Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(13):3965-3975. doi:10.7150/jca.42663 This issue Cite
Review
The role and function of PPARγ in bladder cancer
Tianchen Peng1,5,*, Gang Wang2,3,4,*, Songtao Cheng1,5,*, Yaoyi Xiong1,5, Rui Cao6, Kaiyu Qian2,3,4, Lingao Ju2,3,4, Xinghuan Wang1, ![]() , Yu Xiao1,2,3,4,5,
, Yu Xiao1,2,3,4,5, ![]()
1. Department of Urology, Zhongnan Hospital of Wuhan University, Wuhan, China
2. Department of Biological Repositories, Zhongnan Hospital of Wuhan University, Wuhan, China
3. Human Genetics Resource Preservation Center of Wuhan University, Wuhan, China
4. Human Genetics Resource Preservation Center of Hubei Province, Wuhan, China
5. Cancer Precision Diagnosis and Treatment and Translational Medicine Hubei Engineering Research Center, Wuhan, China
6. Department of Urology, Beijing Friendship Hospital, Capital Medical University, Beijing, China.
*These authors contributed equally to this work.
Received 2019-12-2; Accepted 2020-3-8; Published 2020-4-6
Abstract
Peroxisome proliferator-activated receptor gamma (PPARγ), a member of the nuclear receptor superfamily, participates in multiple physiological and pathological processes. Extensive studies have revealed the relationship between PPARγ and various tumors. However, the expression and function of PPARγ in bladder cancer seem to be controversial. It has been demonstrated that PPARγ affects the occurrence and progression of bladder cancer by regulating proliferation, apoptosis, metastasis, and reactive oxygen species (ROS) and lipid metabolism, probably through PPARγ-SIRT1 feedback loops, the PI3K-Akt signaling pathway, and the WNT/β-catenin signaling pathway. Considering the frequent relapses after chemotherapy, some researchers have focused on the relationship between PPARγ and chemotherapy sensitivity in bladder cancer. Moreover, the feasibility of PPARγ ligands as potential therapeutic targets for bladder cancer has been uncovered. Taken together, this review summarizes the relevant literature and our findings to explore the complicated role and function of PPARγ in bladder cancer.
Keywords: PPARγ, bladder cancer, ROS metabolism, lipid metabolism, chemotherapy sensitivity, ligands
Introduction
Bladder cancer (BCa) is one of the most prevalent malignant tumors in the urinary system [1]. Because of a high recurrence rate, bladder cancer patients require frequent follow-up, which is an extremely heavy burden on patients and their families. Approximately 70% of newly diagnosed patients have non‐muscle‐invasive bladder cancer (NMIBC), and 10-20% of patients will progress to muscle‐invasive bladder cancer (MIBC) [2]. Diagnosis and treatment review of bladder cancer still relies on traditional diagnosis by cystoscopy biopsy. Furthermore, for patients with NMIBC, the preferred therapeutic method is transurethral resection of the bladder cancer [3]. The gold standard treatment for MIBC is radical cystectomy [4]. Considering the invasiveness of the diagnostic method and the unfavorable outcomes of the treatment approach, finding new targets for the diagnosis and therapy of bladder cancer has been an urgent problem. Peroxisome proliferator-activated receptors (PPARs), which are members of the nuclear receptor superfamily, can be divided into three subtypes: PPARα, PPARβ and PPARγ [5]. Our results and previous studies showed that PPARγ plays a significant role in the occurrence and progression of bladder cancer through regulation of proliferation, apoptosis, metastasis, and reactive oxygen species (ROS) and lipid metabolism [6-10]. The purpose of this paper is to provide an overview of the role, function and potential molecular mechanisms of PPARγ in bladder cancer.
PPARγ and Bladder Cancer
The expression of PPARγ in bladder cancer
Previous studies have compared bladder cancer with paracancerous tissues, and showed the controversial consequences of the expression of PPARγ in bladder cancer. Cheng et al. searched the Oncomine database and found that there was amplified mRNA expression of PPARγ in BCa tissues [7]. In another study, PPARγ expression was evaluated in human BCa and normal bladder tissue samples by FISH assay. The results indicated that 8 out of 21 tumors had PPARγ upregulation while merely 1 out of 23 normal bladder samples showed PPARγ amplification [11]. In addition, in a study involving 117 paraffin slice specimens of bladder cancer, researchers found that PPARγ was more likely to be upregulated in the Ta-T1 phase of tumors than in invasive tumors in the T2-T4 phase. Moreover, PPARγ was negatively correlated with the tumor grade, since its expression was higher in tumors of low grade (grade I) than in those of higher grades (grades II and III) [12]. In contrast to the above results, Zhang et al. collected information on patients with BCa from The Cancer Genome Atlas (TCGA) and revealed that PPARγ expression was downregulated in BCa [13]. Furthermore, in a tissue array of 66 volunteers with BCa, the translation level of PPARγ was significantly increased in paracancerous (normal) tissues [14]. Based on the above controversial results, more bladder cancer samples from different races, clinical stages and subtypes are needed to investigate the expression of PPARγ. Regarding cancer prognosis, the results were surprisingly consistent in that amplification of PPARγ mattered greatly in longer survival time and reduced recurrence or progression [12, 14-18].
The cellular mechanisms of PPARγ functions
Gene ontology (GO) enrichment analysis of BCa datasets from The Cancer Genome Atlas indicated that PPARγ was related to the regulation of cell proliferation, apoptosis and enhancement of biosynthetic processes [7]. Using Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, PPARγ seemed to be involved in the cell cycle, cell adhesion, cell death, and the PI3K-AKT signaling pathway [7]. Moreover, a transcriptome analysis of bladder cancer tissue samples suggested a close correlation between PPARs, sirtuins, cell cycle regulation, ROS metabolism, and the forkhead box class O (FOXO) signaling pathway in BCa [19]. PPARγ was reported to display proto-oncogene impacts in metastatic prostate cancer, neuroblastoma and bladder cancer [20, 21]. Reducing the expression of PPARγ inhibited bladder cancer cell viability [22], specifically in cell lines expressing a gain or enhancement of PPARγ [21, 23]. In addition to its definite function in adipocyte differentiation, PPARγ is also related to the differentiation of many tissues including the urothelium [24]. Copy number alterations in PPARγ have also been reported to be associated with luminal tumors [25, 26], which exhibit notable characteristics of strong PPAR pathway activation and amplification of PPARγ and its coactivator and direct transcriptional target, FABP4 [27]. PPARγ activation also promotes the differentiation of basal bladder cancer cells to the luminal subgroup, cooperating with amplification of GATA3 and FOXA1 or activation of estrogen receptor (ER) [28, 29] and displaying downstream enrichment of CD24, ERBB3, ERBB2, FGFR3, ELF3, CDKN1A and TSC1 mutations and overexpression of E-cadherin, HER2/3, Rab-25 and Src [30, 31]. PPARγ also regulates urothelial cell differentiation by inducing the mRNA expression of uroplakin [24]. Changes in the cell phenotypes and related gene expression induced by PPARγ in BCa are summarized in Fig. 1.
The functin of PPARγ in bladder cancer.
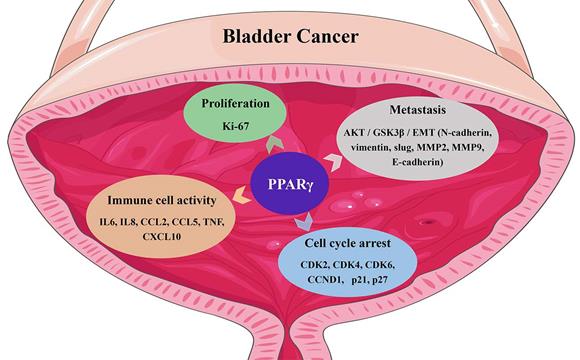
The molecular mechanism of PPARγ functions
Ligand binding activates PPARs to translocate to the DNA and bind to retinoid X receptors (RXRs) to form heterodimers and then bind to specific PPAR response elements (PPREs) [32]. In turn, this binding promotes gene expression by activating gene transcription. A recent study showed that cell cycle arrest and apoptosis caused by PPARγ agonists may be due to inhibition of the PI3K-Akt signaling pathway [14]. PTEN, a nonredundant phosphatase, is of great importance in regulating the PI3K-Akt signaling pathway [33]. It has been widely acknowledged that PPARγ inhibits the proliferation, metastasis and invasion of cancer by activating the expression of genes such as PTEN, c-myc and p27 [34, 35]. Elevated PTEN expression contributes to phosphatidylinositol trisphosphate (PIP3) dephosphorylation, which then suppresses the PIP3 and PI3K/AKT signaling pathways [36-38]. PPARγ agonists also upregulate the expression of PTEN [39], resulting in inhibition of the PI3K-Akt signaling pathway in lung cancer [40]. Based on the TCGA database, Pearson analysis showed a negative correlation of PTEN and PPARγ expression in BCa [13], which greatly different compared with other cancers. In many pathophysiological states, the activation of PPARγ and its target genes induces inhibition of the WNT/β-catenin pathway [41-43]. In nasopharyngeal carcinoma, PPARγ ligands inhibit proliferation and metastasis by regulating E2F2 [44]. Other signaling pathways including NFκB, EGFR and AP1 are also involved in the molecular regulation of PPARγ [45, 46].
The interactions of PPARγ with other molecules
PPARγ inhibits SIRT1 at both the transcriptional and translational levels after directly binding to the SIRT1 promoter [47]. In turn, SIRT1 also suppresses PPARγ [48, 49], thus forming a negative feedback loop. Transmembrane-4-L-Six-Family-1 (TM4SF1) is a member of the L6 family and strongly upregulated in MIBC. The regulatory ability of the PPARγ-SIRT1 feedback loop explains the tumor promoting effect of TM4SF1 on bladder cancer [8]. Holliday junction recognition protein (HJURP) is a centromeric histone chaperone that also acts on the PPARγ-SIRT1 negative feedback loop, impacting on cell proliferation and apoptosis in bladder cancer [9]. TGF-β1 upregulation inhibits PPARγ expression [50, 51], whereas PPARγ agonists also directly inhibit TGF-β1 activity and then suppress the expression of α-SMA [52]. In various tumors, typical WNT/β-catenin signaling is usually negatively correlated with PPARγ [53]. The WNT/β-catenin pathway directly interacts with the catenin-binding domain on PPARγ through the TCF/LEF domain on β-catenin [41]. PPARγ agonists also act on the WNT/β-catenin/PI3K/Akt signaling pathway [54], while Akt plays a part in PPARγ regulatory inhibition [55]. Different from the negative feedback loops (Fig. 2) mentioned above, reciprocal regulation found between Nrf2 and PPARγ pathways shows its unique significance [56, 57]. PPARγ directly regulated by Nrf2 through the newly identified antioxidant response elements (AREs) on its promoter [58]. The presence of putative PPREs has also been found in the promoter regions of Nrf2 [59], which provides possible direct binding for PPARγ on the Nrf2 promoter to regulate the Nrf2 pathway. In addition, numerous inflammatory cytokines, chemokines, or intracellular signaling pathways, such as TNF-α, interleukin (IL)-1, IL-13, connective tissue growth factor (CTGF), leptin and lysophosphatidic acid (LPA) decrease PPARγ expression in many pathophysiological states [60-62].
The interactions of PPARγ with other molecules.
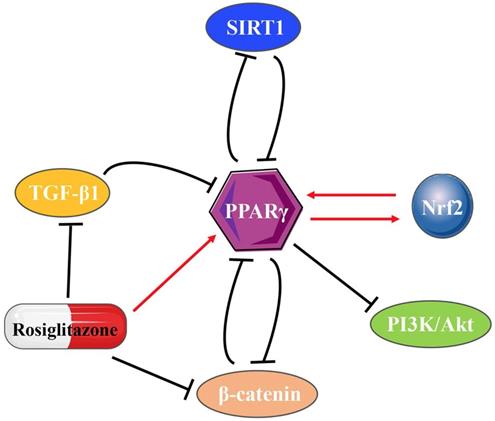
The variant genotype of PPARγ in bladder cancer
As early as 15 years ago, it was shown that the variant genotype of PPARγ indicated a lower recurrence risk among untreated patients with BCa, and MIBC exhibited a significantly higher frequency of variant PPARγ genotypes than NMIBC [63]. Subsequent studies revealed that PPARγ activity was enhanced by the P113S mutation, probably because of the mitogen-activated protein kinases (MAP kinases), which inhibit S112 phosphorylation [64]. Recently, a study showed that three recurrent mutations, PPARγM280I, PPARγT475M and PPARγI290M, modify the conformation and structural dynamics of the protein, and subsequently affect the ligand binding domain of PPARγ, resulting in a change in protein activity in luminal bladder cancer [65]. The PPARγ/RXRα pathway was presumed to be involved in the mechanism of immune escape in bladder cancer, since it was found to be related to the treatment response of NMIBC patients to Bacillus Calmette-Guérin (BCG) [34, 66]. Recurrent mutations in RXRα (S242F/Y) occurred in only 5% of MIBCs, whereas these mutations were enriched in the luminal subgroup of MIBC tumors [25, 67]. RXRα (S427F/Y) mutations drive the proliferation of urothelial organoids in the absence of tumor suppressor genes and strengthen the interaction between RXRα and PPARγ, activating the PPARγ/RXRα pathway [68], and downregulating the expression of a series of proinflammatory chemokines. Consequently, tumors rebuild the immune microenvironment to resist immune targeted therapy [67, 69].
These results revealed that the point mutation that activated PPARγ or RXRα mutation that acted through PPARγ-dependent pathways [22] provide convincing evidence for identifying PPARγ as a proto-oncogene in bladder cancer and highlight PPARγ as a promising therapeutic target for luminal MIBC [21].
PPARγ and ROS Metabolism
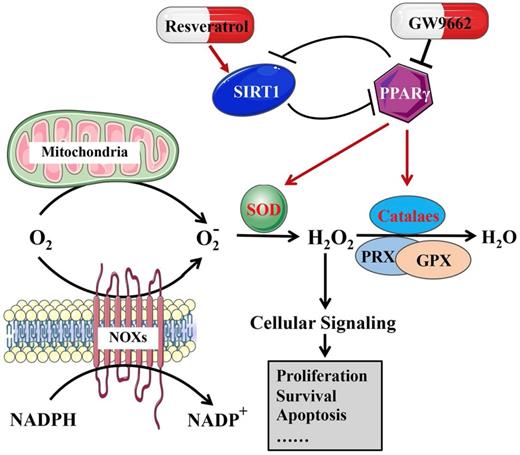
The role of ROS metabolism in cancer biology is complicated, similar to a double-edged sword, and it regulates both cell survival and apoptosis, depending on the dose, duration, type, or location [70]. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidases and mitochondria are two major sources of ROS generation [71, 72]. A moderate amount of reactive oxygen species is very important for the development of tumors, while an excessive amount of ROS suppresses tumors by inducing apoptosis [73-75]. Activation or deactivation of PPARs both affect genes related to lipid peroxidation, biological metabolism and stress reactions, including ROS [76]. PPARγ induces mitochondrial stabilization and protection against oxidative stress [77]. 15d-PGJ2, a PPARγ agonist, induces the production of ROS in bladder cancer cells [78]. Moreover, the PPARγ-SIRT1 feedback loop has been identified as regulating cell death, the antioxidative response and ROS metabolism [79, 80].
The results of our group revealed that when SIRT1 was knocked down in BCa cells, ROS production was reduced, accompanied by an increase in antioxidant enzymes and total/acetylated FOXO3a, a transcription factor that is the most closely connected to SIRT1 among the FOXO family [19]. TM4SF1 is highly expressed in human MIBC. In bladder cancer cell lines, silencing TM4SF1 activated the PPARγ-SIRT1 feedback loop, promoting the formation of ROS and increasing the expression of SOD2 and catalase [8], revealing that excess ROS were produced by disrupting homeostasis, resulting in oxidative stress and death in bladder cancer cells [81]. Both PPARγ antagonists (GW9662) and SIRT1 agonists (resveratrol) reduce the excess level of ROS production, which was induced by the knockdown of TM4SF1 in BCa cells [8]. In addition, our group found that HJURP could have an impact on the PPARγ-SIRT1 feedback loop, regulating ROS metabolism, and resulting in the progression of bladder cancer [9]. Our results, together with previous studies, revealed that the PPARγ-SIRT1 feedback loop mattered greatly in ROS metabolism in bladder cancer (Fig. 3).
The effect of the PPARγ-SIRT1 feedback loop on ROS metabolism in bladder cancer.
PPARγ and Lipid Metabolism
Reprogramming energy metabolism has been recognized as a hallmark of cancer [82]. Otto Warburg first described that cancer cells preferred aerobic glycolysis as the main energy resource even when oxygen was sufficient, which is known as the Warburg effect [83]. Recently, many studies have stressed the crucial role of fatty acid oxidation in providing adenosine triphosphate (ATP), NADPH, and other important anabolic materials in cancer cells [84-86]. Rather than aerobic glycolysis, in some cancers, lipids have been reported as the primary energy supply [87]. As some metabolomics profiling analyses have shown, active fatty acid β-oxidation promotes the development and progression of bladder cancer [88]. PPARs are of great importance in the regulation of cell proliferation, cellular differentiation, tumorigenesis and lipid metabolism [89, 90], and can also promote the expression of genes related to fatty acid oxidation and peroxisomal β-oxidation enzymes [91], which are important regulators that maintain the homeostasis of lipid ligands binding to nuclear receptors [92, 93].
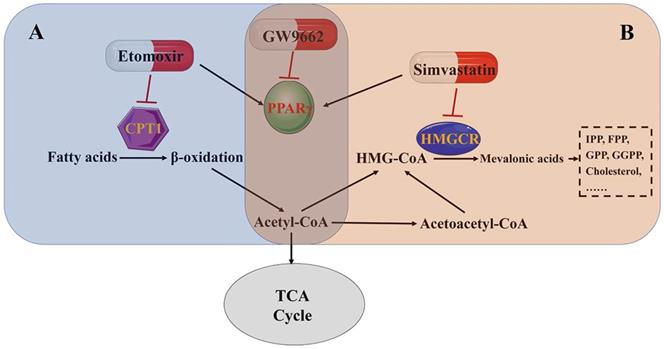
Our group has performed microarray analysis with human bladder samples. The results revealed that the PPAR signaling pathway mattered greatly in fatty acid/lipid metabolism in bladder cancer [10]. Etomoxir, a fatty acid β-oxidation inhibitor, activates PPARγ-mediated signaling, resulting in the cell cycle arrest in BCa cells [6]. Moreover, intracellular cholesterol and fatty acids are important components of the cell membrane [94], especially in the membrane domains, which are enriched with lipid rafts and are important in cell proliferation and biological metabolism [95, 96]. Simvastatin is a widely used statin drug that is important because of its rate-limiting function in cholesterol synthesis. Results from our group showed that simvastatin inhibited cell viability and induced cell cycle arrest, accompanied by PPARγ activation in BCa cell lines. A PPARγ antagonist (GW9662) restored BCa cells from simvastatin-induced cell cycle arrest [10]. These results further demonstrate that lipid metabolism in bladder cancer is regulated by the PPARγ signaling pathway (Fig. 4 A and B).
PPARγ affects lipid metabolism in bladder cancer. (A) Fatty acid β-oxidation pathway. (B) Mevalonate pathway.
PPARγ and Chemotherapy Sensitivity
Of bladder tumors, nearly 70% are low-grade non-muscle invasive cancers, which are not life-threatening but have a distinct tendency to relapse. Maintaining life-long surveillance is very expensive [97]. Since current treatments for bladder cancer are so unsatisfactory, new therapy is urgently needed. In American and European consensus guidelines, it is recommended to use intravesical infusion of chemotherapy drugs to inhibit the progression of NMIBC, and BCG is the preferred treatment [98]. BCG triggers an immune response, inducing in vitro apoptosis of BCa cells [99-101]. Moreover, the function of BCG in inducing the expression of PPARγ in murine BCa has been proven both in vitro and in vivo [34, 66]. In a bladder cancer cell line (MB49), BCG induced PPARγ amplification, nuclear translocation and transcriptional activity, resulting in proliferation inhibition, which was strengthened by 15-d-PGJ2, a PPARγ agonist [34]. The molecular mechanism might be that intravesical perfusion of BCG stimulates the immune response and induces antitumor activity [102], while PPARγ plays a role in the immune response, since rosiglitazone specifically increases natural killer T-lymphocyte cell-specific receptors [103]. In other studies, researchers found that the variant PPARγ allele was associated with a reduced risk of recurrence and progression in BCG-naïve patients and an increased risk of recurrence and progression in those patients who had received prior BCG therapy [63]. According to the above results, the combination of PPARγ agonists with certain chemotherapy drugs might be a potential strategy for the treatment of NMIBC [102].
In addition, presurgical (neoadjuvant) cisplatin-based chemotherapy (NAC) is the preferred therapy for high-risk MIBC [104]. Treatment selection depends heavily on clinical pathological features, unfortunately current staging systems are woefully inaccurate and result in an increased likelihood of disease progression and eventual death because of inadequate treatment [105]. Choi et al. reported that MIBCs can be classified as basal or luminal subtypes. Compared with basal MIBCs, luminal MIBCs are more sensitive to NAC, which can be interpreted as an indication that luminal MIBCs are more likely to have an amplification of PPARγ and activate FGFR3 mutations [31]. Although cisplatin-based chemotherapy is only effective in nearly 30% of MIBC patients, effective identification methods for patients who are sensitive to the therapy have not yet been established [104]. Therefore, to reach the maximum efficacy in bladder cancer, conventional chemotherapy should be combined with targeted therapies.
PPARγ Ligands and Bladder Cancer
Many reports have suggested that the regulation of agonists or antagonists on PPARs in cancer cells may be a potential therapeutic strategy for metabolic diseases and cancers, including bladder cancer [46]. However, there is heterogeneity in the role of PPARγ ligands in different cancers. It was reported that the PPARγ agonist rosiglitazone activates PI3K-Akt through PPARγ-dependent biosynthesis of VEGF and leptin [106, 107]. In another study, PPARγ agonists upregulated PTEN expression and inhibited the PI3K-Akt signaling pathway in lung cancer [39, 40]. Therefore, we summarized the previous studies of PPARγ ligands in bladder cancer in Table 1. Most studies showed that PPARγ activation markedly inhibited cell proliferation, induced cell cycle arrest and promoted apoptosis in bladder cancer cells and suppressed tumor growth or metastasis in vivo [108-112]. Most of the effect induced by PPARγ activation was recovered by using the PPARγ antagonist GW9662 [113]. However, GW9662 did not inhibit the action of thiazolidinedione on cell cycle arrest or apoptosis [114, 115] and failed to affect LC3-II accumulation caused by troglitazone [116], revealing that some PPARγ agonists may take effect through PPARγ activation-independent signaling pathways.
The effect and mechanism of PPARγ ligands in bladder cancer.
| Type of Ligands | Cell Type | Type of Effect | Mechanism | Year | Ref. |
|---|---|---|---|---|---|
| Rosiglitazone, Pioglitazone | 5637, UMUC3 | cell growth inhibition, cell cycle arrest, apoptosis induction | inhibition the phosphorylation of Akt (Thr308 and Ser473) and its down-stream molecules including S6, PRAS40 and GSK-3α | 2019 | [14] |
| 4-nitrophenol | T24, HUC | promote cellular proliferation, migration and invasion, inhibit adhesion and apoptosis | Bax and E‐cadherin were decreased, N‐cadherin, vimentin, snail, and slug were increased. the expression levels of cancer‐promoting genes HIF‐1, IL‐1β, VEGFα and K‐Ras were enhanced, but p53, PTEN and BRCA were decreased | 2019 | [113] |
| Pioglitazone | NUTE, J82 | cell growth inhibition, apoptosis induction | downregulate the protein levels of p53 and cyclin D1 | 2018 | [123] |
| 15d-PGJ2 | T24, 5637 | cell growth inhibition, apoptosis induction, sphere formation inhibiton | downregulate the expression of the stemness-related genes, Oct4 and Nanog. facilitate the generation of ROS | 2014 | [78] |
| Troglitazone | T24 | induces autophagy, apoptosis, necroptosis | increase the ratio of LC3-II and the phosphorylation of AMPKα. attenuate the phosphorylation of both mTOR and S6K1 | 2014 | [116] |
| DIM-Cs | UM-UC1, UM-UC3, UM-UC5, UM-UC6, UM-UC13, RT4、253JP, 253J-BV, KU7 | inhibit cell growth and render the resistant cells sensitive to EGFR inhibition | proximal promoters of PPARγ posses CEBP regulatory elements | 2013 | [46] |
| Beta-eleostearic acid (β-ESA) | T24 | apoptosis induction | ROS-mediated pathway | 2012 | [108] |
| Ciglitazone | RT4, T24 | cell cycle arrest, apoptosis induction | overexpression of p53, p21waf1/CIP1 and p27Kip1. decrease of cyclin B1. increase membrane-bound TRAIL | 2011 | [114] |
| Rosiglitazone, Troglitazone | RT4, T24 | cell cycle arrest, apoptosis induction | upregulation of soluble and/or membrane-bound TRAIL. increased cell surface death receptor 5 expression. downregulation of c-FLIP and survivin | 2010 | [115] |
| Telmisartan | T24, Caki-1 | cell growth inhibition, apoptosis induction | induce DNA fragmentation | 2010 | [109] |
| Troglitazone, 15d-PGJ2 | T24 | apoptosis induction | vascular endothelial cell growth factor, mutated p53 | 2009 | [110] |
| 1,1-bis(3-indolyl)-1-(p-substitutedphenyl) methanes (DIM-Cs) | KU7, 253J-BV | cell growth Inhibition | induced caveolin-1 and p21 expression | 2006 | [111] |
| LY 293111 | MCF7, BT 474, SK-BR-3, LL 86, H460,SW480, COLO320/HSR, RT4, HT1197 | synergistic to additive effects with gemcitabine in bladder cancer cell lines | 2004 | [112] |
Concerns arise when considering PPARγ ligands as potential chemopreventive and therapeutic agents for bladder cancer. Researchers found that PPARγ agonists cause bad results in rats, not only in bladder cancer [117-120] but also in gallbladder and adipose tumors [121]. This might be related to the dose used [122]. However, an in vitro study showed that pioglitazone usage did not increase the risk of bladder cancer [123]. There is a concept that long-term use of PPARγ agonists such as pioglitazone may increase the threat of bladder cancer in patients, while short-term use does not [124, 125]. Another theory recommended that PPARγ agonists induce changes in urine composition, particularly an increase in endogenous urinary solids (urolithiasis), which might explain the relationship between urinary bladder tumors and PPARγ agonists [126]. Increased urinary sediment, crystals, and/or calculi can lead to necrosis, erosion, and ulceration of urothelial cells, resulting in increased cell proliferation and ultimately cancer formation through epigenetic mechanisms. Some researchers confirmed that several factors affect the formation of endogenous urine solids, including the pH of urine [127], while others obtained the opposite result, denying that changes in urinary parameters are to blame [119, 128]. Overall, the mechanism and effect of PPARγ ligands in bladder cancer remain unclear, and basic and clinical research needs to be expanded and deepened in this area.
Conclusions
PPARγ is a member of the PPAR family and is expressed in a variety of cancers [129]. Activation of PPARγ regulates the expression of multiple target genes and inhibits the proliferation or migration of tumor cells [130]. However, its expression in bladder cancer is still controversial. Therefore, more bladder cancer tissue samples from different races, ages, tumor stages and grades need to be analyzed to investigate the clinical relevance of the expression of PPARγ in bladder cancer.
Abnormal metabolism is an important feature of malignant tumors [131]. Investigating the potential biomarkers or pathways involved in tumor metabolic dysfunction may provide new insights into the diagnosis and treatment of bladder cancer. Our group collected bladder cancer tissue and normal tissue for transcriptomics data analysis. The results revealed that the PPAR signaling pathway is closely related to bladder cancer and may be involved in regulating lipid metabolism [6, 10]. Jin et al. reported 12 differential metabolites that contributed to the distinction between the BCa and control groups and many of them were involved in fatty acid β-oxidation [88]. Carnitine palmitoyltransferase-1 (CPT1A) is an important rate-limiting enzyme in carnitine-dependent transport across the mitochondrial inner membrane for fatty acid β-oxidation [132]. The mRNA expression of CPT1A was significantly higher in MIBC patients than in NMIBC patients [133]. In vivo and in vitro experiments by our group showed that etomoxir, a specific inhibitor of CPT1A, inhibited bladder cell proliferation and induced cell cycle arrest in G0/G1 phase by activating the PPARγ signaling pathway [6]. In addition, our results also revealed that simvastatin, an inhibitor of cholesterol synthesis, reduced intracellular cholesterol levels and inhibited cell proliferation and migration through the PPARγ signaling pathway [10]. The above results confirmed that the PPARγ signaling pathway plays an important role in the regulation of bladder cancer lipid metabolism. However, the precise mechanisms affecting the PPARγ signaling pathway in response to lipid metabolism in bladder cancer remain poorly understood. Increased reactive oxygen species leads to an oxidation/antioxidation imbalance in tumor cells. On the one hand, the accumulation of ROS damages biological macromolecules, leading to a variety of diseases including cancer. On the other hand, it can also selectively kill tumor cells by regulating intracellular ROS levels. Our previous results and related studies showed that the PPARγ can disrupt the homeostasis of ROS metabolism in bladder cancer by affecting the expression of antioxidant enzymes (catalase and SOD2), thereby affecting the occurrence and progression of bladder cancer [8, 9].
Other studies have hypothesized that the PTEN and PI3K-Akt signaling pathways may be important mechanisms for the downstream regulation of PPARγ. Moreover, complex interactions between PPARγ and other molecules, such as negative feedback loops with SIRT1, WNT/β-catenin, and TGF-β1 and reciprocal regulation with Nrf2, also exist in a variety of physiological and pathological conditions, but the specific mechanism still needs to be further investigated.
PPARγ is a ligand-activated transcription factor. Previous studies have shown that PPARγ receptor agonists inhibit the proliferation and growth of tumor cells and enhance the sensitivity of tumor cells to chemotherapies in other malignant tumors. Unfortunately, few studies have investigated the synergistic effects of PPARγ agonists and chemotherapy drugs, such as gemcitabine and docetaxel, in bladder cancer. Taken together, basic research and clinical trials are needed to determine whether PPARγ is a promising biomarker for bladder cancer diagnosis and an effective target for bladder cancer treatment.
Abbreviations
AREs: antioxidant response elements; ATP: adenosine triphosphate; BCa: bladder cancer; BCG: bacillus calmette-guérin; CPT1A: carnitine palmitoyltransferase-1; CTGF: connective tissue growth factor; ER: estrogen receptor; FOXO: forkhead box class O; GO: gene ontology; HJURP: holliday junction recognition protein; IL: interleukin; KEGG: kyoto encyclopedia of genes and genomes; LPA: lysophosphatidic acid; MAP kinases: mitogen-activated protein kinases; MIBC: muscle-invasive bladder cancer; NAC: presurgical (neoadjuvant) cisplatin-based chemotherapy; NADPH: nicotinamide adenine dinucleotide phosphate; NMIBC: non-muscle-invasive bladder cancer; PIP3: phosphatidylinositol trisphosphate; PPARγ: peroxisome proliferator-activated receptor gamma; PPARs: peroxisome proliferator-activated receptors; PPREs: PPAR response elements; ROS: reactive oxygen species; RXRs: retinoid X receptors; TCGA: The Cancer Genome Atlas; TM4SF1: transmembrane-4-L-Six-Family-1.
Acknowledgements
Author Contributions
T.P., G.W., S.C., X.W. and Y.X. conceived the idea and designed the manuscript. T.P., G.W., S.C., Y.X., R.C., K.Q. and L.J. wrote the manuscript. All authors commented on the manuscript and approved the final form of manuscript.
Funding
This study was supported in part by grants from the National Natural Science Foundation of China (grant number 81902603), the Health commission of Hubei Province scientific research project (grant number WJ2019H013) and the Fundamental Research Funds for the Central Universities (grant number 2042018kf1040 and 2042019kf0176).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7-30
2. Yun SJ, Kim SK, Kim WJ. How do we manage high-grade T1 bladder cancer? Conservative or aggressive therapy? Investig Clin Urol. 2016;57(Suppl 1):S44-51
3. Babjuk M, Bohle A, Burger M. et al. EAU Guidelines on Non-Muscle-invasive Urothelial Carcinoma of the Bladder: Update 2016. Eur Urol. 2017;71:447-61
4. Witjes JA, Comperat E, Cowan NC. et al. EAU guidelines on muscle-invasive and metastatic bladder cancer: summary of the 2013 guidelines. Eur Urol. 2014;65:778-92
5. Lemberger T, Braissant O, Juge-Aubry C. et al. PPAR tissue distribution and interactions with other hormone-signaling pathways. Ann N Y Acad Sci. 1996;804:231-51
6. Cheng S, Wang G, Wang Y. et al. Fatty acid oxidation inhibitor etomoxir suppresses tumor progression and induces cell cycle arrest via PPARgamma-mediated pathway in bladder cancer. Clin Sci. 2019;133:1745-58
7. Cheng S, Qian K, Wang Y. et al. PPARgamma inhibition regulates the cell cycle, proliferation and motility of bladder cancer cells. J Cell Mol Med. 2019;23:3724-36
8. Cao R, Wang G, Qian K. et al. TM4SF1 regulates apoptosis, cell cycle and ROS metabolism via the PPARgamma-SIRT1 feedback loop in human bladder cancer cells. Cancer Lett. 2018;414:278-93
9. Cao R, Wang G, Qian K. et al. Silencing of HJURP induces dysregulation of cell cycle and ROS metabolism in bladder cancer cells via PPARgamma-SIRT1 feedback loop. J Cancer. 2017;8:2282-95
10. Wang G, Cao R, Wang Y. et al. Simvastatin induces cell cycle arrest and inhibits proliferation of bladder cancer cells via PPARgamma signalling pathway. Sci Rep. 2016;6:35783
11. Yang DR, Lin SJ, Ding XF. et al. Higher expression of peroxisome proliferator-activated receptor gamma or its activation by agonist thiazolidinedione-rosiglitazone promotes bladder cancer cell migration and invasion. Urology. 2013;81:1109 e1-6
12. Mylona E, Giannopoulou I, Diamantopoulou K. et al. Peroxisome proliferator-activated receptor gamma expression in urothelial carcinomas of the bladder: association with differentiation, proliferation and clinical outcome. Eur J Surg Oncol. 2009;35:197-201
13. Zhang Z, Xu H, Ji J. et al. Heterogeneity of PTEN and PPAR-gamma in cancer and their prognostic application to bladder cancer. Exp Ther Med. 2019;18:3177-83
14. Lv S, Wang W, Wang H. et al. PPARgamma activation serves as therapeutic strategy against bladder cancer via inhibiting PI3K-Akt signaling pathway. BMC cancer. 2019;19:204
15. Sandes EO, Lodillinsky C, Langle Y. et al. Inducible nitric oxide synthase and PPARgamma are involved in bladder cancer progression. J Urol. 2012;188:967-73
16. Dubosq F, Ploussard G, Soliman H. et al. Identification of a three-gene expression signature of early recurrence in non-muscle-invasive urothelial cell carcinoma of the bladder. Urol Oncol. 2012;30:833-40
17. Boiteux G, Lascombe I, Roche E. et al. A-FABP, a candidate progression marker of human transitional cell carcinoma of the bladder, is differentially regulated by PPAR in urothelial cancer cells. Int J Cancer. 2009;124:1820-8
18. Possati L, Rocchetti R, Talevi S. et al. The role of peroxisome proliferator-activated receptor gamma in bladder cancer in relation to angiogenesis and progression. Gen Pharmacol. 2000;35:269-75
19. Hu Q, Wang G, Peng J. et al. Knockdown of SIRT1 Suppresses Bladder Cancer Cell Proliferation and Migration and Induces Cell Cycle Arrest and Antioxidant Response through FOXO3a-Mediated Pathways. Biomed Res Int. 2017;2017:3781904
20. Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat Rev Cancer. 2012;12:181-95
21. Goldstein JT, Berger AC, Shih J. et al. Genomic Activation of PPARG Reveals a Candidate Therapeutic Axis in Bladder Cancer. Cancer Res. 2017;77:6987-98
22. Biton A, Bernard-Pierrot I, Lou Y. et al. Independent component analysis uncovers the landscape of the bladder tumor transcriptome and reveals insights into luminal and basal subtypes. Cell Rep. 2014;9:1235-45
23. Ahmad I, Mui E, Galbraith L. et al. Sleeping Beauty screen reveals Pparg activation in metastatic prostate cancer. Proc Natl Acad Sci U S A. 2016;113:8290-5
24. Varley CL, Stahlschmidt J, Lee WC. et al. Role of PPARgamma and EGFR signalling in the urothelial terminal differentiation programme. J Cell Sci. 2004;117:2029-36
25. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315-22
26. Robertson AG, Kim J, Al-Ahmadie H. et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell. 2017;171:540-56 e25
27. Ayers SD, Nedrow KL, Gillilan RE. et al. Continuous nucleocytoplasmic shuttling underlies transcriptional activation of PPARgamma by FABP4. Biochemistry. 2007;46:6744-52
28. Warrick JI, Walter V, Yamashita H. et al. FOXA1, GATA3 and PPAR Cooperate to Drive Luminal Subtype in Bladder Cancer: A Molecular Analysis of Established Human Cell Lines. Sci Rep. 2016;6:38531
29. Kiselyov A, Bunimovich-Mendrazitsky S, Startsev V. Key signaling pathways in the muscle-invasive bladder carcinoma: Clinical markers for disease modeling and optimized treatment. Int J Cancer. 2016;138:2562-9
30. Dadhania V, Zhang M, Zhang L. et al. Meta-Analysis of the Luminal and Basal Subtypes of Bladder Cancer and the Identification of Signature Immunohistochemical Markers for Clinical Use. EBioMedicine. 2016;12:105-17
31. Choi W, Porten S, Kim S. et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer cell. 2014;25:152-65
32. Agarwal S, Yadav A, Chaturvedi RK. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem Biophys Res Commun. 2017;483:1166-77
33. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403-14
34. Langle Y, Lodillinsky C, Belgorosky D. et al. Role of peroxisome proliferator activated receptor-gamma in bacillus Calmette-Guerin bladder cancer therapy. J Urol. 2012;188:2384-90
35. Lin MS, Huang JX, Chen WC. et al. Expression of PPARgamma and PTEN in human colorectal cancer: An immunohistochemical study using tissue microarray methodology. Oncol Lett. 2011;2:1219-24
36. Strand DW, DeGraff DJ, Jiang M. et al. Deficiency in metabolic regulators PPARgamma and PTEN cooperates to drive keratinizing squamous metaplasia in novel models of human tissue regeneration. Am J Pathol. 2013;182:449-59
37. Aboseif S, El-Sakka A, Young P. et al. Mesenchymal reprogramming of adult human epithelial differentiation. Differentiation. 1999;65:113-8
38. Li Y, Liu W, Hayward SW. et al. Plasticity of the urothelial phenotype: effects of gastro-intestinal mesenchyme/stroma and implications for urinary tract reconstruction. Differentiation. 2000;66:126-35
39. Patel L, Pass I, Coxon P. et al. Tumor suppressor and anti-inflammatory actions of PPARgamma agonists are mediated via upregulation of PTEN. Curr Biol. 2001;11:764-8
40. To KKW, Wu WKK, Loong HHF. PPARgamma agonists sensitize PTEN-deficient resistant lung cancer cells to EGFR tyrosine kinase inhibitors by inducing autophagy. Eur J Pharmacol. 2018;823:19-26
41. Liu J, Wang H, Zuo Y. et al. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol Cell Biol. 2006;26:5827-37
42. Moldes M, Zuo Y, Morrison RF. et al. Peroxisome-proliferator-activated receptor gamma suppresses Wnt/beta-catenin signalling during adipogenesis. Biochem J. 2003;376:607-13
43. Sharma C, Pradeep A, Wong L. et al. Peroxisome proliferator-activated receptor gamma activation can regulate beta-catenin levels via a proteasome-mediated and adenomatous polyposis coli-independent pathway. J Biol Chem. 2004;279:35583-94
44. Yang PL, Wang JS, Cheng XM. et al. PPAR-gamma Ligand Inhibits Nasopharyngeal Carcinoma Cell Proliferation and Metastasis by Regulating E2F2. PPAR Res. 2019;2019:8679271
45. Fucci A, Colangelo T, Votino C. et al. The role of peroxisome proliferator-activated receptors in the esophageal, gastric, and colorectal cancer. PPAR Res. 2012;2012:242498
46. Mansure JJ, Nassim R, Chevalier S. et al. A novel mechanism of PPAR gamma induction via EGFR signalling constitutes rational for combination therapy in bladder cancer. PLoS One. 2013;8:e55997
47. Han L, Zhou R, Niu J. et al. SIRT1 is regulated by a PPAR{gamma}-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010;38:7458-71
48. Picard F, Kurtev M, Chung N. et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771-6
49. Qiang L, Wang L, Kon N. et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Ppargamma. Cell. 2012;150:620-32
50. Wei J, Ghosh AK, Sargent JL. et al. PPARgamma downregulation by TGFss in fibroblast and impaired expression and function in systemic sclerosis: a novel mechanism for progressive fibrogenesis. PloS one. 2010;5:e13778
51. Zheng S, Chen A. Disruption of transforming growth factor-beta signaling by curcumin induces gene expression of peroxisome proliferator-activated receptor-gamma in rat hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. 2007;292:G113-23
52. Wei J, Zhu H, Komura K. et al. A synthetic PPAR-gamma agonist triterpenoid ameliorates experimental fibrosis: PPAR-gamma-independent suppression of fibrotic responses. Ann Rheum Dis. 2014;73:446-54
53. Vallee A, Lecarpentier Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/beta-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front Immunol. 2018;9:745
54. Farshbaf MJ, Ghaedi K, Shirani M. et al. Peroxisome proliferator activated receptor gamma (PPARgamma) as a therapeutic target for improvement of cognitive performance in Fragile-X. Med Hypotheses. 2014;82:291-4
55. Kim SP, Ha JM, Yun SJ. et al. Transcriptional activation of peroxisome proliferator-activated receptor-gamma requires activation of both protein kinase A and Akt during adipocyte differentiation. Biochem Biophys Res Commun. 2010;399:55-9
56. Polvani S, Tarocchi M, Galli A. PPARgamma and Oxidative Stress: Con(beta) Catenating NRF2 and FOXO. PPAR Res. 2012;2012:641087
57. Huang J, Tabbi-Anneni I, Gunda V. et al. Transcription factor Nrf2 regulates SHP and lipogenic gene expression in hepatic lipid metabolism. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1211-21
58. Lee C. Collaborative Power of Nrf2 and PPARgamma Activators against Metabolic and Drug-Induced Oxidative Injury. Oxid Med Cell Longev. 2017;2017:1378175
59. Kvandova M, Majzunova M, Dovinova I. The role of PPARgamma in cardiovascular diseases. Physiol Res. 2016;65:S343-S63
60. Simon MF, Daviaud D, Pradere JP. et al. Lysophosphatidic acid inhibits adipocyte differentiation via lysophosphatidic acid 1 receptor-dependent down-regulation of peroxisome proliferator-activated receptor gamma2. J Biol Chem. 2005;280:14656-62
61. Tan JT, McLennan SV, Song WW. et al. Connective tissue growth factor inhibits adipocyte differentiation. Am J Physiol Cell Physiol. 2008;295:C740-51
62. Yamasaki S, Nakashima T, Kawakami A. et al. Cytokines regulate fibroblast-like synovial cell differentiation to adipocyte-like cells. Rheumatology. 2004;43:448-52
63. Leibovici D, Grossman HB, Dinney CP. et al. Polymorphisms in inflammation genes and bladder cancer: from initiation to recurrence, progression, and survival. J Clin Oncol. 2005;23:5746-56
64. Ristow M, Muller-Wieland D, Pfeiffer A. et al. Obesity associated with a mutation in a genetic regulator of adipocyte differentiation. N Engl J Med. 1998;339:953-9
65. Rochel N, Krucker C, Coutos-Thevenot L. et al. Recurrent activating mutations of PPARgamma associated with luminal bladder tumors. Nat Commun. 2019;10:253
66. Lodillinsky C, Umerez MS, Jasnis MA. et al. Bacillus Calmette-Guerin induces the expression of peroxisome proliferator-activated receptor gamma in bladder cancer cells. Int J Mol Med. 2006;17:269-73
67. Korpal M, Puyang X, Jeremy Wu Z. et al. Evasion of immunosurveillance by genomic alterations of PPARgamma/RXRalpha in bladder cancer. Nat Commun. 2017;8:103
68. Halstead AM, Kapadia CD, Bolzenius J. et al. Bladder-cancer-associated mutations in RXRA activate peroxisome proliferator-activated receptors to drive urothelial proliferation. Elife. 2017 6
69. Hedegaard J, Lamy P, Nordentoft I. et al. Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer cell. 2016;30:27-42
70. Gupta SC, Hevia D, Patchva S. et al. Upsides and downsides of reactive oxygen species for cancer: the roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid Redox Signal. 2012;16:1295-322
71. Alexandre J, Hu Y, Lu W. et al. Novel action of paclitaxel against cancer cells: bystander effect mediated by reactive oxygen species. Cancer Res. 2007;67:3512-7
72. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245-313
73. Trachootham D, Zhou Y, Zhang H. et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer cell. 2006;10:241-52
74. Halliwell B. Oxidative stress and cancer: have we moved forward? Biochem J. 2007;401:1-11
75. Lin MH, Lee YH, Cheng HL. et al. Capsaicin Inhibits Multiple Bladder Cancer Cell Phenotypes by Inhibiting Tumor-Associated NADH Oxidase (tNOX) and Sirtuin1 (SIRT1). Molecules. 2016 21
76. Vecchione G, Grasselli E, Voci A. et al. Silybin counteracts lipid excess and oxidative stress in cultured steatotic hepatic cells. World J Gastroenterol. 2016;22:6016-26
77. Fuenzalida K, Quintanilla R, Ramos P. et al. Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J Biol Chem. 2007;282:37006-15
78. Wang Y, Tan H, Xu D. et al. The combinatory effects of PPAR-gamma agonist and survivin inhibition on the cancer stem-like phenotype and cell proliferation in bladder cancer cells. Int J Mol Med. 2014;34:262-8
79. Xiong F, Hu L, Zhang Y. et al. miR-22 inhibits mouse ovarian granulosa cell apoptosis by targeting SIRT1. Biol Open. 2016;5:367-71
80. Wang Y, Liang X, Chen Y. et al. Screening SIRT1 Activators from Medicinal Plants as Bioactive Compounds against Oxidative Damage in Mitochondrial Function. Oxid Med Cell Longev. 2016;2016:4206392
81. Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48:749-62
82. Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532
83. Bensinger SJ, Christofk HR. New aspects of the Warburg effect in cancer cell biology. Semin Cell Dev Biol. 2012;23:352-61
84. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. 2013;13:227-32
85. Hossain F, Al-Khami AA, Wyczechowska D. et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol Res. 2015;3:1236-47
86. Ricciardi MR, Mirabilii S, Allegretti M. et al. Targeting the leukemia cell metabolism by the CPT1a inhibition: functional preclinical effects in leukemias. Blood. 2015;126:1925-9
87. Massari F, Ciccarese C, Santoni M. et al. Metabolic phenotype of bladder cancer. Cancer Treat Rev. 2016;45:46-57
88. Jin X, Yun SJ, Jeong P. et al. Diagnosis of bladder cancer and prediction of survival by urinary metabolomics. Oncotarget. 2014;5:1635-45
89. Belfiore A, Genua M, Malaguarnera R. PPAR-gamma agonists and their effects on IGF-I receptor signaling: Implications for cancer. PPAR Res. 2009;2009:830501
90. Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409-35
91. Nakamura MT, Yudell BE, Loor JJ. Regulation of energy metabolism by long-chain fatty acids. Prog Lipid Res. 2014;53:124-44
92. Latruffe N, Cherkaoui Malki M, Nicolas-Frances V. et al. Regulation of the peroxisomal beta-oxidation-dependent pathway by peroxisome proliferator-activated receptor alpha and kinases. Biochem Pharmacol. 2000;60:1027-32
93. Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9:162-76
94. Mouritsen OG, Bagatolli LA. Lipid domains in model membranes: a brief historical perspective. Essays Biochem. 2015;57:1-19
95. Chichili GR, Rodgers W. Cytoskeleton-membrane interactions in membrane raft structure. Cell Mol Life Sci. 2009;66:2319-28
96. George KS, Wu S. Lipid raft: A floating island of death or survival. Toxicol Appl Pharmacol. 2012;259:311-9
97. Botteman MF, Pashos CL, Redaelli A. et al. The health economics of bladder cancer: a comprehensive review of the published literature. Pharmacoeconomics. 2003;21:1315-30
98. Sylvester RJ, van der MA, Lamm DL. Intravesical bacillus Calmette-Guerin reduces the risk of progression in patients with superficial bladder cancer: a meta-analysis of the published results of randomized clinical trials. J Urol. 2002;168:1964-70
99. Sandes E, Lodillinsky C, Cwirenbaum R. et al. Cathepsin B is involved in the apoptosis intrinsic pathway induced by Bacillus Calmette-Guerin in transitional cancer cell lines. Int J Mol Med. 2007;20:823-8
100. Riemensberger J, Bohle A, Brandau S. IFN-gamma and IL-12 but not IL-10 are required for local tumour surveillance in a syngeneic model of orthotopic bladder cancer. Clin Exp Immunol. 2002;127:20-6
101. Suttmann H, Riemensberger J, Bentien G. et al. Neutrophil granulocytes are required for effective Bacillus Calmette-Guerin immunotherapy of bladder cancer and orchestrate local immune responses. Cancer Res. 2006;66:8250-7
102. Inamoto T, Azuma H, Ubai T. et al. Association of PPAR- gamma with BCG immunotherapy. Med Hypotheses. 2009;72:231
103. Kukreja A, Costi G, Marker J. et al. NKT cell defects in NOD mice suggest therapeutic opportunities. J Autoimmun. 2002;19:117-28
104. Shah JB, McConkey DJ, Dinney CP. New strategies in muscle-invasive bladder cancer: on the road to personalized medicine. Clin Cancer Res. 2011;17:2608-12
105. Svatek RS, Shariat SF, Novara G. et al. Discrepancy between clinical and pathological stage: external validation of the impact on prognosis in an international radical cystectomy cohort. BJU Int. 2011;107:898-904
106. Ku YH, Cho BJ, Kim MJ. et al. Rosiglitazone increases endothelial cell migration and vascular permeability through Akt phosphorylation. BMC Pharmacol Toxicol. 2017;18:62
107. Terrasi M, Bazan V, Caruso S. et al. Effects of PPARgamma agonists on the expression of leptin and vascular endothelial growth factor in breast cancer cells. J Cell Physiol. 2013;228:1368-74
108. Sun Z, Wang H, Ye S. et al. Beta-eleostearic acid induce apoptosis in T24 human bladder cancer cells through reactive oxygen species (ROS)-mediated pathway. Prostaglandins Other Lipid Mediat. 2012;99:1-8
109. Matsuyama M, Funao K, Kuratsukuri K. et al. Telmisartan inhibits human urological cancer cell growth through early apoptosis. Exp Ther Med. 2010;1:301-6
110. Matsuyama M, Yoshimura R. A novel approach to anticancer therapies: peroxisome proliferator activator-receptor-gamma as a new target therapy in the treatment of human urological cancer. Endocr Metab Immune Disord Drug Targets. 2009;9:76-83
111. Kassouf W, Chintharlapalli S, Abdelrahim M. et al. Inhibition of bladder tumor growth by 1,1-bis(3'-indolyl)-1-(p-substitutedphenyl)methanes: a new class of peroxisome proliferator-activated receptor gamma agonists. Cancer Res. 2006;66:412-8
112. Budman DR, Calabro A. Studies of synergistic and antagonistic combinations of conventional cytotoxic agents with the multiple eicosanoid pathway modulator LY 293111. Anticancer Drugs. 2004;15:877-81
113. Dong F, Chen L, Wang R. et al. 4-nitrophenol exposure in T24 human bladder cancer cells promotes proliferation, motilities, and epithelial-to-mesenchymal transition. Environ Mol Mutagen. 2019
114. Plissonnier ML, Fauconnet S, Bittard H. et al. The antidiabetic drug ciglitazone induces high grade bladder cancer cells apoptosis through the up-regulation of TRAIL. PloS one. 2011;6:e28354
115. Plissonnier ML, Fauconnet S, Bittard H. et al. Insights on distinct pathways of thiazolidinediones (PPARgamma ligand)-promoted apoptosis in TRAIL-sensitive or -resistant malignant urothelial cells. Int J Cancer. 2010;127:1769-84
116. Yan S, Yang X, Chen T. et al. The PPARgamma agonist Troglitazone induces autophagy, apoptosis and necroptosis in bladder cancer cells. Cancer Gene Ther. 2014;21:188-93
117. Long GG, Reynolds VL, Dochterman LW. et al. Neoplastic and non-neoplastic changes in F-344 rats treated with Naveglitazar, a gamma-dominant PPAR alpha/gamma agonist. Toxicol Pathol. 2009;37:741-53
118. Lubet RA, Fischer SM, Steele VE. et al. Rosiglitazone, a PPAR gamma agonist: potent promoter of hydroxybutyl(butyl)nitrosamine-induced urinary bladder cancers. Int J Cancer. 2008;123:2254-9
119. Long GG, Reynolds VL, Lopez-Martinez A. et al. Urothelial carcinogenesis in the urinary bladder of rats treated with naveglitazar, a gamma-dominant PPAR alpha/gamma agonist: lack of evidence for urolithiasis as an inciting event. Toxicol Pathol. 2008;36:218-31
120. Oleksiewicz MB, Southgate J, Iversen L. et al. Rat Urinary Bladder Carcinogenesis by Dual-Acting PPARalpha + gamma Agonists. PPAR Res. 2008;2008:103167
121. Tannehill-Gregg SH, Sanderson TP, Minnema D. et al. Rodent carcinogenicity profile of the antidiabetic dual PPAR alpha and gamma agonist muraglitazar. Toxicol Sci. 2007;98:258-70
122. Waites CR, Dominick MA, Sanderson TP. et al. Nonclinical safety evaluation of muraglitazar, a novel PPARalpha/gamma agonist. Toxicol Sci. 2007;100:248-58
123. Yang SL, Wang JJ, Chen M. et al. Pioglitazone Use and Risk of Bladder Cancer: an In Vitro Study. Int J Med Sci. 2018;15:228-37
124. Lewis JD, Ferrara A, Peng T. et al. Risk of bladder cancer among diabetic patients treated with pioglitazone: interim report of a longitudinal cohort study. Diabetes Care. 2011;34:916-22
125. Dormandy J, Bhattacharya M, van Troostenburg de Bruyn AR. et al. Safety and tolerability of pioglitazone in high-risk patients with type 2 diabetes: an overview of data from PROactive. Drug Saf. 2009;32:187-202
126. Cohen SM. Effects of PPARgamma and combined agonists on the urinary tract of rats and other species. Toxicol Sci. 2005;87:322-7
127. Dominick MA, White MR, Sanderson TP. et al. Urothelial carcinogenesis in the urinary bladder of male rats treated with muraglitazar, a PPAR alpha/gamma agonist: Evidence for urolithiasis as the inciting event in the mode of action. Toxicol Pathol. 2006;34:903-20
128. Achanzar WE, Moyer CF, Marthaler LT. et al. Urine acidification has no effect on peroxisome proliferator-activated receptor (PPAR) signaling or epidermal growth factor (EGF) expression in rat urinary bladder urothelium. Toxicol Appl Pharmacol. 2007;223:246-56
129. Shu L, Huang R, Wu S. et al. PPARgamma and Its Ligands: Potential Antitumor Agents in the Digestive System. Curr Stem Cell Res Ther. 2016;11:274-81
130. Yousefnia S, Momenzadeh S, Seyed Forootan F. et al. The influence of peroxisome proliferator-activated receptor gamma (PPARgamma) ligands on cancer cell tumorigenicity. Gene. 2018;649:14-22
131. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
132. Qu Q, Zeng F, Liu X. et al. Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. 2016;7:e2226
133. Kim WT, Yun SJ, Yan C. et al. Metabolic Pathway Signatures Associated with Urinary Metabolite Biomarkers Differentiate Bladder Cancer Patients from Healthy Controls. Yonsei Med J. 2016;57:865-71
Author contact
![]() Corresponding author: Dr. Yu Xiao, Email: yu.xiaoedu.cn, Tel. +86-27-6781-2689, Fax: +86-27-6781-2892 and Dr. Xinghuan Wang, Email: wangxinghuanedu.cn, Tel. +86-27-6781-3104, Fax: +86-27-6781-2892.
Corresponding author: Dr. Yu Xiao, Email: yu.xiaoedu.cn, Tel. +86-27-6781-2689, Fax: +86-27-6781-2892 and Dr. Xinghuan Wang, Email: wangxinghuanedu.cn, Tel. +86-27-6781-3104, Fax: +86-27-6781-2892.