N2, stage III+IV and no new tumor event. Although the risk score is not statistically significant in male, T1+T2, N0, stage I+II and new tumor event, we can also find that the high risk score group has a poorer survival. The result suggests that four-mRNA signature have the potential to be used as a combined biomarker for CRC prognosis. The GO and KEGG enrichment analysis shows that genes up-regulated in the high risk score group are mainly associated with receptor ligand activity, neuroactive ligand-receptor interactions and estrogen signaling pathway. Neuroactive ligand-receptor interactions have been found to be associated with multiple cancers [32], estrogen signaling pathway plays an important role in the development of CRC [33], receptor ligand activity affects invasion and metastasis of CRC. This result provides new insights and research ideas for the four-mRNA signatures affecting the prognosis of patients with CRC.
EphA8 is one of the receptors in Ephs subfamily of receptor tyrosine kinases, it is associated with angiogenesis, cell adhesion and migration. Overexpression of EphA8 enhances the invasive ability of oral squamous cell carcinoma [34], miR-10a/EphA8 pathway can affect glioma invasion and migration through epithelial-mesenchymal transition [35]. In our study, the HR of EPHA8 > 1, this means that patients with high expression of EphA8 have a poorer prognosis. Gammaaminobutyric acid (GABA) is an inhibitory neurotransmitter, and Gabra3 is a subunit of GABA type A receptor [36]. In previous research, Gumireddy K et al. found that GABRA3 can promote cell invasion, migration and metastasis through AKT pathway in breast cancer [37], Recent studies have shown that miR-92b-3p targets and reduces GABRA3 expression and thus inhibits pancreatic cancer cell invasion and migration [38], GABRA3 also plays an important role in the occurrence and development of liver cancer and lung cancer [39, 40]. However, the role of GABRA3 in CRC is still unclear. Interestingly, in this work, GABRA3 is a protective factor, but the role of GABRA3 in CRC needs further research.
For the first time, our study reported a four-mRNA signature associated with LNM for prognosis of CRC using bioinformatics methods. In this work, patients with high risk scores have a poorer prognosis. Independent of other clinic pathological parameters, four-mRNA signature may become a combined biomarker for predicting the prognosis of CRC patients. However, the four-mRNA signature should be validated in a larger sample size database as well as in clinical samples. Moreover, our results warrant further studies of the mechanisms by which the four-mRNA signature affects prognosis of CRC.
Conclusions
The results of this study indicate that a four-mRNA signature related with LNM can effectively predict the prognosis of patients with CRC, but the specific molecular mechanism of the four-mRNA acting on colorectal cancer and whether this four-mRNA signature can be successfully applied to the clinic still need further research.
Abbreviations
CRC: colorectal cancer; LNM: lymph node metastasis; NLNM: no lymph node metastasis; DEGs: differentially expressed genes; EPHA8: EPH receptor A8; KRT85: keratin 85; GABRA3: gamma-aminobutyric acid (GABA) A receptor, subunit alpha 3; CLPSL1: colipase like 1.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by National Natural Science Foundation of China and Liaoning joint fund key program (No. U1608281), National Natural Science Foundation of China (No. 81573462), Liaoning Revitalization Talents Program (No. XLYC1807201), Shenyang S&T Projects (19-109-4-09).
Author Contributions
LZ, LYJ, ASQ and LW collected the data; QW, LWZ, XML, YYZ and XYS performed the statistical analysis; XPL and QZ wrote the paper; LZ and MJW conceived the study. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Xu P, Fan W, Zhang Z. et al. The Clinicopathological and Prognostic Implications of FoxP3(+) Regulatory T Cells in Patients with Colorectal Cancer: A Meta-Analysis. Front Physiol. 2017;8:950
2. Bray F, Ferlay J, Soerjomataram I. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394-424
3. Jin M, Frankel WL. Lymph Node Metastasis in Colorectal Cancer. Surg Oncol Clin N Am. 2018;27(2):401-12
4. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7-30
5. Gao JY, Song BR, Peng JJ. et al. Correlation between mitochondrial TRAP-1 expression and lymph node metastasis in colorectal cancer. World J Gastroentero. 2012;18(41):5965-71
6. Yiu AJ, Yiu CY. Biomarkers in Colorectal Cancer. Anticancer Res. 2016;36(3):1093-102
7. Falchook GS, Kurzrock R. VEGF and dual-EGFR inhibition in colorectal cancer. Cell Cycle. 2015;14(8):1129-30
8. Ren Q, Jin B. The clinical value and biological function of PTTG1 in colorectal cancer. Biomed Pharmacother. 2017;89:108-15
9. Zhang JS, Yang LQ, Du BR. et al. Higher RABEX-5 mRNA predicts unfavourable survival in patients with colorectal cancer. Eur Rev Med Pharmacol Sci. 2017;21(10):2372-6
10. Pereira AL, Magalhaes L, Moreira FC. et al. Epigenetic Field Cancerization in Gastric Cancer: microRNAs as Promising Biomarkers. J Cancer. 2019;10(6):1560-9
11. Rokavec M, Kaller M, Horst D. et al. Pan-cancer EMT-signature identifies RBM47 down-regulation during colorectal cancer progression. Sci Rep. 2017;7(1):4687
12. Angius A, Uva P, Pira G. et al. Integrated Analysis of miRNA and mRNA Endorses a Twenty miRNAs Signature for Colorectal Carcinoma. Int J Mol Sci. 2019;20(16):4067
13. Dong C, Cui D, Liu G. et al. Cancer stem cell associated eight gene-based signature predicts clinical outcomes of colorectal cancer. Oncol Lett. 2019;17(1):442-9
14. Zuo S, Dai G, Ren X. Identification of a 6-gene signature predicting prognosis for colorectal cancer. Cancer Cell Int. 2019;19:6
15. Baba H, Ishikawa T, Mogushi K. et al. Identification of SATB1 as a Specific Biomarker for Lymph Node Metastasis in Colorectal Cancer. Anticancer Res. 2016;36(8):4069-76
16. Verbeke H, Struyf S, Laureys G. et al. The expression and role of CXC chemokines in colorectal cancer. Cytokine Growth Factor Rev. 2011;22(5-6):345-58
17. Lech G, Slotwinski R, Slodkowski M. et al. Colorectal cancer tumour markers and biomarkers: Recent therapeutic advances. World J Gastroenterol. 2016;22(5):1745-55
18. Sun AJ, Gao HB, Liu G. et al. Identification of MSX1 and DCLK1 as mRNA Biomarkers for Colorectal Cancer Detection Through DNA Methylation Information. J Cell Physiol. 2017;232(7):1879-84
19. Zong Z, Zhou TC, Rao LJ. et al. Musashi2 as a novel predictive biomarker for liver metastasis and poor prognosis in colorectal cancer. Cancer Med-Us. 2016;5(4):623-30
20. Song GH, Xu SF, Zhang H. et al. TIMP1 is a prognostic marker for the progression and metastasis of colon cancer through FAK-PI3K/AKT and MAPK pathway. J Exp Clin Canc Res. 2016;35(1):148
21. Zhang Z, Song N, Wang Y. et al. Analysis of differentially expressed circular RNAs for the identification of a coexpression RNA network and signature in colorectal cancer. J Cell Biochem. 2019;120(4):6409-19
22. Dai W, Feng Y, Mo S. et al. Transcriptome profiling reveals an integrated mRNA-lncRNA signature with predictive value of early relapse in colon cancer. Carcinogenesis. 2018;39(10):1235-44
23. Dai W, Li Y, Mo S. et al. A robust gene signature for the prediction of early relapse in stage I-III colon cancer. Mol Oncol. 2018;12(4):463-75
24. Xing Y, Zhao Z, Zhu Y. et al. Comprehensive analysis of differential expression profiles of mRNAs and lncRNAs and identification of a 14-lncRNA prognostic signature for patients with colon adenocarcinoma. Oncol Rep. 2018;39(5):2365-75
25. Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352(6282):169-75
26. Cao H, Xu EP, Liu H. et al. Epithelial-mesenchymal transition in colorectal cancer metastasis: A system review. Pathol Res Pract. 2015;211(8):557-69
27. Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Gene Dev. 2013;27(20):2192-206
28. Ma Y, Li YF, Wang T. et al. Identification of proteins associated with lymph node metastasis of gastric cancer. J Cancer Res Clin Oncol. 2014;140(10):1739-49
29. Qi L, Ding Y. Analysis of metastasis associated signal regulatory network in colorectal cancer. Biochem Biophys Res Commun. 2018;501(1):113-8
30. Weitz J, Koch M, Debus J. et al. Colorectal cancer. Lancet. 2005;365(9454):153-65
31. Ozawa T, Kandimalla R, Gao F. et al. A MicroRNA Signature Associated With Metastasis of T1 Colorectal Cancers to Lymph Nodes. Gastroenterology. 2018;154(4):844-8.e7
32. Zhang HY, Liu JM, Fu XY. et al. Identification of Key Genes and Pathways in Tongue Squamous Cell Carcinoma Using Bioinformatics Analysis. Med Sci Monitor. 2017;23:5924-32
33. Barzi A, Lenz AM, Labonte MJ. et al. Molecular pathways: Estrogen pathway in colorectal cancer. Clin Cancer Res. 2013;19(21):5842-8
34. Liu LJ, Wang XF, Ge WZ. EphA8 is a Prognostic Factor for Oral Tongue Squamous Cell Carcinoma. Med Sci Monitor. 2018;24:7213-22
35. Yan Y, Wang Q, Yan XL. et al. miR-10a controls glioma migration and invasion through regulating epithelial-mesenchymal transition via EphA8. Febs Lett. 2015;589(6):756-65
36. Vithlani M, Terunuma M, Moss SJ. The dynamic modulation of GABA(A) receptor trafficking and its role in regulating the plasticity of inhibitory synapses. Physiol Rev. 2011;91(3):1009-22
37. Gumireddy K, Li A, Kossenkov AV. et al. The mRNA-edited form of GABRA3 suppresses GABRA3-mediated Akt activation and breast cancer metastasis. Nat Commun. 2016;7:10715
38. Long MM, Zhan M, Xu SW. et al. miR-92b-3p acts as a tumor suppressor by targeting Gabra3 in pancreatic cancer. Mol Cancer. 2017;16(1):167
39. Liu Y, Li YH, Guo FJ. et al. Gamma-aminobutyric acid promotes human hepatocellular carcinoma growth through overexpressed gamma-aminobutyric acid A receptor alpha 3 subunit. World J Gastroentero. 2008;14(47):7175-82
40. Liu Y, Guo FJ, Dai M. et al. Gammaaminobutyric Acid A Receptor Alpha 3 Subunit is Overexpressed in Lung Cancer. Pathol Oncol Res. 2009;15(3):351-8
Author Biography
Minjie Wei, PhD, Dean of the School of Pharmacy,China Medical University, Director of the Department of Pharmacology.Mainly engaged in molecular tumor pharmacology, neuroimmunological pharmacology and pharmacogenomics research,she has undertaken more than 30 various types of vertical scientific research projects including 7 national natural science funds and 4 major special sub-projects of the Ministry of Science and Technology; The research papers were published in more than 200 academic journals such as "Cancer Research" and "Cancer Cell", among which nearly 70 papers were included in SCI; Received five achievements awards from the Ministry of Education and the Second Prize of Liaoning Province Science and Technology Progress Award;She holds 12 national invention patents.
Lin Zhao, PhD, Department of Pharmacology, China Medical University, mainly engaged in the study of tumor pathogenesis and drug resistance mechanisms, she has presided over six projects including the National Natural Science Foundation of China and the National Natural Science Youth Fund, and published more than 40 research papers at home and abroad, including more than 20 articles in SCI source journals. Received scientific and technological achievements from the Ministry of Education and Liaoning Science and Technology Awards, She holds three patents.
![]() Corresponding authors: Minjie Wei and Lin Zhao, PhD, Department of Pharmacology, School of Pharmacy, China Medical University, No.77 Puhe Road, Shenyang North New Area, Shenyang 110122, Liaoning, China. Email: weiminjiecmucom; zl_cmucom.
Corresponding authors: Minjie Wei and Lin Zhao, PhD, Department of Pharmacology, School of Pharmacy, China Medical University, No.77 Puhe Road, Shenyang North New Area, Shenyang 110122, Liaoning, China. Email: weiminjiecmucom; zl_cmucom.