Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(15):4474-4494. doi:10.7150/jca.44313 This issue Cite
Review
Angiogenesis in Breast Cancer Progression, Diagnosis, and Treatment
Chikezie O. Madu1, Stephanie Wang2, Chinua O. Madu3, Yi Lu4 ![]()
1. Departments of Biological Sciences, University of Memphis, Memphis, TN 38152. USA.
2. Departments of Biology and Advanced Placement Biology, White Station High School, Memphis, TN 38117. USA.
3. Departments of Biology and Advanced Placement Biology, White Station High School, Memphis, TN 38117. USA.
4. Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Memphis, TN 38163. USA.
Received 2020-1-27; Accepted 2020-4-4; Published 2020-5-18
Abstract
Angiogenesis is a significant event in a wide range of healthy and diseased conditions. This process frequently involves vasodilation and an increase in vascular permeability. Numerous players referred to as angiogenic factors, work in tandem to facilitate the outgrowth of endothelial cells (EC) and the consequent vascularity. Conversely, angiogenic factors could also feature in pathological conditions.
Angiogenesis is a critical factor in the development of tumors and metastases in numerous cancers. An increased level of angiogenesis is associated with decreased survival in breast cancer patients. Therefore, a good understanding of the angiogenic mechanism holds a promise of providing effective treatments for breast cancer progression, thereby enhancing patients' survival. Disrupting the initiation and progression of this process by targeting angiogenic factors such as vascular endothelial growth factor (Vegf)-one of the most potent member of the VEGF family- or by targeting transcription factors, such as Hypoxia-Inducible Factors (HIFs) that act as angiogenic regulators, have been considered potential treatment options for several types of cancers.
The objective of this review is to highlight the mechanism of angiogenesis in diseases, specifically its role in the progression of malignancy in breast cancer, as well as to highlight the undergoing research in the development of angiogenesis-targeting therapies.
Keywords: angiogenesis, VEGF, Breast cancer, metastasis, angiogenesis, hypoxia, vascular diseases, anti-angiogenic therapies
Introduction
According to the American Cancer Society in 2018, breast cancer (BCa) is the most common invasive malignancy and the second leading cause of tumor-related death among women globally. It is estimated that about 270,000 new cases of invasive breast cancer are projected to be diagnosed in women in the U.S. in 2018. (1) In the U.S., breast cancer death rates are higher than those of any other cancer, except for lung and skin cancer. Furthermore, it is the most commonly diagnosed cancer among American women, (2) with 40,290 women estimated to die from breast cancer annually. In contrast, only about 2,500 new cases of invasive breast cancer are expected to be diagnosed in men; 440 of that number will die from the disease. In women under 45, breast cancer is more prevalent in African-American women than in Caucasian women. African-American women overall, are more likely to die of breast cancer than women of other ethnicities, although the five-year relative survival rate for women with invasive breast cancer surged from 75 percent to 90 percent in a twenty-year period. (3) The mortality rate has dropped nearly 40% in the past 25 years due to a combination of improved early diagnosis and advanced medical treatment. (2)
Angiogenesis, the rapid increase in the formation of blood vessels, is required for supply of sufficient oxygen and nutrition for breast tumor growth. Breast cancer cells, like all body tissues, need constant nourishment and oxygen supply through the vascular network of capillaries in the system. (4) These capillaries usually do not proliferate because the cells that line the interior surface of blood vessels, endothelial cells (ECs), do not multiply. Low levels of O2 (hypoxia) triggers numerous transcriptional responses, mediated by transcription factors, referred to as hypoxia-inducible factors (HIFs). HIFs are highly conserved transcription factors that regulate the expression of multiple genes responsible for stimulating specific physiological responses, such as metabolism, angiogenesis, and cell division. Local angiogenesis is one of the tumor's microenvironment long-term primary adaptation to low O2 levels. (5) It involves the convergence of EC precursors that give rise to capillary plexus, subsequently developing into blood vessels. Angiogenesis is a crucial player in normal processes, such as embryonic development, growth, and wound healing. (6)
Angiogenesis under physiological circumstances involves the generation of novel ECs and the subsequent structural morphing of them into tubes. (7) Angiogenesis is critical in the development, progression, and metastasis of solid tumor cells. (8) During its onset, the tumor does not stimulate angiogenesis, and with low nutrient and oxygen supply, will remain limited in its growth to about 1-2 mm in diameter. (9,10) In this early phase, the tumor may reside in a dormant state, where the rate of cell death counterbalances cell proliferation, partly due to the hypoxia and, hence, insufficiently available nutrients in the microenvironment. This condition is due to the demand created by metabolites produced by the tumor cells. (11)
Consequently, the tumor activates an angiogenic switch and evolves irreversibly to an active angiogenic state. (12) This newly attained status by the tumor confers upon it the ability to recruit new capillaries, thereby resuming oxygen and nutrients supplies to both the angiogenic cells and the surrounding non-angiogenic cells, leading to rapidly increasing tumor growth. (13) Although surgically removing tumors is the current primary treatment of breast cancer, adjuvant treatment such as anti-angiogenic therapy has been employed after surgery, in advanced disease stages, when the option of surgery is no longer available. (14,15)
The initiation and progression of tumor angiogenesis are mainly due to angiogenic growth factors, such as vascular endothelial growth factor (VEGF) and fibroblast growth factors (FGF). (16-19) Several studies have shown that levels of angiogenic factors, and the subsequent number of vascular networks formed, is a predictive factor for breast cancer survival. (20-22) In other words, elevated levels are symptomatic of the aggressive nature of the respective tumor cells and correlate to a relatively poor prognosis. (23 -25) Coupled with activating angiogenesis, these factors also dictate the rate and extent to which the blood vessels permeate. To this end, compounds that target the angiogenesis pathway have increasingly attracted attention in research in breast cancer therapy. (26)
The most extensively studied compound is the drug, Bevacizumab, a humanized anti-VEGF monoclonal antibody. The FDA approved bevacizumab in 2008 for treatment of metastatic HER2-negative breast cancer after promising results in targeting VEGF were observed in preclinical trials. (27) Following that, several anti-angiogenic drugs targeting VEGF or blocking the activity of its receptor, have been approved, and are commonly used in the treatment of different cancers. (28-29) In 2011, however, the FDA rescinded its approval due to contradictory results from previous studies and reports of resultant elevated toxicity. (31,32)
While the discovery of these anti-angiogenic drugs and small molecules were hailed as a breakthrough and potential victory in one aspect of the fight against cancer, this celebration was quelled by the modest activities of these agents, such as their inability to arrest recurrent tumors in a latent state, and the moderate improvement they provide in overall patient survival.
Mechanisms of Angiogenesis
The growth and metastasis of tumors largely depend on angiogenesis. (13,17) When blood supply is deficient, tumors are incapable of growing, necrosis sets. After a while, a subsequent metastatic spread to the systemic circulation is prevented. (8, 33) Research of the mechanism and the various factors surrounding angiogenesis have helped scientists understand its impact on breast cancer and mount a countermeasure against tumor progression. Due to the dual nature of this process, however, it is vital to carefully analyze and distinguish between the mechanism that leads to normal angiogenesis, such as wound repair, normal growth, and embryo nourishment, and that of tumor-related angiogenesis.
Certain substances, referred to as angiogenic activators due to their capability of stimulating proliferation of different cells in vitro, are responsible for the initiation of angiogenesis, (17, 25) which involves signaling between tumor cells and numerous other cell types within the tumor microenvironment.
The induction of this process has been shown to depend on the production of pro-angiogenic growth factors by the tumor cells, which affect the existing vessels. (13) During these tightly regulated processes, a complex signal balance, between pro- and anti-angiogenic factors, is aggressively sustained in the microenvironment, to develop and stabilize the newly formed blood vessels. (34) Numerous studies, therefore, have confirmed that these angiogenic activators play an essential role in the development of tumors. (35)
Studies that were done earlier revealed that specific tumor cells produce both pro- and anti-angiogenic proteins that stimulate and inhibits angiogenesis, respectively. (11) (Figure 1) (36) Scientists believe that tumors activate the angiogenic switch by altering the balance between angiogenesis inducers and inhibitors exerting opposing action. (13) This switch can be accomplished by changing the transcription of the genes as observed in several tumors where an increase in the levels of VEGF and/or FGFs is recorded when compared to healthy tissue. Conversely, in other tumors, the levels of endogenous inhibitors are reduced. (37) However, the complex mechanism that directs these shifts in the balances between the regulators is still a subject of fascinating study.
Angiogenesis, the physiological process by which development of new blood vessels from preexisting vessels. New blood vessels form out of pre-existing capillaries. The new blood vessels, near and in the tumor, provide it with essential nutrients for growth. Angiogenesis in healthy tissues is regulated by the balance between anti- and pro-angiogenic factors (bottom), and this balance is destroyed by the prevalence of angiogenic factors in tumors, resulting in abnormal structure and function of blood vessels and leading to hypoxia. This reverts the balance and normalizes the vasculature. (37)
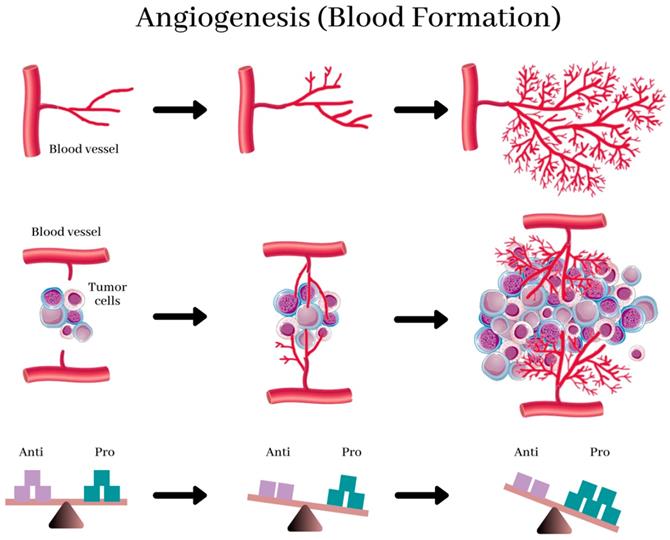
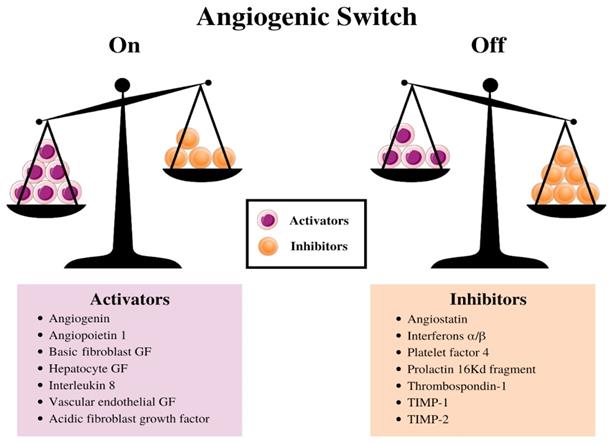
The balance between this switch determines whether the tumor will switch on angiogenesis. (13) (Figure 2). (38) Further experiments indicated a decrease in the production of the anti-angiogenic proteins turns on the tumor angiogenic switch, (39) and consequently, promotes the tumor growth and metastases. (40-42) Stimulating angiogenesis in a tumor and creating the subsequent endothelial tubes involves a multistep process that is regulated by hypoxia at every step. This process relies extensively on ECs expressing the heterodimeric transcription factor, HIF-1α (43). HIF-1α protein is stabilized and forms a heterodimer with HIF-1β under hypoxic conditions, (43,44) and this duo activates the transcription of several target genes to adapt to the hypoxic environment in human cancer cells. (45)
Schematic diagram illustrating the balance hypothesis of the angiogenic switch. It is speculated that an angiogenesis switch mechanism tightly regulates normal angiogenesis (formation of new capillaries). This balance can be disrupted to favor increased blood vessel formation through inducers and inhibitors of angiogenesis, which activates the switch. Reducing the inhibitor concentration, e.g., thrombospondin-1, 16kD prolactin, Interferon αιβ, Platelet factor-4, Angiostatin, etc. or increasing the activator levels, e.g., aFGF, bFGF, VEGF, etc., can change the balance and activate the switch, which could lead to the growth of new blood vessels. (13)
Some studies have shown that HIF-1α, working in tandem with other members of the HIF family, regulates nearly every aspect of angiogenesis, thereby making the HIF pathway a master regulator of angiogenesis. HIF-1α and HIF-2α expression have also been associated with poor prognosis and metastatic disease in several cancers (46). As a result, it is considered an attractive therapeutic target for many diseases. (47,48)
For the new blood vessels to sprout and grow, hypoxia and the HIF pathway activation in the tumor cells are critical, since they regulate the expression of a collection of pro-angiogenic genes These includes the potent cytokines, vascular endothelial growth factor (VEGF)—an endothelial mitogen and pro-angiogenic factor, (39,49,50) angiopoietin-1, angiopoietin-2, platelet-derived growth factor (PDGF), and basic fibroblast growth factor (bFGF). (51)
Additional research has centered more on the FGF and VEGF families (52) than all other angiogenetic growth factors. (53) Vascular endothelial growth factor (VEGF-A) was discovered in 1983 and sequenced completely in 1989. It was the first cytokine characterized as a major contributor to tumor angiogenesis, (52,54,55) was originally purified from tumor cell ascites as vascular permeability factor (VPF), (53) and also reported to have some biological effects on EC mitogenesis; thus, VPF is generally referred to as VEGF. (54,56,57)
VEGF is now described as a multifunctional peptide, capable of inducing receptor-mediated endothelial cell proliferation and angiogenesis both in vivo and in vitro. (54, 56-58) The VEGF family is made up of at least five members whose effects are mediated via three VEGF receptors (VEGFR), (Figure 3).
Schematic diagram of the receptor-binding specificity of vascular endothelial growth factor (VEGF) family members and their signaling pathways. VEGF family members bind to specific receptor tyrosine kinases: VEGFR-1, VEGFR-2, and VEGFR-3 respectively and through these signaling pathways activate different cascades and exert their various biologic effects. (59)
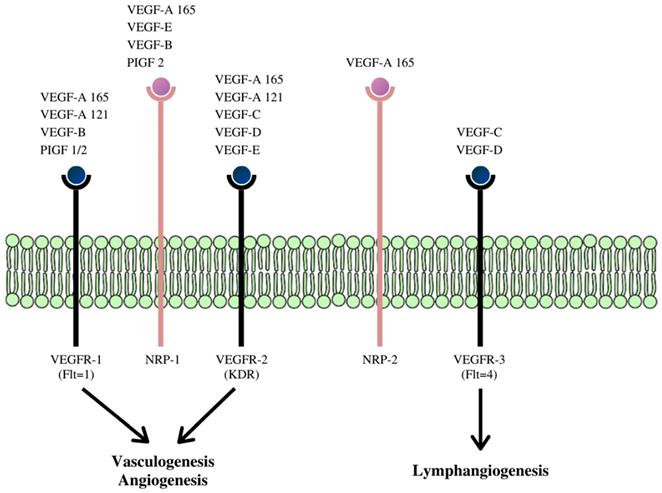
These receptors communicate with the cell's interior via transmembrane receptor tyrosine kinases (RTKs). The VEGF gene is under intricate transcriptional regulation, (60) and due to alternative splicing of its pre-mRNAs, four different RNA isoforms are expressed with diverse biological properties. This process gives rise to the other family members of this class of cytokine- VEGF-B, VEGF-C, VEGF-D, VEGF -E, and platelet-derived growth factor (PDGF). (61-64)
Pro-angiogenic factors, for example, VEGF, excites angiogenesis through the VEGF receptors (VEGFRs) and ligands (Figure 3). (65,66) The induction and progression of angiogenesis proceed in distinct steps during tumor development and can be observed through the action of vascular endothelial growth factor (VEGF) and acidic and basic fibroblast growth factors (FGF1/2). After it is expressed, VEGF binds to its receptor (VEGFR) and ligands located on the surface of ECs (Figure 3). (13,65,67,68) After binding to, and consequent activating the transmembrane tyrosine kinase receptors on the surface of the cell, it leads to dimerization, autophosphorylation, and activation of the downstream signaling pathway. This process is followed by the survival, proliferation, migration of the ECs, inhibition of apoptosis, and after several cascading processes, tube formation and sprouting. This process eventually, but slowly transforms into a developed network of new blood vessels. VEGF also induces vasodilation and stimulates vascular permeability, an underlying characteristic of tissue inflammation and the tumor microenvironment. (69)
Increase in the production of pro-angiogenic factors, such as VEGF and proteolytic enzymes, (71) and the decrease in anti-angiogenic factors, (5,71,72) results in the activity of the ECs discussed above. Ultimately, a capillary network is successfully laid down that supplies the growing tumor with sufficient nutrients and oxygen. (73) Through this new vascular bed, the tumor cell, taking advantage of this trail, may enter the systemic circulation, and promote distant metastases. Therefore, the number of metastasis sites is positively correlated with the number of cancer cells entering the circulation initially. (74)
Since its discovery, the number of angiogenic inducers identified has increased over the last decade and implicated in the regulatory process of angiogenesis in tumors. (75-77) Conceivably, tumor angiogenesis presents a uniquely attractive target therapy in all types of human tumors, and by interfering with the intracellular signaling of VEGF and VEGFR, anti-angiogenic therapy inhibits the growth of tumor vessels. (50,78-80)
Incidence of Angiogenesis in Breast Cancer
An angiogenic disease is described as either an excessive or deficient growth of microvessels. Initially, angiogenesis was implicated in cancer, arthritis, and psoriasis. However, the effect it has in several other diseases has been described. (81) The nature and composition of tumors make them inherently prime for effective angiogenic growth. An active vascular system is made up of adipose tissue, enveloped by stromal cells which gives it a supporting framework for the tumor's vascular system to emerge. White adipose tissue sustains the growth of the new vasculature and supports the development and progression of breast cancer in mouse models, (82) and the brown adipose tissue (made up of cells with numerous mitochondria) supports the tumor growth by providing a constant supply of oxygen and nutrients. (83) Both types of adipose tissues are responsible for producing angiogenic factors, most commonly VEGF A, B, and C, basic fibroblast growth factor (bFGF)/FGF-2; matrix metalloproteinases (MMPs); and IL-8, factors associated with breast cancer. (83,84) This aberrant growth of blood vessel has now been implicated in many life-threatening and disabling diseases conditions, such as cardiovascular disease, cancer, blindness, and diabetic ulcers. (9,49, 85-89)
As already discussed, abnormal angiogenesis is also critical for cancer metastasis including breast cancer metastasis. (12, 90-93) Angiogenesis involves a coordinate regulation of some vascular growth factors, such as basic fibroblast growth factor (bFGF), transforming growth factor beta-1 (TGFβ-1), platelet-derived EC growth factor, placenta growth factor, and some other growth factors, (94-96) and clinical studies have shown that it plays a critical role in breast cancer progression and metastasis. (97) These growth factors are expressed and/or upregulated in aggressive human breast cancers, and among these growth factors, the expression of VEGF and its different isoforms has been characterized as the most significant in breast cancer, (98) although low-level protein expression has been detected in a healthy human mammary gland. (99)
VEGF and IL-8 are the most studied growth factors. Breast cancer cell lines with high VEGF expression have been reported to also express high levels of interleukin-8 (IL-8), suggesting that they play very crucial roles in the promotion of angiogenesis in breast cancer angiogenesis, (100-103) A high level of VEGF receptor-3 has been detected in invasive breast cancer and also found to be upregulated in the endothelium of angiogenic blood vessels. (104) The interaction between VEGF-A and VEGFR-1 or 2 is intricately involved in breast cancer development, progression, and metastasis. (50,105-108)
One of the prognostic indicators for survival is the level of angiogenesis in breast cancer. (21,22)
An increased level of angiogenic growth factors in the breast cancer cells correlates with the aggressiveness and risk of the invasive breast cancer, (13, 23,24) and this has also been linked to p53 genes inactivation. (26) Furthermore, the number of microvessels in an invasive breast carcinoma from surgical samples may be a predictor of metastasis or relapse. (22)
Studies reveal that, for tumor development and metastasis to occur, angiogenesis in the tumors must involve an interplay of some or all these growth factors- VEGF, interleukin 8 (IL-8), basic fibroblast growth factor (bFGF/FGF-2), and matrix metalloproteinases (MMPs). (35,109,110)
Interleukins are a group of proteins and signal molecules, generally called cytokines, and first discovered in leukocytes. (111) They are secreted by cells as an immune response to various pathological stimuli. IL-8 is a member of the IL family that is produced by macrophages, airway smooth muscle cells, tumor cells, and other cell types. (112-115) and has been reported to excite the production of VEGF in ECs by binding with its receptor, and thereby activating VEGF receptors. (116) IL-8 also has a direct influence on angiogenesis by enhancing the proliferation and survival of EC, upregulating matrix metalloproteinases in certain EC lines, (110) and stimulating the formation of capillary tubes in vitro. All these are critical features of breast cancer progression and metastasis. (100-102,117-119) Furthermore, breast tumors with upregulated IL-8 levels have been observed to be more aggressive and invasive, making IL-8 levels an attractive target for anti-angiogenic treatments, (35,103) and a potential prognostic biomarker for various cancers, including breast cancer. (119)
Fibroblast growth factors (bFGF/FGF-2) are collectively a family of powerful angiogenic stimulators linked to breast cancer risk. (120-123) Substances can modulate the interactions between FGF-2 and its receptor in the extracellular environment leading to regulation of angiogenesis, and subsequent tumor progression, and metastasis. (121,124-126) MMPs belong to a larger family of proteases (126) involved in angiogenesis due to their ability to degrade extracellular matrix proteins, and thereby remodel the extracellular matrix. They are primarily involved in destabilization of the existing blood vessel wall, degradation of matrix proteins, and migration of ECs-stages that have been described as the initiation process of angiogenesis. (127-129)
Several antiangiogenic treatments that have been approved for clinical use target these pro-angiogenic growth factors, and/or their receptors, cytokines, and proteases associated with them. (130-134) Some of the examples of compounds/drugs that have gone through clinical research and approval will be covered in the next section.
Inhibitors of Angiogenesis (in Breast Cancer) and their Modes of Action
Inhibiting the proliferation of the vasculature was proposed several years ago by Judah Folkman as a model for cancer treatment, (135) and this, researchers later discovered, would entail treatment with anti-angiogenic factors, or/and compounds that can decrease the release of pro-angiogenic factors, prevent their binding to receptors, or inhibit their actions. As a result, research has made the inhibition of the VEGF pathway a central focus of angiogenesis therapy. Some of the strategies that have been formulated, other than inhibiting the VEGF pathway, include employing antibodies targeting VEGF or VEGFR, use of soluble VEGFR/VEGFR hybrids, and use of inhibitors directed against tyrosine kinase. (136,137)
Endogenous Inhibitors of Angiogenesis
As discussed above, endogenous stimulators and inhibitors regulate angiogenic processes in the body. These are found in cells or systemic circulation as protein, glycoproteins, proteoglycans, or small proteins, where they interfere with specific activities in and by the EC, such as down-regulation of genes expression, cell formation and migration, and tube morphogenesis. (138)
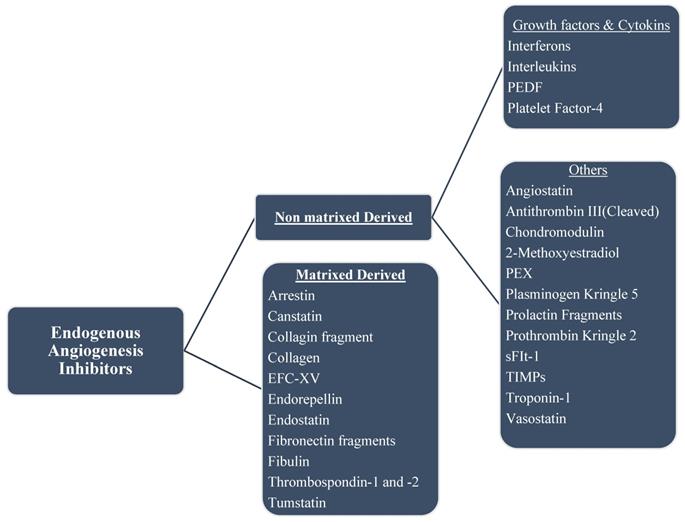
Some of the endogenous inhibitors and a brief description of their mechanism of actions are discussed below; a list of several others is shown in Figure 4.
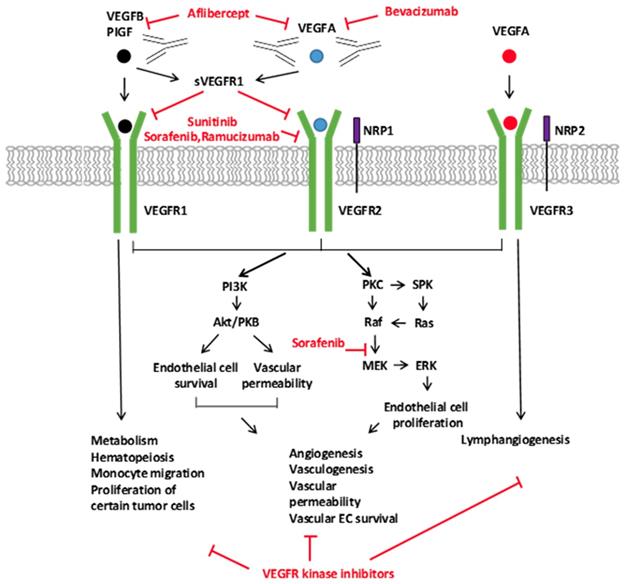
Signaling of the VEGF Ligands, Receptors, and the Inhibitors of the VEGFA Pathway. The VEGF family of ligands and their receptor-binding patterns are shown at the top. VEGF ligand family members, VEGFA, -B, -C, -D, and placental growth factor (PIGF), selectively bind to three tyrosine kinase receptors, VEGFR1, VEGFR2, and VEGFR3. As co-receptors of VEGFR2 and VEGFR3, neuropilin 1 and 2 (NRP1 and NRP2) modulate their signaling pathways. The soluble VEGFR1 (sVEGFR1) inhibits the signaling of VEGFR1 and VEGFR2 by sequestering free ligands. The different VEGF pathway inhibitors and their targets are indicated with red. Downstream VEGFR signaling pathways are shown on the bottom. VEGFR activates many proteins through PKC or PI3K. The activation of downstream signal transduction molecules leads to numerous distinct biological processes as indicated in the diagram. (139)
Thrombospondins (TSP). The thrombospondins (TSP) are a family of calcium-binding glycoproteins, composed of five highly conserved structurally related ECM proteins. They are secreted from the α granules of stimulated platelets and play a critical role in regulating in coagulation, cell adhesion, angiogenesis, and inflammation. In vetebrtates, this family is divided into two subgroups-A and B, and thrombospondins in subgroup: A (-1 and -2) are homotrimers. (140,141)
Interestingly, Thrombospondin-1 (TSP-1) was the first protein to be identified as a naturally occurring angiogenic inhibitor. (41) Studies have proposed that TSP-1, depending on proteases that produce the fragments of TSP-1, may display a dual nature, i.e., antiangiogenic and proangiogenic. (142,143) TSP-1 has been reported to inhibit tumor growth and metastasis, therefore qualifying it a powerful inhibitor of neovascularization and tumorigenesis in vivo. (144-146). Numerous research indicates that TSPs found in breast cancer, can function as strong endogenous antiangiogenic factors, consequently leading to tumor suppression; (144,147-151) TSP-2, which is similar structurally to TSP-1, has a comparable antiangiogenic and antitumor property. (152,153)
Scientists have proposed, as possible mechanisms for this antiangiogenic activity, the inhibition of VEGF using thrombospondin-2, which consequently prevents EC migration, tube formation, and EC-specific apoptosis. (154)
Endostatin. Endostatin, a fragment of collagen XVIII produced by tumor cell proteases, blocks EC proliferation and migration. (155-158) It is considered a potent antagonist of angiogenesis and inhibitor of tumor growth in mouse models. In experimental animal models, recombinant endostatin effectively inhibits angiogenesis and suppresses primary tumor growth and metastasis without obvious side effects or toxicity. (155,159,160) It is reported to have several possible mechanisms of action in relation to the inhibition of tumor angiogenesis including inhibition of tumor necrosis factor alpha (TNFα), initiation of the JNK signaling pathway, (160) interacting with and antagonizing alpha (5)- and alpha(v)-integrins on human endothelial cells surface, (158) and inhibiting EC cycle progression. (157)
Endostatin inhibits signal transduction that is stimulated by FGF-2, thus, among other actions, blocks EC motility, (162) and inhibits signaling mediated by VEGF through direct interaction with the subfamily of receptor tyrosine kinases (RTKs), VEGF-R2/KDR/Flk-1, in HUVECs. (163-165) It was found to also reduce the growth of certain breast cancer cells in vivo and thereby might prove effective in the treatment of breast cancer. (166)
Interferons. (IFNs) are a group of cytokines that mount a cellular immune response to a range of pathogens including viruses, bacteria, and tumors. (167) Studies show that they inhibit angiogenesis, induced by tumor cells in mice, by significantly lowering the secretion of the major angiogenic factor produced by tumors: interleukin (IL)-8. (168,169) Previous research reveals that interferons-α/β can inhibit tumor angiogenesis by downregulating translational expression of bFGF/FGF-2 and reducing expression of the MMP-2 gene, making them potential candidates for antiangiogenic treatments. (166,170-175) Other research, however, suggests the mechanism of IFN-α is not mediated by bFGF or VEGF. (176-178)
Interleukins. (ILs) are a family of leukocyte-derived cytokines, involved in cell signaling, predominantly serving as modulators of the immune responses. (179,180) They also have broad-ranging effects on various physiologic activities, including angiogenesis. Members of this family exert their antiangiogenic properties in diverse ways. IL-1β inhibits FGF-stimulated angiogenesis via an autocrine pathway, (181) IL-4 induced angiogenesis by inhibiting bFGF, (182) while IL-12 blocks angiogenesis through the downstream chemokines, such as IFN-inducible protein-10 and IFN-γ- induced monokine. (183-185)
Angiostatin. Angiostatin is an endogenous angiogenesis inhibitor found in humans and several other animal species. It is a proteolytic fragment of plasminogen that was isolated from tumor-bearing mice. (186) Studies show that angiostatin suppresses tumor metastasis by inhibiting the formation of blood vessels and is believed to inhibit endothelial cell migration and block tumor progression, (157, 186-188) although the fundamental mechanisms remain uncertain. (187)
Continous exposure of cells to angiostatin has been revealed to block the activation of the MAPK, extracellular-signal-regulated kinases-1 (ERK1) and ERK2, by FGF-2 or VEGF in human in endothelial and smooth muscle cells, (188,189) which ultimately leads to disruption of angiogenesis.
Decorin. Decorin is a member of a small leucine-rich proteoglycans family that is secreted by mesenchymal cells, connective tissue cells, and tumor stromal cells. It is involved in numerous cellular processes including wound healing, matrix organization, the formation of collagen fibrils, and maintenance of cell proliferation by interacting with growth factors and other ECM proteins. (190-195) It influences the balance of anti- and proangiogenic proteins, thereby creating a pathological environment. It is an inhibitor of tumor angiogenesis, progression, and metastasis through its association with certain receptors and proteins such as EGFRs, TGF-β, VEGF and VEGFR-2, and bFGF/FGF-2. (196)
Decorin also inhibits tumor cell-mediated production of hypoxia-inducible factor-1α (HIF-1α), and c-Met, and concurrently stimulates the rapid production of the antiangiogenic, angiostatic molecules thrombospondin-1 and tissue inhibitor of metalloproteinases 3 (TIMP3). (196-198) It induces Peg3-dependent autophagy in both microvascular and macrovascular endothelial cells, which leads to angiogenesis suppression. (199) Also, decorin significantly inhibits VEGFR2 by binding directly to a site that incompletely overlaps with the site for VEGF-A. (200, 201)
Pharmaceutical Inhibitors of Angiogenesis
Molecules targeting angiogenesis pathway have been extensively researched in the treatment of breast cancer and other forms of cancer. (26) Few key clinical strategies in combating angiogenesis have been adopted, including- the use of monoclonal antibodies binding VEGF, the use of decoy receptors, the use tyrosine kinase inhibitors, and the use of monoclonal antibodies targeting VEGF receptors. (202-206) Bevacizumab and ramucirumab are monoclonal humanized antibodies designed to inhibit the interaction between VEGF ligands and receptors. (207,208)
These agents are used in breast and other types of cancer therapy. (209) Several anti-angiogenesis drugs have been approved by the FDA and are currently used in cancer treatment. Still, other agents are in different stages of clinical development or preclinical evaluation. (210)
In 1993, results showed that bevacizumab, a humanized monoclonal antibody, and called Avastin, binds specifically to all isoforms of VEGF-A and disrupts its activity and interactions with VEGFR-1 and -2, leading to a decrease in tumor growth. (211,212) (Figure 4). (213)
Shortly, after that in 2004, it was approved by first US Food and Drug Administration (FDA) for the treatment of advanced colorectal cancer, then other forms of cancer, either alone or together with other chemotherapeutics, (214,215) and then extended for use in several others, including breast cancer. (216-218)
In 2007, the European Medicines Agency (EMA) granted the use of bevacizumab in metastatic HER2-negative breast cancer, followed by the approval of the FDA in 2008 for the same indication. (219)
The clinical breakthrough of bevacizumab inspired the development of several VEGFA pathway inhibitors, including a monoclonal antibody against VEGFR2 that inhibits the binding to a ligand, a recombinant chimeric receptor that sequesters various ligands in the VEGF family, and several small-molecule tyrosine kinases that inhibits the action of VEGF receptors (Figure 5). (220)
Chart of endogenous angiogenesis inhibitors. Several are fragments of a naturally occurring extracellular matrix (ECM) and basement membrane proteins. The endogenous inhibitors of angiogenesis are divided into two major categories.
Several of these compounds have been proven to have positive results with patients with various forms of cancers, (28, 29, 30) and have been approved by the FDA and/or EMA. (28-30) While we briefly discuss several of these anti-angiogenesis drugs, we particularly highlight the current findings that show the activities of antiangiogenic agents in the treatment of breast cancer.
The drugs and treatment strategies highlighted here do not represent standard therapies for only breast cancer patients, nor does it seek to exhaust all currently used drugs or regimen in the treatment.
The FDA lists the following as approved angiogenesis inhibitors:
Axitinib (Inlyta®)
Bevacizumab (Avastin®)
Cabozantinib (Cometriq®)
Everolimus (Afinitor®)
Lenalidomide (Revlimid®)
Lenvatinib mesylate (Lenvima®)
Pazopanib (Votrient®)
Ramucirumab (Cyramza®)
Regorafenib (Stivarga®)
Sorafenib (Nexavar®)
Sunitinib (Sutent®)
Temsirolimus
Thalidomide (Synovir, Thalomid®)
Vandetanib (Caprelsa®)
Ziv-aflibercept (Zaltrap®) (221)
Anti-VEGFR antibodies
One of the earliest and extensively researched strategies in combatting antiangiogenic treatment is the use of a monoclonal antibody neutralizing circulating VEGF. They involve monoclonal antibodies such as bevacizumab, antibody derivatives such as ranibizumab (Lucentis), or orally-available small molecules that inhibit the tyrosine kinases stimulated by VEGF: lapatinib, sunitinib, sorafenib, axitinib, and pazopanib. Some target VEGF receptors rather than the VEGFs and have been especially attractive in the triple-negative breast cancer subtype because they were shown to increase VEGF expression and enhance angiogenesis. (26)
Bevacizumab (Avastin). Bevacizumab is a humanized, monoclonal antibody that binds VEGF-A and blocks its activity and interactions with VEGFR-1 and -2, reducing tumor growth. (163, 211,222-224) Incidentally, it was the first FDA approved angiogenesis inhibitor and the first commercially available anti-angiogenesis drug, used in combination with additional chemotherapeutics for treatment of metastatic colon cancer. (215,225) Later, this was expanded with its single and combinatorial use for non-small cell lung cancer, renal cell carcinoma, pancreatic cancer, ovarian cancer, advanced kidney cancer, glioma, leukemia, and breast cancer. (215,218, 226-228)
In phase, I study, bevacizumab was tested in combination with chemotherapy and reported a good safety profile, (228) then got expanded as the first line of treatment when combined with paclitaxel, a chemotherapy medication, in breast cancer patients due to its ability to double the median progression-free survival (PFS). (216) In combination with other chemotherapy therapies, it was shown to have significant positive outcomes in patients with different respective tumor types. (217,229,230)
However, the therapeutic value of bevacizumab in this indication was called into question when it failed to show an effect on overall survival. Furthermore, it produced an array of side effects, including high blood pressure, significant excreted protein levels, bleeding, and blood clots. (132, 231), which seemed to increase when combined with chemotherapy. (215,232,233)
Eventually, in 2011, the FDA revoked the approval for the therapeutic use of bevacizumab for metastatic breast cancer due to lack of substantial improvements in response and survival. (27)
Cetuximab (Erbitux®). Cetuximab is a monoclonal antibody that binds to the extracellular domain of epidermal growth factor receptor (EGFR), blocking the subsequent binding of the ligand, and stimulation of the receptor. This leads to the degradation of the receptor and consequent inhibition of cell proliferation and angiogenesis. It is the first monoclonal antibody that was approved by the FDA as a second-line treatment for metastatic colorectal cancer (234, 235) and was shown to downregulate the VEGF in human colorectal carcinoma (CRC) cell line and in human CRC mouse xenografts. (236)
Ramucirumab.Ramucirumab is a human monoclonal antibody that binds to VEGFR2, blocking the binding of the receptor and its ligand VEGF. Presently, ramucirumab has EMA and FDA approval for treating advanced gastric cancer, metastatic colorectal cancer, and advanced non-small cell lung cancer. (237-239) It presents itself as a receptor antagonist blocking the binding of vascular endothelial growth factor (VEGF) to VEGFR2 by binding to VEGFR2. Preclinical and clinical evidence suggests that Ramucirumab may critical roles in angiogenesis in breast cancer growth, invasion, and metastasis. (240)
Ranibizumab. Ranibizumab is a recombinant, humanized, monoclonal antibody Fab fragment against VEGF-A. The antibody fragment was generated from the same parent mouse antibody as bevacizumab and approved as an anti-angiogenic drug for the treatment of the "wet" type of age-related macular degeneration. (241, 242)
Tyrosine kinase inhibitors
Another strategy employed in targeting the VEGF signaling is inhibiting the signal transduction, particularly of the VEGF-receptor, with tyrosine kinase inhibitors (TKI). (204,205)
Tyrosine kinases play a vital role in modifying the signaling of growth factor. When forms of these enzymes are activated, it can cause increases in proliferation and growth of tumor cell, induction of antiapoptotic effects, and promotion of angiogenesis and metastasis. (243)
TKIs are small-molecular-weight drugs that block the kinase activity of various receptors. TKIs cross the cellular membrane, where they can interfere with downstream signaling pathways. (244) The FDA has approved almost 30 small-molecule kinase inhibitors, including agents that target VEGFR, e.g., sunitinib, sorafenib, axitinib, and pazopanib.
Unlike VEGF neutralizing antibodies, TKI does not interfere with the binding of VEGF to its receptors, and they usually target other kinases, besides, such as PDGFR, FGFR, and c-KIT. (245) Currently, they collectively serve more as a secondary or tertiary line of therapies, rather than as primary therapy, and probably more useful in combination with traditional cytotoxic chemotherapy. (246) Some of these small molecules have been discussed below.
Axitinib (Inlyta). Axitinib is the first receptor tyrosine kinase inhibitors (TKI) compound with established antitumor activity. In clinical trials, it was shown to incite some response in treating renal cell carcinoma, (247) and several other types of tumor, (248) either alone or used in combination with chemotherapeutic drugs. (249-254)
Axitinib is a small molecule inhibits VEGF and platelet-derived growth factor receptor (PDGF), thereby inciting an anti-angiogenic effect. It was reported to be a potent and selective inhibitor of VEGFR-1, -2, and -3, and has been reported to reduce vascular permeability, tumor vascularization, and tumor volume. (251) In animal (xenograft) models, it was shown to significantly inhibit the growth of breast cancer. (255)
Clinical studies, however, indicates that Axitinib has shown significant benefits only in patients who have previously received paclitaxel. (256)
Sorafenib (Nexavar®). Sorafenib is a TKI with a different mechanism of action that acts downstream targeting VEGFR-1, -2, and -3, PDGFR-β, and Raf-. (257,258) It received FDA approval for the treatment of inoperable hepatocellular carcinoma, (259) advanced renal cell carcinoma, (260, 261) and for recurrent or metastatic thyroid carcinomas. (262,263)
However, the results from the treatment were mixed, since the tumors progressed in some patients. (263) Based on the NCI clinical trials search results, there have been at least 168 active clinical trials involving sorafenib in different cancers, and some are still ongoing, making it one of the recurrently studied drugs. (264-266)
The clinical result of sorafenib in breast cancer regimen is encouraging, although limited, with completed phase I/II trial of the combination of sorafenib and anastrozole, suggesting that sorafenib may be able to delay the initiation of chemotherapy in some patients, although this combination nevertheless, had significant toxicity. (267)
However, the combination of sorafenib with capecitabine, either as a primary or secondary line of treatment, did not improve PFS, OS, or ORR, and furthermore, the rates of toxicities were significantly higher with sorafenib alone. (268)
Sunitinib (Sutent®). Sunitinib is another TKI that was approved by the FDA and EMA to treat renal cell carcinoma (RCC), and imatinib-resistant gastrointestinal stromal tumors, and well-differentiated pancreatic neuroendocrine tumors (pNET). (269) When activated, it inhibits VEGFR and PDGFR, (270) and has been evaluated in about 150 clinical trials for several cancer conditions, such as ovarian, (270) breast, (271) and non-small cell lung cancer, (272) among others, (273) with mixed results. (274-277)
Currently, the benefit of sunitinib in breast cancer is still not very clear, due to some disappointing results observed. It displayed a single-agent reaction in the treatment of metastatic breast cancer, (278) while in other trials, it failed to show a positive therapeutic result in either first-line or refractory breast cancer. (279-281)
Also, three phase III studies investigating the effect of using a combination of sunitinib with chemotherapy, (280-282) and one comparing sunitinib versus capecitabine as monotherapy for patients, did not show improvement in PFS or OS. (284)
Cabozantinib (Cometriq®). Cabozantinib is a TKI, a small molecule that targets and binds to the tyrosine kinase receptors, inhibiting the activity of multiple tyrosine kinases, including RET, MET, and VEGF, and consequently inhibiting tumorigenesis, angiogenesis, metastasis, and drug resistance. (285,286)
Some studies suggest that cabozantinib is a potential agent for inhibiting tumor angiogenesis and metastasis in cancers, and the observed antitumor activity is the result of mechanisms affecting tumor angiogenesis and the inhibition of invasive tumor growth rather than the result of directly targeting cellular proliferation, (287) and clinical studies with patients with metastatic breast carcinoma patients revealed very promising data. (285)
Inhibitors of mTOR
mTOR, the mammalian target of rapamycin or the mechanistic target of rapamycin, plays a role in tumor cell proliferation and angiogenesis. (286) Rapamycin and related mTOR inhibitors block the expression of VEGF on endothelial cell, together with VEGF-induced endothelial cell proliferation. (288) mTOR inhibitors are a vital group of anti-angiogenic compounds, some of which include deforolimus, everolimus, rapamycin (sirolimus), and temsirolimus. (289,290)
Research and preclinical work conducted earlier have shown that stimulation of the mTOR signaling pathway is linked with resistance to hormonal therapy in ER+ breast cancer, and drugs that inhibit mTOR signaling can contribute in overcoming this resistance. (291-293)
Temsirolimus (Toricel®). Temsirolimus (Toricel®) is a mTOR inhibitor, approved for treating advanced renal cell carcinoma, (294) and treatment with this compound inhibits tumor angiogenesis, among other activities, by downregulating the synthesis of VEGF. (295)
Other Angiogenic Agents
Bortezomib (Velcade®). Bortezomib is a proteasome inhibitor that causes cell death and tumor regression by interfering with cancer cells signaling. (296-298) It has indirect anti-angiogenic properties, (299) and FDA-approved for the treatment of myeloma that has relapsed following two previous therapies. In a clinical trial, patients with metastatic breast cancer were treated with bortezomib, and although it was well tolerated, it showed limited clinical activity against metastatic breast cancer when used as a single agent. (300)
The VEGF-Trap
Ziv-aflibercept (Zaltrap®). Aflibercept is a drug, approved by the FDA and EMA for the treatment of wet macular degeneration for metastatic colorectal cancer as an inhibitor of VEGF. (301-303), It binds to VEGF in circulation, functioning like a soluble VEGF decoy receptor, inhibits the activity of the VEGF-A and VEGF-B, as well as PGF, (304) and consequently inhibiting the growth of new blood vessels in the choriocapillaris or the tumor, respectively. (305-307)
Wu, et al, in their research suggest aflibercept as another potential VEGF pathway-targeted antiangiogenic agent in combination with neoadjuvant-plus-adjuvant chemotherapy regimens in triple-negative breast cancer (TNBC). (308)
Natural Inhibitor of Angiogenesis
Neovastat. Neovastat is an extract from shark cartilage that inhibits angiogenesis by selectively competing for the binding of VEGF and its receptor, inhibits matrix metalloproteinases, incites tissue plasminogen activator enzymatic activities, and induces apoptotic activity in ECs. (309) Neovastat was found to have significant antitumor activity, (310,311) and in vivo experiments revealed that it inhibits the formation of blood vessels in Matrigel implants containing basic fibroblast growth factor (bFGF). (312)
Anti-Angiogenesis Therapies in Bca: Past, Current, Future Explorations
The reasoning underlying antiangiogenic research and motivation as an active area of investigation was the idea that inhibiting the formation of a blood vessel in tumors would induce tumor dormancy or regression by depriving them of necessary oxygen and nutrients. Although clinical results provided sufficient evidence that anti-angiogenic treatment is a valid therapeutic strategy, the full potential of this approach seems to have been inadequate. The use of these agents (discussed above and several others) in combination with standard protocols or neoadjuvant therapy targeting angiogenesis -driven tumor growth have shown average clinical results.
One of the earliest preclinical research revealed that treatment with an anti-VEGF monoclonal antibody yielded a significant decrease in vascular density and tumor growth delay in mice. (313) However, these clinical trials did not translate into the same expected results in solid human tumors-reporting just moderate response rates and little survival gains for patients. (313)
Following several years of clinical research, anti-VEGF agents seem to be effective as single agents only in a few types of cancers, while effective in others, such as breast cancer, when combined with chemotherapy. (313,314)
So far, studies have reported that a combination of antiangiogenetic compounds with standard chemotherapy treatments in metastatic breast cancer has produced a limited clinical impact on overall survival in several forms of cancer, including breast cancer.
Few years after its approval, several controversies arose regarding the use of bevacizumab in the treatment of metastatic breast cancer. Although the more than 2000 trials revealed that combining bevacizumab with paclitaxel improved PFS, as well as overall survival rates compared to paclitaxel alone, it revealed a range of side effects including bleeding and blood clots, abnormal excreted protein levels, and high blood pressure. (31,314) However, after the FDA eventually withdrew the approval of bevacizumab for metastatic HER2 negative breast cancer, (32,315) the EMA did not follow suit immediately because of the increased PFS the drug in combination with capecitabine or paclitaxel showed. (316)
Enormous challenges persist in this area of research, in the ensuing clinical trials, and eventual treatment. The challenges that surround developing antiangiogenic agents and determining their efficacy, however, are not unique to breast cancer treatment. If the goal is to optimize the possibility of these agents ultimately making an impact in the treatment of breast cancer, then researchers must anticipate and address the potential obstacles. Several factors may explain the low efficacies observed so far, including toxicity, drug resistance, alternative mechanisms of angiogenesis, and the challenge of identifying the right patient who will benefit from anti-VEGF therapy in breast cancer. Some of these challenges and possible strategies to overcome are discussed next.
Toxicity. Since angiogenesis is not a very common process in adults, VEGF-targeted treatments were expected to be toxicity free. Several clinical trials, however, revealed that anti-angiogenic therapy had numerous adverse side effects. (202) While some of these can be appropriately managed with proper care, increasing levels of toxicity could lead to an intermittent treatment regimen, reduction in dosage, or treatment cessation, ultimately or possibly minimizing the efficiency of the treatment followed by rapid regrowth of the tumor. However, several studies have proposed using some of these side effects as a predictive biomarker for treatment efficiency, due to observation such as one reported between hypertension and longer PFS and/or OS in patients undergoing anti-angiogenic treatment. (317-320)
Resistance. Resistance to antiangiogenic treatment is a critical issue. Since normal endothelial cells are genetically stable, it was presumed initially that antiangiogenic treatment would not face the problem of resistance. (321) However, some forms of cancer have been reported to manifest both evasive and intrinsic resistance to anti-angiogenic therapies. (206,v322) This problem is one of the contributing factors to the low efficiency of the anti-VEGF therapy in breast cancer and understanding the mechanisms behind this process could enhance the care and contribute to treatment.
Heterogeneity. Within the endothelium vessels involved in angiogenesis, significant heterogeneity and frequent mutations have been described. (323) This has presented several challenges for effective and proper treatment; for instance, different mechanisms may be involved in the vascularization within the same tumor at a specific time and between different tumors in the same patient, (324) which has been reported as responsible for the variation in drug response/ drug metabolism or transport. (323)
This heterogeneity has been described in invasive cancers, which also commonly express multiple angiogenic factors. Several different pro-angiogenic proteins have been identified in primary breast tumors. (99) and the heterogeneity may also be a major contributing factor to drug resistance and low efficacy of the anti-angiogenic therapy in breast cancer and all other cancer therapeutics. (325,326)
Biomarkers. A biomarker can be described as "a characteristic objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention." (327,328) Due to the complexity in the mechanisms in determining response and resistance to anti-angiogenic drugs, it has been difficult to come up with predictive biomarkers for this class of agent. Several pro-angiogenic proteins mentioned above, including VEGF, has been speculated to be used indirectly as biomarkers to measure angiogenic activity in breast cancers. One of the early biomarkers to be examined was the plasma concentration of VEGF-A, (329) and higher levels of some of these proteins have been found in the serum and urine of cancer patients. (330)
Therefore, plasma levels of VEGF-A or VEGFR-2 continue to serve as promising biomarkers. (331-333) Some of the ways biomarkers can be used include for prognosis (for assessing the overall disease outcome), (334) predictive (to provide data about the response or survival of a certain patient under a specific treatment before therapy), (335) screening, and diagnostic.
Furthermore, the increase in the discovery of more biomarkers uncovers an equally important challenge of standardizing procedures for assessing the biomarkers across different centers for validation and routine adoption. (202) Consequently, the probability appears low that a single biomarker would suffice to effectively predict the efficiency of anti-angiogenic agents, particularly in cases with multiple metastases, where challenges posed by the tumor heterogeneity will complicate the attempt. (202) Extensive studies conducted is yet to produce a biomarker for the efficacy of bevacizumab in different patients, which may explain in part its inability to produce significant benefits clinically. (336,337)
Ongoing research into factors such as levels of pretreatment serum VEGF is attracting some attention as having a prognostic role. (31, 338, 339) Patients that had higher than normal levels at baseline were reported to have a poorer prognosis and an increased treatment effect of bevacizumab when compared with patients with low levels. (31,338) The proposal that VEGF level, in serum or tumor biopsies, could fulfill the requirements of a predictive biomarker has so far remained inconclusive. (340,341)
Although circulating VEGF levels in cancer patients revealed the importance of VEGF, not as a predictive biomarker, but a prognostic biomarker. (202) Additionally, as mentioned above, some of the adverse side effects related to antiangiogenic drugs seems to be positively linked with response to treatment. For instance, hypertension associated with the bevacizumab or TKIs has been reported to be correlated with clinical response in patients with breast and some other form of cancer. (342)
The use of biomarkers offers some hope as a possible solution to the problem of indiscriminate anti-angiogenic therapy, by providing physicians a better way of selecting patients with the most significant possibility for a positive response to a treatment, which will also serve to address the problem of the overall high variability in the response of patient to anti-angiogenic drugs and the quick development of resistance by predicting patients' response to VEGF-inhibition and help individualize drug and determine dosage. (78) The search for such biomarkers is still ongoing, and numerous prospective candidates have been considered.
Alternative approaches
Alternative strategies have been proposed in targeting vascularity of tumors. One such idea is to improve the delivery of chemotherapy by designing treatment that normalizes the tumor vasculature explicitly. (343-345) Preclinical and clinical studies have revealed that there is quick regrowth of the tumor due to rapid revascularization when antiangiogenic therapy is stopped abruptly. (346-348)
An argument has been put forward that to develop a successful anti-angiogenic therapy in the future; a better appreciation will be needed of how different tumors attain vascularization and avoid the action of anti-angiogenic therapy. This could lead to the development of new anti-angiogenic strategies directed towards specific cancer. (202)
However, the precise approach of how this would be effectively done in different cancers to suppress the tumor, prevent resistance, and prolong the survival of the patient is still being determined. (202)
Conclusion and Future Perspective
The increased angiogenesis, a measure of levels of VEGF or microvessel density, is an independent adverse prognostic factor in early breast cancer. (33) Tumors that are starved of blood supply stop growing and become necrotic, but if the connection to systemic circulation is established and sustained, it leads to hematogenous metastatic spread cancer. (8,33) Therefore, there is a practical justification for the use of anti-VEGF (specifically) and antiangiogenic therapies (in general) in primary, locally advanced and metastatic breast cancer.
Anti-VEGF treatment in metastatic breast cancer came with high hopes and excitement initially. One of the prominent results of the research was bevacizumab, which was received FDA approval based on very promising clinical trials. However, conflicting subsequent results and reports on adverse side effect led to the FDA and EMA revoking this approval. (349)
The problem of drug resistance in angiogenesis treatments of many cancer types has been a persistent challenge, and several proposals have been offered including dugs or strategies targeting microenvironment tumor heterogeneity and microvascular heterogeneity. (350) Targeting the multifaceted nature of the disease, would require as many paths as can be unraveled in the developmental stage. However, several agents that multi target this disease has not revealed much effectiveness. (351) Some scientists have suggested that nanotechnology provide the best prospects of discovering agents that will enhance the treatment of these cancer types, and Nanoparticles (particles less than 100 nm) is expected to achieve the positive results that anti-angiogenic agents have struggled to attain, and to serve as strategy to reduce the challenge of drug resistance (352-353)
Although most of the antiangiogenic drugs have not been very effective against breast cancer, the research leading to their discovery and subsequent clinical trials have provided a good foundation and direction for identifying prospective drugs, drug targets, and a better understanding certain mechanisms involved in the impairment of tumor angiogenesis, growth, and migration.
So, currently, the best strategy may be to take advantage of what we understand from the activities of the angiogenic agents in the early clinical studies and redirect our efforts from designing comprehensive treatments for a population, to targeted therapies for specific patients or groups of patients. The excitement observed some years ago may appear to be on a downward slope, but some exciting discoveries, however, still get often published that keeps a cautious optimism alive.
Recently, immune checkpoint inhibitors (ICIs) have been increasingly studied alongside angiogenesis. Combination treatment with these two approaches have promising results in a wide variety of cancer types, including hepatocellular carcinoma, NSCLC, and melanoma [354-356]. ICIs are molecules that are able to activate anti-tumor responses by blocking negative regulatory immune signals [357]. Some common molecules include programmed cell death protein 1 (PD-1) and cytotoxic T-lymphocyte antigen-4 (CTLA-4) [354]. Pembrolizumab is an inhibitory antibody that targets PD-1, and the FDA has approved its use in 2017 as the first drug to be categorized not by cancer type, otherwise known as 'histology agnostic' [358]. Another drug that targets immune checkpoint inhibitors and their signaling pathway is nivolumab, which has also been approved by the FDA [356].
Immune checkpoint molecules serve an integral role in downregulating the magnitude of immune responses [357]. Upregulation in immune checkpoint signaling pathways like the PD-1/PD-L1 pathway can shield cancer cells for immune detection. Thus, immune checkpoint molecules are ideal targets for anti-cancer treatment. The PD-1 pathway is normally a T-cell inhibitory pathway, which is induced by binding of the PD-1 receptor on the cytotoxic T-cell plasma membrane to PD-L1 on the tumor [356]. The anti-PD-1/PD-L1 therapy enhances cytotoxic ability by restoring T cells from exhausted status [357].
There have been numerous studies done on the synergistic effect of anti-angiogenesis and ICI combination treatment in colon adenocarcinoma, kidney tumors, and mammary tumors in mice [357]. Several mechanisms have been found that relate to synergistic effect. VEGF can prevent lymphocytes from mobilizing via its effect on the Fas ligand [356]. Using a combination treatment of anti-VEGF and anti-PD-L1 in mice, a high proportion of exhausted T cells can be reversed [357]. In another study, bevacizumab (anti-VEGF) and atezolizumab (anti-PD-L1) were used in a renal cell carcinoma model [354]. This combination treatment increased the expression of MHCI in tumor cells and effector T cells, resulting in increased antitumor effects [354].
A strategy must be developed to select those patients that would benefit more from one particular antiangiogenic agent and determine when would be the most effective window of opportunity they are likely going benefit from the treatment. In this regard, although several scientists believe that more efficient drugs or treatment regimens designed to combat breast cancer angiogenesis may be further in the future, it is necessary to pursue the discoveries of potential new drugs and to determine the possibility finding new functions for previous ones.
Acknowledgements
We thank Sara Liu and Pamela Madu for reviewing the manuscript. We also thank Ava Wang for drawing some figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. American Cancer Society. https://www.cancer.org/research/cancer-facts-statistics/breast-cancer-facts-figures.html
2. DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J. 2014;64:52-62
3. American Cancer Society. Breast Cancer Facts & Figures 2015-2016. Atlanta: American Cancer Society, Inc. 2015
4. Castañeda-Gill JM, Vishwanatha JK. Antiangiogenic mechanisms and factors in breast cancer treatment. J Carcinogenesis. 2016;15:1
5. Coelho AL, Gomes MP, Catarino RJ. et al. Angiogenesis in NSCLC: is vessel co-option the trunk that sustains the branches? Oncotarget. 2017;8:39795-804
6. Sim EK, Zhang L, Shim WS, Lim YL, Ge R. Therapeutic angiogenesis for coronary artery disease. J Card Surg. 2002;17:350-4
7. Risau W, Flamme I. "Vasculogenesis". Annual Review Cell Devel Biol. 1995;11:73-91
8. Hanahan D, Weinberg RA. Weinberg, Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
9. Folkman J, Browder T, Palmblad J. Angiogenesis research: guidelines for translation to clinical application. Thromb Haemost. 2001;86:23-33
10. Tannock IF. The relation between cell proliferation and the vascular system in a transplanted mouse mammary tumour. British J Cancer. 1968;22:258-73
11. Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932-6
12. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27-31
13. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353-64
14. Grimm D., Wehland M., Pietsch J.. et al. Drugs interfering with apoptosis in breast cancer. Curr. Pharm. Des. 2011;17:272-83
15. Grimm D., Bauer J., Schönberger J. Blockade of neoangiogenesis, a new and promising technique to control the growth of malignant tumors and its metastases. Curr. Vasc. Pharmacol. 2009;7:347-57
16. Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931-4
17. Folkman J. Tumour angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182-6
18. Fong G, Rossant J, Gartsenstein M. et al. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:67-70
19. Shalably F, Rossant J, Yamaguchi TP. et al. Failure of blood island formation and vasculogenesis in FLK-1 deficient mice. Nature. 1995;376:62-6
20. Horak ER, Klenk N, Leek R. et al. Angiogenesis, assessed by platelet/EC adhesion molecule antibodies, as indicator of node metastases and survival in breast cancer. Lancet. 1992;340:1120-4
21. Vartanian RK, Weidner N. Correlation of intratumoral EC proliferation with microvessel density (tumor angiogenesis) and tumor cell proliferation in breast carcinoma. Am J Pathol. 1994;144:1188-94
22. Weidner N, Semple JP, Welch WR. et al. Tumor angiogenesis and metastasis-correlation in invasive breast carcinoma. N Engl J Med. 1991;324:1-8
23. Linderholm B, Tavelin B, Grankvist K. et al. Does vascular endothelial growth factor (VEGF) predict local relapse and survival in radiotherapytreated node-negative breast cancer? Br J Cancer. 1999;81:727-32
24. George ML, Tutton MG, Janssen F. et al. VEGF-A, VEGF-C, and VEGF-D in colorectal cancer progression. Neoplasia. 2001;3:420-7
25. Holmgren L, O'Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149-53
26. Linderholm BK, Hellborg H, Johansson U. et al. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann Oncol. 2009;20:1639-46
27. FDA Approval for Bevacizumab n.d. 2015, Sep 16. http://www.cancer.gov/cancertopics/druginfo/fda-bevacizumab
28. Tarallo V, De Falco S. The vascular endothelial growth factors and receptors family: up to now the only target for anti-angiogenesis therapy. Int. J. Biochem. Cell Biol. 2015;64:185-9
29. Bellou S, Pentheroudakis G, Murphy C. et al. Anti-angiogenesis in cancer therapy: Hercules and Hydra. Cancer Lett. 2013;338:219-28
30. Ivy SP, Wick JY, Kaufman BM. An overview of small-molecule inhibitors of VEGFR signaling. Nat Rev Clin Oncol. 2009;6:569-79
31. Miles DW, Chan A, Dirix LY. et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2010;28:3239-47
32. Robert NJ, Dieras V, Glaspy J. et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29:1252-60
33. Fidler I, Ellis L. The implications of angiogenesis for the biology and therapy of cancer metastasis. Cell. 1994;79:185-188
34. Papetti M, Herman IM. Mechanisms of normal and tumor-derived angiogenesis. Am J Physiol Cell Physiol. 2002;282:947-70
35. Liekens S, De Clercq E, Neyts J. Angiogenesis: regulators and clinical applications. Biochem Pharmacol. 2001Feb1;61:253-70
36. Maj E, Papiernik D, Wietrzyk J. Antiangiogenic cancer treatment: The great discovery and greater complexity (Review). International Journal of Oncology. 2016;49:1773-84
37. Singh R.K, Gutman M, Bucana C.D, et el. Sequential development of an angiogenic phenotype by human fibroblasts progressing to tumorigenicity. Proc Natl Acad Sci. USA. 1995;92:4562-6
38. Kandel J, Bossy-Wetzel E, Radvanyi F. et al. Neovascularization is associated with a switch to the export of bFGF in the multistep development of fibrosarcoma. Cell. 1991;66:1095-104
39. Folkman J, Hanahan D. Switch to the angiogenic phenotype during tumorigenesis. Princess Takamatsu Symp. 1991;22:339-47
40. Good DJ, Polverini PJ, Rastinejad F. et al. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci. USA. 1990;87:6624-8
41. Rastinejad F, Polverini PJ, Bouck NP. Regulation of the activity of a new inhibitor of angiogenesis by a cancer suppressor gene. Cell. 1989;56:345-55
42. Tang N, Wang L, Esko J. et al. Loss of HIF-1alpha in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell. 2004;6:485-95
43. Harris AL. Hypoxia-A key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38-47
44. Semenza GL. HIF-1 and tumor progression: Pathophysiology and therapeutics. Trends Mol Med. 2002;8:S62-S67
45. Zhong H, De Marzo AM, Laughner E. et al. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res. 1999;59:5830-5
46. Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8:967-75
47. Mazure NM, Brahimi-Horn MC, Berta MA, Benizri E, Bilton RL, Dayan F. et al. HIF-1: master and commander of the hypoxic world. A pharmacological approach to its regulation by siRNAs. Biochem Pharmacol. 2004;68:971-80
48. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721-32
49. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298-307
50. Claesson-Welsh L, Welsh M J VEGFA, tumour angiogenesis. Intern Med. 2013; 273: 114-27.
51. Mendelsohn J, Howley P, Israel M, Liotta L. The molecular basis of cancer. Philadelphia: W. B. Saunders. 1995 p. 206-32
52. Senger DR, Galli SJ, Dvorak AM. et al. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983-5
53. Felmeden DC, Blann AD, Lip GYH. Angiogenesis: basic pathophysiology and implications for disease. European Heart Journal. 2003;24:586-603
54. Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306-9
55. Keck PJ, Hauser SD, Krivi G. et al. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309-12
56. Connolly DT, Heuvelman DM, Nelson R. et al. Tumor vascular permeability factor stimulates endothelial cell growth and angiogenesis. J Clin Invest. 1989;84:1470-8
57. Tischer E, Gospodarowicz D, Mitchell R. et al. Vascular endothelial growth factor: a new member of the platelet derived growth factor gene family. Biochem Biophys Res. 1989;165:1198-206
58. Ferrara N, Houck K, Jakeman L. et al. Molecular and biological properties of vascular endothelial growth factor family of protein. Endocr Rev. 1992;13:18-32
59. Kristensen TB, Knutsson MLT, Wehland M. et al. Anti-Vascular Endothelial Growth Factor Therapy in Breast Cancer. International Journal of Molecular Sciences. 2014;15:23024-41
60. Maxwell PH, Wiesener MS, Chang G-W. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271-5
61. Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4:1317-26
62. Li B, Leung DW. et al. The vascular endothelial growth factor family: identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol Endocr. 1991;5:1806-14
63. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669-76
64. Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling—In control of vascular function. Nat. Rev. Mol. Cell Biol. 2006;7:359-71
65. Bottaro DP, Liotta LA. Cancer: out of air is not out of action. Nature. 2003;423:593-5
66. Zhao Y, Adjei AA. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. The Oncologist. 2015;20:660-73
67. Koch S, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb Perspect Med. 2012 2 (7)
68. Pepper MS, Ferrara N, Orci L, Montesano R. Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem Biophys Res Commun. 1992;189:824-31
69. Korpanty G, Smyth E, Carney DN. Update on anti-angiogenic therapy in non-small cell lung cancer: Are we making progress? J. Thorac. Dis. 2011;3:19-29
70. Heusschen R, van Gink M, Griffioen AW, Thijssen VL. MicroRNAs in the tumor endothelium: novel controls on the angioregulatory switchboard. Biochim Biophys Acta. 2010;1805:87-96
71. Wahl ML, Moser TL, Pizzo SV. Angiostatin and anti-angiogenic therapy in human disease. Recent Prog Horm Res. 2004;59:73-104
72. Lee SH, Jeung IC, Park TW. et al. Extension of the in vivo half-life of endostatin and its improved anti-tumor activities upon fusion to a humanized antibody against tumor-associated glycoprotein 72 in a mouse model of human colorectal carcinoma. Oncotarget. 2015;6:7182-94
73. Caporali A, Emanueli C. MicroRNA regulation in angiogenesis. Vascul Pharmacol. 2011;55:79-86
74. Butler TP, Gullino PM. Quantitation of cell shedding into efferent blood of mammary adenocarcinoma. Cancer Res. 1975;35:512-6
75. Bouck N, Stellmach V, Hsu S. How tumors become angiogenic. Adv. Cancer Res. 1996;69:135-74
76. Folkman J. Tumor angiogenesis. In The Molecular Basis of Cancer. 1995
77. Folkman J. Clinical applications of research on angiogenesis. N. Engl. J. Med. 1995;333:1757-63
78. Wehland M, Bauer J, Magnusson NE. et al. Biomarkers for anti-angiogenic therapy in cancer. Int J Mol Sci. 2013;14:9338-64
79. Leite de Oliveira R, Hamm A, Mazzone M. Growing tumor vessels: More than one way to skin a cat—Implications for angiogenesis targeted cancer therapies. Mol. Aspects Med. 2011;32:71-87
80. Veikkola T, Alitalo K. Semin. Growth factors. Fedi, P, Tronick, SR, and Aaronson, SA. VEGFs, receptors and angiogenesis. Cancer Biol. 1999;9:211-20
81. Tímár J, Döme B, Fazekas K, Janovics A, Paku S. Angiogenesis-dependent diseases and angiogenesis therapy. Pathol Oncol Res. 2001;7:85-94
82. Bertolini F, Petit JY, Kolonin MG. Stem cells from adipose tissue and breast cancer: Hype, risks and hope. Br J Cancer. 2015;112:419-23
83. Christiaens V, Lijnen HR. Angiogenesis and development of adipose tissue. Mol Cell Endocrinol. 2010;318:2-9
84. Yoshida S, Ono M, Shono T, Izumi H, Ishibashi T, Suzuki H. et al. Involvement of interleukin-8, vascular endothelial growth factor, and basic fibroblast growth factor in tumor necrosis factor alpha-dependent angiogenesis. Mol Cell Biol. 1997;17:4015-23
85. Miller JW, Adamis AP, Shima DT. et al. Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a primate model. American Journal of Pathology. 1994;145:574-84
86. Wang L, Lee AY, Wigg JP. et al. miRNA involvement in angiogenesis in age-related macular degeneration. J Physiol Biochem. 2016;72:583-92
87. Wang L, Lee AY, Wigg JP. et al. miR-126 regulation of angiogenesis in age-related macular degeneration in CNV mouse model. Int J Mol Sci. 2016
88. Ozcan Cenksoy P, Oktem M, Erdem O. et al. A potential novel treatment strategy: inhibition of angiogenesis and inflammation by resveratrol for regression of endometriosis in an experimental rat model. Gynecol Endocrinol. 2015;31:219-24
89. Soysal D, Kizildag S, Saatli B. et al. A novel angiogenesis inhibitor bevacizumab induces apoptosis in the rat endometriosis model. Balkan J Med Genet. 2014;17:73-80
90. Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003Jun;9:653-60
91. Li Y, Wu Z, Yuan J. et al. Long non-coding RNA MALAT1 promotes gastric cancer tumorigenicity and metastasis by regulating vasculogenic mimicry and angiogenesis. Cancer Lett. 2017;395:31-44
92. Shen J, Zhou S, Shi L. et al. DUSP1 inhibits cell proliferation, metastasis and invasion and angiogenesis in gallbladder cancer. Oncotarget. 2017;8:12133-44
93. Xu H, Zhang Y, Pena MM. et al. Six1 promotes colorectal cancer growth and metastasis by stimulating angiogenesis and recruiting tumor-associated macrophages. Carcinogenesis. 2017;38:281-92
94. Qi J.H. et al. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat. Med. 2003;9:407-15
95. Voelkel N.F. et al. Janus face of vascular endothelial growth factor: the obligatory survival factor for lung vascular endothelium controls precapillary artery remodeling in severe pulmonary hypertension. Crit. Care Med. 2002;30:251-6
96. Meyer M. et al. A novel vascular endothelial growth factor encoded by Orf virus, VEGF-E, mediates angiogenesis via signalling through VEGFR-2 (KDR) but not VEGFR-1 (Flt-1) receptor tyrosine kinases. EMBO J. 1999;18:363-74
97. Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, Saksela O, Kalkkinen N, Alitalo K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996;15:290-8
98. Relf M, LeJeune S, Scott PA. et al. Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res. 1997;57:963-9
99. Berse B, Brown LF, Van de Water L. et al. Vascular permeability factor (vascular endothelial growth factor) gene is expressed differentially in normal tissues, macrophages, and tumors. Mol Biol Cell. 1992;3:211-20
100. Lin Y, Huang R, Chen L. et al. Identification of interleukin-8 as estrogen receptor-regulated factor involved in breast cancer invasion and angiogenesis by protein arrays. Int J Cancer. 2004;109:507-15
101. Marjon PL, Bobrovnikova-Marjon EV, Abcouwer SF. Expression of the pro-angiogenic factors vascular endothelial growth factor and interleukin-8/CXCL8 by human breast carcinomas is responsive to nutrient deprivation and endoplasmic reticulum stress. Mol Cancer. 2004;3:4
102. Razmkhah M, Jaberipour M, Hosseini A. et al. Expression profile of IL-8 and growth factors in breast cancer cells and adipose-derived stem cells (ASCs) isolated from breast carcinoma. Cell Immunol. 2010;265:80-5
103. Chelouche-Lev D, Miller CP, Tellez C. et al. Different signaling pathways regulate VEGF and IL-8 expression in breast cancer: Implications for therapy. Eur J Cancer. 2004;40:2509-18
104. Valtola R, Salven P, Heikkilä P. et al. VEGFR-3 and its ligand VEGF-C are associated with angiogenesis in breast cancer. Am J Pathol. 1999;154:1381-90
105. Srabovic N, Mujagic Z, Mujanovic-Mustedanagic J. et al. Vascular endothelial growth factor receptor-1 expression in breast cancer and its correlation to vascular endothelial growth factor A. Int J Breast Cancer. 2013;2013:746-9
106. Guo S, Colbert LS, Fuller M. et al. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim Biophys Acta. 2010;1806:108-21
107. Lushnikova AA, Nasunova IB, Parokonnaya AA. et al. VEGFR-2 expression in tumor tissue of breast cancer patients. Dokl Biol Sci. 2010;434:363-7
108. Linardou H, Kalogeras KT, Kronenwett R. et al. The prognostic and predictive value of mRNA expression of vascular endothelial growth factor family members in breast cancer: A study in primary tumors of high-rish early breast cancer patients participating in a randomized Hellenic Cooperative Oncology Group trial. Breast Cancer Res. 2012;14:R145
109. Fidler IJ, Ellis LM. Chemotherapeutic drugs - More really is not better. Nat Med. 2000;6:500-2
110. Li A, Dubey S, Varney ML. et al. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol. 2003;170:3369-76
111. Brocker C, Thompson D, Matsumoto A. et al. Evolutionary divergence and functions of the human interleukin (IL) gene family. Human Genomics. 2010;5:30-55
112. Hedges JC, Singer CA, Gerthoffer WT. Mitogen-activated protein kinases regulate cytokine gene expression in human airway myocytes. Am J Respi. Cell Mol Biol. 2000;23:86-94
113. Wolff B, Burns AR, Middleton J, Rot A. Endothelial cell "memory" of inflammatory stimulation: human venular endothelial cells store interleukin 8 in Weibel-Palade bodies. J Exp Med. 1998;188:1757-62
114. Koch AE, Polverini PJ, Kunkel SL. et al. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science. 1992;258:1798-801
115. Norgauer J, Metzner B, Schraufstatter I. Expression and growth-promoting function of the IL-8 receptor beta in human melanoma cells. J Immunol. 1996;156:1132-7
116. Martin D. Galisteo R. Gutkind JS. CXCL8/IL8 stimulates vascular endothelial growth factor (VEGF) expression and the autocrine activation of VEGFR2 in endothelial cells by activating NFkappaB through the CBM (Carma3/Bcl10/Malt1complex). J Biol Chem. 2009;284:6038-42
117. Bar-Eli M. Role of interleukin-8 in tumor growth and metastasis of human melanoma. Pathobiology. 1999;67:12-8
118. Akdis M, Burgler S, Crameri R. et al. Interleukins, from 1 to 37, and interferon-g: Receptors, functions, and roles in diseases. J Allergy Clin Immunol. 2011;127:701-70
119. Shahzad A. Knapp M. Lang I. Köhler G. Interleukin 8 (IL-8)—a universal biomarker? Int Arch Med. 2010;3:11
120. Slattery ML, John EM, Stern MC. et al. Associations with growth factor genes (FGF1, FGF2, PDGFB, FGFR2, NRG2, EGF, ERBB2) with breast cancer risk and survival: The Breast Cancer Health Disparities Study. Breast Cancer Res Treat. 2013;140:587-601
121. Jianwu F, Siluo H, Huisheng L. Role of FGF-2/FGFR signaling pathway in cancer and its signification in breast cancer. Chin Sci Bull. 2003;48:1539-47
122. Sørensen V, Nilsen T, Wiedlocha A. Functional diversity of FGF-2 isoforms by intracellular sorting. Bioessays. 2006;28:504-14
123. Vlodavsky I, Korner G, Ishai-Michaeli R. et al. Extracellular matrix-resident growth factors and enzymes: Possible involvement in tumor metastasis and angiogenesis. Cancer Metastasis Rev. 1990;9:203-26
124. Yiangou C, Gomm JJ, Coope RC. et al. Fibroblast growth factor 2 in breast cancer: Occurrence and prognostic significance. Br J Cancer. 1997;75:28-33
125. Jain VK, Turner NC. Challenges and opportunities in the targeting of fibroblast growth factor receptors in breast cancer. Breast Cancer Res. 2012;14:208
126. Brady N, Chuntova P, Bade LK, Schwertfeger KL. The FGF/FGFR axis as a therapeutic target in breast cancer. Expert Rev Endocrinol Metab. 2013;8:391-402
127. Woessner JF Jr The family of matrix metalloproteinases. Ann N Y Acad Sci. 1994; 732: 11-21.
128. Heppner KJ, Matrisian LM, Jensen RA, Rodgers WH. Expression of most matrix metalloproteinase family members in breast cancer represents a tumor-induced host response. Am J Pathol. 1996;149:273-82
129. Bartsch JE, Staren ED, Appert HE. Matrix metalloproteinase expression in breast cancer. J Surg Res. 2003;110:383-92
130. Nishida N, Yano H, Nishida T. et al. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2:213-9
131. Gasparini G, Harris AL. Clinical importance of the determination of tumor angiogenesis in breast carcinoma: Much more than a new prognostic tool. J Clin Oncol. 1995;13:765-82
132. Miller KD. Recent translational research: Antiangiogenic therapy for breast cancer - Where do we stand? Breast Cancer Res. 2004;6:128-32
133. Samant RS, Shevde LA. Recent advances in anti-angiogenic therapy of cancer. Oncotarget. 2011;2:122-34
134. Fakhrejahani E, Toi M. Antiangiogenesis therapy for breast cancer: An update and perspectives from clinical trials. Jpn J Clin Oncol. 2014;44:197-207
135. Folkman J, Bach M, Rowe JW. et al. Tumor angiogenesis: Therapeutic implications. N Engl J Med. 1971;285:1182-6
136. Folkman J. "Endogenous angiogenesis inhibitors". Apmis. 2004;112:496-507
137. Cao Y. Endogenous angiogenesis inhibitors and their therapeutic implications. Int J Biochem Cell Biol. 2001;33:357-69
138. Nyberg P, Xie L, Kalluri R. Endogenous inhibitors of angiogenesis. Cancer Research. 2005;65:3967-79
139. Phillips DR, Jennings LK, Prasanna HR. Ca2+-mediated association of glycoprotein G (thrombinsensitive protein, thrombospondin) with human platelets. J Biol Chem. 1980;255:11629-32
140. Ylikarppa R, Eklund L, Sormunen R. et al. Double knockout mice reveal a lack of major functional compensation between collagens XV and XVIII. Matrix Biol. 2003;22:443-8
141. Chen H, Herndon ME, Lawler J. The cell biology of thrombospondin-1. Matrix Biol. 2000;19:597-614
142. Iruela-Arispe ML, Luque A, Lee N. Thrombospondin modules and angiogenesis. Int J Biochem Cell Biol. 2004;36:1070-8
143. Agah A, Kyriakides TR, Lawler J, Bornstein P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am J Pathol. 2002;161:831-9
144. Streit M, Velasco P, Brown LF. et al. Overexpression of thrombospondin-1 decreases angiogenesis and inhibits the growth of human cutaneous squamous cell carcinomas. Am J Pathol. 1999;155:441-52
145. Rodriguez-Manzaneque JC, Lane TF, Ortega MA. et al. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc Natl Acad Sci. 2001;98:12485-90
146. Stellmach V, Volpert OV, Crawford SE. et al. Tumour suppressor genes and angiogenesis: the role of TP53 in fibroblasts. Eur J Cancer. 1996;32A:2394-400
147. Wong SY, Purdie AT, Han P. Thrombospondin and other possible related matrix proteins in malignant and benign breast disease. An immunohistochemical study. Am J Pathol. 1992;140:1473-82
148. Zabrenetzky V, Harris CC, Steeg PS, Roberts DD. Expression of the extracellular matrix molecule thrombospondin inversely correlates with malignant progression in melanoma, lung and breast carcinoma cell lines. Int J Cancer. 1994;59:191-5
149. Volpert OV, Stellmach V, Bouck N. The modulation of thrombospondin and other naturally occurring inhibitors of angiogenesis during tumor progression. Breast Cancer Res Treat. 1995;36:119-26
150. Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002;6:1-12
151. Rice A, Quinn CM. Angiogenesis, thrombospondin, and ductal carcinoma in situ of the breast. J Clin Pathol. 2002;55:569-74
152. Volpert OV, Tolsma SS, Pellerin S. et al. Inhibition of angiogenesis by thrombospondin-2. Biochem Biophys Res Commun. 1995;217:326-32
153. Streit M, Riccardi L, Velasco P. et al. Thrombospondin-2: A potent endogenous inhibitor of tumor growth and angiogenesis. Proc Natl Acad Sci. 1999;96:14888-93
154. Noh YH, Matsuda K, Hong YK. et al. An N-terminal 80 kDa recombinant fragment of human thrombospondin-2 inhibits vascular endothelial growth factor induced endothelial cell migration in vitro and tumor growth and angiogenesis in vivo. J Invest Dermatol. 2003;121:1536-43
155. O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS. et al. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277-85
156. Dhanabal M, Ramchandran R, Waterman MJ. et al. Endostatin induces endothelial cell apoptosis. J Biol Chem. 1999;274:11721-6
157. Scappaticci FA, Smith R, Pathak A. et al. Combination angiostatin and endostatin gene transfer induces synergistic antiangiogenic activity in vitro and antitumor efficacy in leukemia and solid tumors in mice. Mol Ther. 2001;3:186-96
158. Rehn M, Veikkola T, Kukk-Valdre E. et al. Interaction of endostatin with integrins implicated in angiogenesis. Proc Natl Acad Sci. 2001;98:1024-9
159. Marneros AG, Olsen BR. The role of collagen-derived proteolytic fragments in angiogenesis. Matrix Biol. 2001;20:337-45
160. Boehm T, Folkman J, Browder T, O'Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404-7
161. Yin G, Liu W, An P. et al. Endostatin gene transfer inhibits joint angiogenesis and pannus formation in inflammatory arthritis. Mol Ther. 2002;5:547-54
162. Dixelius J, Cross M, Matsumoto T. et al. Endostatin regulates endothelial cell adhesion and cytoskeletal organization. Cancer Res. 2002;62:1944-7
163. Roskoski R Jr Vascular endothelial growth factor (VEGF) signaling in tumor progression. Crit Rev Oncol Hematol. 2007; 62: 179-213.
164. Longatto Filho A, Lopes JM, Schmitt FC. Angiogenesis and breast cancer. J Oncol. 2010;2010:1-7
165. Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial targe t for anti- and pro-angiogenic therapies. Genes Cancer. 2011;2:1097-105
166. Indraccolo S, Gola E, Rosato A. et al. Differential effects of angiostatin, endostatin and interferon-alpha (1) gene transfer on in vivo growth of human breast cancer cells. Gene Ther. 2002;9:867-78
167. Parkin J, Cohen B. An overview of the immune system. Lancet. 2001;357:1777-89
168. Lingen MW, Polverini PJ, Bouck NP. Retinoic acid and interferon α act synergistically as antiangiogenic and antitumor agents against human head and neck squamous cell carcinoma. Cancer Res. 1998;58:5551-8
169. Mitsuyasu RT. Interferon α in the treatment of AIDS-related Kaposi's sarcoma. Br J Haematol. 1991;79:69-73
170. Singh RK, Gutman M, Bucana CD, Sanchez R, Llansa N, Fidler IJ. Interferons alpha and beta down-regulate the expression of basic fibroblast growth factor in human carcinomas. Proc Natl Acad Sci. 1995;92:4562-6
171. Albini A, Marchisone C, Del Grosso F. et al. Inhibition of angiogenesis and vascular tumor growth by interferon-producing cells: A gene therapy approach. Am J Pathol. 2000;156:1381-93
172. Ma Z, Qin H, Benveniste EN. Transcriptional suppression of matrix metalloproteinase-9 gene expression by IFN-γ and IFN-β: critical role of STAT-1α. J Immunol. 2001;167:5150-9
173. Dinney CP, Bielenberg DR, Perrotte P. et al. Inhibition of basic fibroblast growth factor expression, angiogenesis, and growth of human bladder carcinoma in mice by systemic interferon-α administration. Cancer Res. 1998;58:808-14
174. Ribatti D, Crivellato E, Candussio L. et al. Angiogenic activity of rat mast cells in the chick embryo chorioallantoic membrane is down-regulated by treatment with recombinant human α-2a interferon and partly mediated by fibroblast growth factor-2. Haematologica. 2002;87:465-71
175. Lopez-Ocejo O, Perea SE, Bequet-Romero M. et al. Impaired angiogenic balance and suppression of tumorigenicity in HeLa cells chronically exposed to interferon-α. Biochem Biophys Res Commun. 2000;277:410-6
176. Nilsson A, Janson ET, Eriksson B, Larsson A. Levels of angiogenic peptides in sera from patients with carcinoid tumours during α-interferon treatment. Anticancer Res. 2001;21:4087-90
177. Sasamura H, Takahashi A, Miyao N. et al. Inhibitory effect on expression of angiogenic factors by antiangiogenic agents in renal cell carcinoma. Br J Cancer. 2002;86:768-73
178. Wang L, Wu WZ, Sun HC. et al. Mechanism of interferon α on inhibition of metastasis and angiogenesis of hepatocellular carcinoma after curative resection in nude mice. J Gastrointest Surg. 2003;7:587-94
179. Paul W. E, Seder R. A. Lymphocyte responses and cytokines. Cell. 1994;76:241-51
180. Kishimoto T, Taga T, Akira S. Cytokine signal transduction. Cell. 1994;76:253-62
181. Cozzolino F, Torcia M, Aldinucci D. et al. Interleukin 1 is an autocrine regulator of human endothelial cell growth. Proc Natl Acad Sci. 1990;87:6487-91
182. Volpert OV, Fong T, Koch AE. et al. Inhibition of angiogenesis by interleukin 4. J Exp Med. 1998;188:1039-46
183. Voest EE, Kenyon BM, O'Reilly MS, Truitt G, D'Amato RJ, Folkman J. Inhibition of angiogenesis in vivo by interleukin 12. J Natl Cancer Inst. 1995;87:581-6
184. Sgadari C, Angiolillo AL, Tosato G. Inhibition of angiogenesis by interleukin-12 is mediated by the interferon-inducible protein 10. Blood. 1996;87:3877-82
185. Yao L, Sgadari C, Furuke K. et al. Contribution of natural killer cells to inhibition of angiogenesis by interleukin-12. Blood. 1999;93:1612-21
186. O'Reilly MS, Holmgren L, Shing Y. et al. Angiostatin: a novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell. 1994;79:315-28
187. Redlitz A, Daum G, Sage EH. Angiostatin diminishes activation of the mitogen-activated protein kinases ERK-1 and ERK-2 in human dermal microvascular endothelial cells. Journal of Vascular Research. 1999;36:28-34
188. Wajih N, Sane DC. Angiostatin selectively inhibits signaling by hepatocyte growth factor in endothelial and smooth muscle cells. Blood. 2003;101:1857-63
189. Harris AL, Generali D. Biological therapies for metastatic breast cancer: Antiangiogenesis. In: Piccart M, Wood WC, Hung MC, Solin LJ, Cardoso F, editors. Breast Cancer Management and Molecular Medicine: Towards Tailored Approaches. Berlin, Germany: Springer Science & Business Media. 2007:685-7
190. Iozzo RV, Moscatello DK, McQuillan DJ, Eichstetter I. Decorin is a biological ligand for the epidermal growth factor receptor. J Biol Chem. 1999;274:4489-92
191. Grant DS, Yenisey C, Rose RW. et al. Decorin suppresses tumor cell-mediated angiogenesis. Oncogene. 2002;21:4765-77
192. Reed CC, Iozzo RV. The role of decorin in collagen fibrillogenesis and skin homeostasis. Glycoconj J. 2002;19:249-55
193. Reed CC, Waterhouse A, Kirby S. et al. Decorin prevents metastatic spreading of breast cancer. Oncogene. 2005;24:1104-10
194. Sofeu Feugaing DD, Götte M, Viola M. More than matrix: The multifaceted role of decorin in cancer. Eur J Cell Biol. 2013;92:1-11
195. Järveläinen H, Sainio A, Wight TN. Pivotal role for decorin in angiogenesis. Matrix Biol. 2015;43:15-26
196. Neill T, Schaefer L, Iozzo R. V. Instructive roles of extracellular matrix on autophagy. The American Journal of Pathology. 2014;184(8):2146-53
197. Neill T, Jones HR, Crane-Smith Z. et al. Decorin induces rapid secretion of thrombospondin-1 in basal breast carcinoma cells via inhibition of Ras homolog gene family, member A/Rho-associated coiled-coil containing protein kinase 1. The FEBS Journal. 2013;280:2353-68
198. Neill T, Painter, Buraschi S. et al. Decorin antagonizes the angiogenic network: concurrent inhibition of met, hypoxia inducible factor 1α, vascular endothelial growth factor A, and induction of thrombospondin-1 and tiMP3. J Biological Chemistry. 2012;287:5492-506
199. Buraschi S, Neill T, Goyal A. et al. Decorin causes autophagy in endothelial cells via Peg3. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2582-E2591
200. Khan GA, Girish GV, Lala N. et al. Decorin is a novel VEGFR-2-binding antagonist for the human extravillous trophoblast. Mol Endocrinol. 2011;25:1431-43
201. Santra M, Reed CC, Iozzo RV. Decorin binds to a narrow region of the epidermal growth factor (EGF) receptor, partially overlapping but distinct from the EGF-binding epitope. J Biol Chem. 2002;277:35671-81
202. Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: Current progress, unresolved questions and future directions. Angiogenesis. 2014;17:471-494
203. Jain RK. Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell. 2014;26:605-22
204. Aparicio-Gallego G, Blanco M, Figueroa A. et al. New insights into molecular mechanisms of sunitinib-associated side effects. Mol Cancer Ther. 2011;10:2215-23
205. Gotink KJ, Verheul HMW. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. 2010;13:1-14
206. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer. 2008;8:592-603
207. Miller K.D, Chap L.I, Holmes F.A. et al. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J. Clin. Oncol. 2005;23:792-9
208. Mackey J, Gelmon K, Martin M. et al. TRIO-012: A multicenter, multinational, randomized, double-blind phase III study of IMC-1121B plus docetaxel vs. placebo plus docetaxel in previously untreated patients with HER2-negative, unresectable, locally recurrent or metastatic breast cancer. Clin. Breast Cancer. 2009;9:258-61
209. Wang Z, Dabrosin C, Yin X, Fuster MM, Arreola A, Rathmell WK, Generali D, Nagaraju GP, El-Rayes B, Ribatti D. et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin Cancer Biol. 2015;35(Suppl):224-43
210. National Cancer Institute. https://www.cancer.gov/about-cancer/treatment/types/immunotherapy/angiogenesis-inhibitors-fact-sheet
211. Kim KJ, Li B, Winer J. et al. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour. growth in vivo. Nature. 1993;362:841-4
212. Hurwitz H, Fehrenbacher L, Novotny W. et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335-42
213. Kowanetz M, Ferrara N. Vascular endothelial growth factor signaling pathways: therapeutic perspective. Clin Cancer Res. 2006Sep1;12:5018-22
214. FDA Approval for Bevacizumab n.d. Accessed September 16, 2015. http://www.cancer.gov/cancertopics/druginfo/fda-bevacizumab
215. Kabbinavar F, Hurwitz HI, Fehrenbacher L, Meropol NJ, Novotny WF, Lieberman G. et al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol. 2003;21:60-5
216. Sachdev JC, Jahanzeb M. Evolution of bevacizumab-based therapy in the management of breast cancer. Clin Breast Cancer. 2008;8:402-10
217. Torrisi R, Bagnardi V, Cardillo A. et al. Preoperative bevacizumab combined with letrozole and chemotherapy in locally advanced ER- and/or PgR-positive breast cancer: Clinical and biological activity. Br J Cancer. 2008;99:1564-71
218. Miller K, Wang M, Gralow J. et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666-76
219. EMA Approval for Bevacizumab. n.d
220. Ye W. The Complexity of Translating Anti-angiogenesis Therapy from Basic Science to the Clinic. Developmental Cell. 2016;37:114-25
221. NIH: National Cancer Institute. Angiogenesis Inhibitors. https://www.cancer.gov/about-cancer/treatment/types/immunotherapy/angiogenesis-inhibitors-fact-sheet
222. Presta LG, Chen H, O'Connor SJ, Chisholm V, Meng YG, Krummen L. et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593-9
223. Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391-400
224. Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005Jul29;333:328-35
225. Rini BI. Vascular endothelial growth factor-targeted therapy in renal cell carcinoma: current status and future directions. Clinical Cancer Research. 2007;13:1098-106
226. Johnson DH, Fehrenbacher L, Novotny WF. et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2004;22:2184-9
227. de Gramont A, Van Cutsem E. Investigating the potential of bevacizumab in other indications: Metastatic renal cell, non-small cell lung, pancreatic and breast cancer. Oncology. 2005;69(Suppl 3):46-56
228. Herbst RS, Johnson DH, Mininberg E. et al. Phase i/ii trial evaluating the anti-vascular endothelial growth factor monoclonal antibody bevacizumab in combination with the her-1/epidermal growth factor receptor tyrosine kinase inhibitor erlotinib for patients with recurrent non-small-cell lung cancer. J Clin Oncol. 2005;23:2544-55
229. Groot JF, Yung WK. Bevacizumab and irinotecan in the treatment of recurrent malignant gliomas. Cancer J. 2008;14:279-85
230. Blagosklonny MV. How avastin potentiates chemotherapeutic drugs: Action and reaction in antiangiogenic therapy. Cancer Biol Ther. 2005;4:1307-10
231. Gordon MS, Margolin K, Talpaz M. et al. Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J Clin Oncol. 2001;19:843-50
232. Giantonio B.J, Levy D.E, O'Dwyer P.J. et al. A phase II study of high-dose bevacizumab in combination with irinotecan, 5-fluorouracil, leucovorin, as initial therapy for advanced colorectal cancer: Results from the Eastern Cooperative Oncology Group study E2200. Ann. Oncol. 2006;17:1399-403
233. Zhou MY, Yu P, Qu XJ. et al. Phase III trials of standard chemotherapy with or without bevacizumab for ovarian cancer: A meta-analysis. PLoS ONE. 2013;8:e81858
234. Vincenzi B, Santini D, Rabitti C, Coppola R, Beomonte Zobel B, Trodella L, Tonini G. Cetuximab and irinotecan as third-line therapy in advanced colorectal cancer patients: A single centre phase ii trial. Br J Cancer. 2006;94:792-7
235. Gebbia V, Del Prete S, Borsellino N. et al. Efficacy and safety of cetuximab/irinotecan in chemotherapy-refractory metastatic colorectal adenocarcinomas: A clinical practice setting, multicenter experience. Clin Colorectal Cancer. 2006;5:422-8
236. Petit AM, Rak J, Hung MC. et al. Neutralizing antibodies against epidermal growth factor and erbb-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: Angiogenic implications for signal transduction therapy of solid tumors. Am J Pathol. 1997;151:1523-30
237. Larkins E, Scepura B, Blumenthal GM. et al. U.S. Food and Drug Administration approval summary: ramucirumab for the treatment of metastatic non-small cell lung cancer following disease progression on or after platinum-based chemotherapy. Oncologist. 2015;20:1320-5
238. Fuchs CS, Tomasek J, Yong CJ. et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): an international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. 2014;383:31-9
239. Tabernero J, Yoshino T, Cohn AL. et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015;16:499-508
240. Vahdat LT, Layman R, Yardley, DA. et al. Randomized Phase II Study of Ramucirumab or Icrucumab in Combination with Capecitabine in Patients with Previously Treated Locally Advanced or Metastatic Breast Cancer. The Oncologist. 2017;22:245-54
241. Gaudreault J, Fei D, Rusit J. et al. Preclinical pharmacokinetics of Ranibizumab (rhuFabV2) after a single intravitreal administration. Invest Ophthalmol Vis Sci. 2005;46:726-33
242. Bakri SJ, Snyder MR, Reid JM. et al. Pharmacokinetics of intravitreal ranibizumab (Lucentis). Ophthalmology. 2007;114(12):2179-82
243. Pawson T. Regulation and targets of receptor tyrosine kinases. Eur J Cancer. 2002;38(Suppl 5):3-10
244. Ribatti D. History of research on tumor angiogenesis. Springer; New York, NY. 2009
245. Goel S, Duda DG, Xu L. et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071-121
246. Amit Arora, Eric M. Scholar. Role of Tyrosine Kinase Inhibitors in Cancer Therapy. Journal of Pharmacology and Experimental Therapeutics. 2005;315(3):971-979
247. Rini B, Rixe O, Bukowski R. et al. AG-013736, a multi-target tyrosine kinase receptor inhibitor, demonstrates anti-tumor activity in a Phase 2 study of cytokine-refractory, metastatic renal cell cancer (RCC). J Clinical Oncol. 2005
248. Rugo HS, Herbst RS, Liu G. et al. Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: pharmacokinetic and clinical results. Journal of Clinical Oncology. 2005;23:5474-83
249. Choueiri TK. Axitinib, a novel anti-angiogenic drug with promising activity in various solid tumors. Curr Opin Investig Drugs. 2008;9:658-71
250. Rixe O, Bukowski RM, Michaelson MD. et al. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol. 2007;8:975-84
251. Escudier B, Gore M. Axitinib for the Management of Metastatic Renal Cell Carcinoma. Drugs in R&D. 2011;11:113-26
252. Rini B, Grünwald V, Fishman MN. et al. Axitinib for first-line metastatic renal cell cancer (mRCC): overall efficacy and pharmacokinetic (PK) analyses from a randomized phase II study. J Clin Oncol. 2012;30(Suppl):4503
253. Rini B, Escudier B, Tomczak P. et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomized phase 3 trial. Lancet. 2011;378:1931-9
254. Hutson T, Gallardo J, Lesovoy V. et al. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal cell carcinoma (mRCC). J Clin Oncol. 2013 31(Suppl) Abstr LBA348
255. Wilmes LJ, Pallavicini MG, Fleming LM. et al. AG-013736, a novel inhibitor of VEGF receptor tyrosine kinases, inhibits breast cancer growth and decreases vascular permeability as detected by dynamic contrast-enhanced magnetic resonance imaging. Magnetic Resonance Imaging. 2007;25:319-27
256. Rugo HS, Stopeck AT, Joy AA. et al. Randomized, placebo-controlled, double-blind, phase II study of axitinib plus docetaxel versus docetaxel plus placebo in patients with metastatic breast cancer. J Clin Oncol. 2011;29:2459-65
257. Wilhelm S, Carter C, Lynch M. et al. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5:835-44
258. Wehland M, Bauer J, Infanger M, Grimm D. Target-based anti-angiogenic therapy in breast cancer. Curr. Pharm. Des. 2012;18:4244-57
259. Kane RC, Farrell AT, Madabushi R. et al. Sorafenib for the treatment of unresectable hepatocellular carcinoma. Oncologist. 2009;14:95-100
260. Kane RC, Farrell AT, Saber H. et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin Cancer Res. 2006;12:7271-8
261. Escudier B, Eisen T, Stadler WM. et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol. 2009Jul10;27:3312-8
262. FDA Approval for Sorafenib Tosylate n.d
263. EMA Approval for Sorafenib n.d. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000690/human_med_000929.jsp&
264. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M. et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125-34
265. Llovet JM, Ricci S, Mazzaferro V. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Medl. 2008;359:378-90
266. Wilhelm SM, Adnane L, Newell P. et al. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008;7:3129-40
267. Isaacs C, Herbolsheimer P, Liu, MC. et al. Phase I/II Study of Sorafenib with Anastrozole in Patients with Hormone Receptor Positive Aromatase Inhibitor Resistant Metastatic Breast Cancer. Breast Cancer Research and Treatment. 2011;125:137-43
268. Baselga J, Zamagni C, Gomez P. et al. LBA8A phase III randomized, double-blind, trial comparing sorafenib plus capecitabine versus placebo plus capecitabine in the treatment of locally advanced or metastatic HER2-negative breast cancer (RESILIENCE). Ann Oncol. 2014;25:438-5
269. FDA Approval for Sunitinib Malate n.d. EMA Approval for Sunitinib n.d. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/
270. Taran A, Ignatov A, Smith B. et al. Acute hepatic failure following monotherapy with sunitinib for ovarian cancer. Cancer Chemother Pharmacol. 2009;63:971-2
271. Park IH, Kwon Y, Kim EA. et al. Major response to sunitinib (sutene(r)) in metastatic malignant phyllodes tumor of breast. Invest New Drugs. 2009;27:387-8
272. Socinski MA. The current status and evolving role of sunitinib in non-small cell lung cancer. J Thorac Oncol. 2008;3:119-23
273. Rini BI. Sunitinib. Expert Opin Pharmacother. 2007;8:2359-69
274. Kulke M, Lenz HJ, Meropol NJ, et al A phase 2 study to evaluate the efficacy, safety of SU 11248 in patients (pts) with unresectable neuroendocrine tumors (NETs) J. Clin. Oncol. 2005;23:310-6
275. Socinski MA, Novello S, Sanchez JM. et al. Efficacy and safety of sunitinib in previously treated, advanced non-small cell lung cancer (NSCLC): Preliminary results of a multicenter phase II trial. J Clin Oncol. 2006;24:364-4
276. Miller KD, Burstein HJ, Elias AD. et al. Phase II study of SU11248, a multitargeted receptor tyrosine kinase inhibitor (TKI), in patients (pts) with previously treated metastatic breast cancer (MBC); Proceedings of the San Antonio Breast Cancer Symposium 28th Annual Meeting; San Antonio, TX, USA. 8-11 December. 2005
277. Lenz H, Marshall J, Rosen L. et al. Phase II trial of SU11248 in patients with metastatic colorectal cancer (MCRC) after failure of standard chemotherapy; Proceedings of the American Society of Clinical Oncology Gastrointestinal 4th Annual Meeting; San Francisco, CA, USA. 27-29 January. 2006
278. Burstein HJ, Elias AD, Rugo HS. et al. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2008;26(11):1810-6
279. Bergh J, Greil R, Voytko N. et al. Sunitinib (SU) in combination with docetaxel (D) versus D alone for the first-line treatment of advanced breast cancer (ABC). J Clin Oncol. 2010;30:921-9
280. Crown J, Dieras V, Staroslawska E. et al. Phase III trial of sunitinib (SU) in combination with capecitabine (C) versus C in previously treated advanced breast cancer (ABC). J Clin Oncol. 2013;31:2870-8
281. Motzer RJ, Hutson TE, Tomczak P. et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115-24
282. Hutson TE, Escudier B, Esteban E. et al. Randomized Phase III Trial of Temsirolimus Versus Sorafenib As Second-Line Therapy After Sunitinib in Patients With Metastatic Renal Cell Carcinoma. Journal of Clinical Oncology. 2014;32:760-7
283. Robert NJ. et al. Sunitinib plus paclitaxel versus bevacizumab plus paclitaxel for first-line treatment of patients with advanced breast cancer: a phase III, randomized, open-label trial. Clin Breast Cancer. 2011;11:82-92
284. Barrios CH, Liu MC, Lee SC. et al. Phase III randomized trial of sunitinib versus capecitabine in patients with previously treated HER2-negative advanced breast cancer. Breast Cancer Res Treat. 2010;121:121-31
285. Yakes FM, Chen J, Tan J. et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10:2298-308
286. Katayama R, Kobayashi Y, Friboulet L. et al. Cabozantinib overcomes crizotinib resistance in ROS1 fusion-positive cancer. Clin Cancer Res. 2015;21:166-74
287. Gao N, Zhang Z, Jiang BH, Shi X. Role of pi3k/akt/mtor signaling in the cell cycle progression of human prostate cancer. Biochem Biophys Res Commun. 2003;310:1124-32
288. Dormond O, Madsen JC, Briscoe DM. The effects of mtor-akt interactions on anti-apoptotic signaling in vascular endothelial cells. J Biol Chem. 2007;282:23679-86
289. Giles FJ, Albitar M. Mammalian target of rapamycin as a therapeutic target in leukemia. Curr Mol Med. 2005;5:653-61
290. Martelli AM, Tazzari PL, Evangelisti C. et al. Targeting the phosphatidylinositol 3-kinase/akt/mammalian target of rapamycin module for acute myelogenous leukemia therapy: From bench to bedside. Curr Med Chem. 2007;14:2009-23
291. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A. et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084-91
292. Miller TW, Hennessy BT, González-Angulo AM. et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest. 2010;120:2406-13
293. Crowder RJ, Phommaly C, Tao Y. et al. PIK3CA and PIK3CB inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res. 2009;69:3955-62
294. Temsirolimus C. Cci 779, cci-779, cell cycle inhibitor-779. Drugs R D. 2004;5:363-7
295. Wan Xiaolin, Shen Na, Mendoza et al. CCI-779 Inhibits Rhabdomyosarcoma Xenograft Growth by an Antiangiogenic Mechanism Linked to the Targeting of mTOR/Hif-1α/VEGF Signaling. Neoplasia. 2006;8:394-401
296. Richardson PG, Sonneveld P, Schuster MW. et al. Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators.Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med. 2005;352:2487-98
297. Adams J, Palombella VJ, Sausville EA. et al. Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999;59:2615-22
298. Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4:349-60
299. Roccaro AM, Hideshima T, Richardson PG, Russo D, Ribatti D, Vacca A, Dammacco F, Anderson KC. Bortezomib as an antitumor agent. Curr Pharm Biotechnol. 2006;7:441-8
300. Yang CH, Gonzalez-Angulo AM, Reuben J. et al. Cristofanilli.Bortezomib (VELCADE®) in metastatic breast cancer: pharmacodynamics, biological effects, and prediction of clinical benefits. Annals of Oncology. 2006;17:813-7
301. FDA Approval for Ziv-Aflibercept. Available online: https://. www.cancer.gov/about-cancer/treatment/drugs/fda-ziv-aflibercept
302. Van Cutsem E, Tabernero J, Lakomy R. et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. 2012;30:3499-506
303. Duncan WC, van den Driesche S, Fraser HM. Inhibition of Vascular Endothelial Growth Factor in the Primate Ovary Up-Regulates Hypoxia-Inducible Factor-1{alpha} in the Follicle and Corpus Luteum. Endocrinology. 2008;149:3313-20
304. Ciombor KK, Berlin J, Chan E. Aflibercept. Clin. Cancer Res. 2013;19:1920-5
305. Gass JD. Pathogenesis of disciform detachment of the neuroepithelium. Am J Ophthalmol. 1967 63(3, Suppl): 1-139
306. Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman Lecture. Ophthalmology. 1993;100:1519-35
307. Green WR. Histopathology of age-related macular degeneration. Mol Vis. 1999;5:27
308. Wu FTH, Paez-Ribes M, Xu P. et al. Aflibercept and Ang1 supplementation improve neoadjuvant or adjuvant chemotherapy in a preclinical model of resectable breast cancer. Scientific Reports. 2016;6:36694
309. Falardeau P1, Champagne P, Poyet P. et al. Semin Oncol. Neovastat, a naturally occurring multifunctional antiangiogenic drug, in phase III clinical trials. 2001;28:620-5
310. Gingras D1, Batist G, Béliveau R. AE-941 (Neovastat): a novel multifunctional antiangiogenic compound. Expert Rev Anticancer Ther. 2001;1:341-7
311. Gingras D1, Batist G, Béliveau R. Oral administration of Neovastat in mice with subcutaneous grafted breast cancer (DA3) cells showed a significant reduction in tumor volume. Semin Oncol. 2001;28:620-5
312. Gingras D, Kruger EA, Lamy S. et al. The antiangiogenic agent neovastat (AE-941) inhibits vascular endothelial growth factor-mediated biological effects. Béliveau R1. Clin Cancer Res. 2002;8(4):1242-50
313. Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006;3:24-40
314. Jayson GC, Hicklin DJ, Ellis LM. Antiangiogenic therapy-evolving view based on clinical trial results. Nat Rev Clin Oncol. 2012;9:297-303
315. European Medicines Agency: European Medicines Agency completes its review of Avastin used in breast cancer [Press release 16/12/2010], 2010
316. Dienstmann R. et al. Toxicity as a biomarker of efficacy of molecular targeted therapies: focus on EGFR and VEGF inhibiting anticancer drugs. Oncologist. 2011;16:1729-40
317. Schuster C. et al. Clinical efficacy and safety of bevacizumab monotherapy in patients with metastatic melanoma: predictive importance of induced early hypertension. PLoS ONE. 2012;7(6):e38364
318. Rini BI. et al. Hypertension as a biomarker of efficacy in patients with metastatic renal cell carcinoma treated with sunitinib. J Natl Cancer Inst. 2011;103:763-73
319. Osterlund P. et al. Hypertension and overall survival in metastatic colorectal cancer patients treated with bevacizumab-containing chemotherapy. Br J Cancer. 2011;104:599-604
320. Kerbel RS. A cancer therapy resistant to resistance. Nature. 390; 335: 1997-336.
321. Loges S, Schmidt T, Carmeliet P. Mechanisms of resistance to anti-angiogenic therapy and development of third-generation anti-angiogenic drug candidates. Genes Cancer. 2010;1:12-25
322. Streubel B, Chott A, Huber D. et al. Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphomas. N Engl J Med. 2004;351(3):250-9
323. Yap TA. et al. Intratumor heterogeneity: seeing the wood for the trees. Sci Transl Med. 2012;4:127ps10
324. Chaudhry SR, Muhammad S, Eidens M. et al. Pharmacogenetic prediction of individual variability in drug response based on CYP2D6, CYP2C9 and CYP2C19 genetic polymorphisms. Curr Drug Metab. 2014;15:711-8
325. Turner NC, Reis-Filho JS. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012;13:178-85
326. Atkinson AJ, Colburn WA, DeGruttola VG. et al. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework*. Clin Pharmacol Ther. 2001;69:89-95
327. De Gruttola VG, Clax P, DeMets DL. et al. Considerations in the evaluation of surrogate endpoints in clinical trials. Summary of a National Institutes of Health workshop. Control. Clin. Trials. 2001;22:485-502
328. Dowlati A, Gray R, Sandler AB. et al. Cell adhesion molecules, vascular endothelial growth factor, and basic fibroblast growth factor in patients with non-small cell lung cancer treated with chemotherapy with or without bevacizumab - an Eastern Cooperative Oncology Group Study. Clin Cancer Res. 2008;14:1407-12
329. Nguyen M, Watanabe H, Budson AE. et al. Elevated levels of an angiogenic peptide, basic fibroblast growth factor, in the urine of patients with a wide spectrum of cancers. Journal of the National Cancer Institute. 1994;86:356-61
330. Cameron D. et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): primary results of a randomised, phase 3 trial. Lancet Oncol. 2013;14:933-42
331. Miles DW. et al. Biomarker results from the AVADO phase 3 trial of first-line bevacizumab plus docetaxel for HER2-negative metastatic breast cancer. Br J Cancer. 2013;108:1052-60
332. Miles D. et al. Bevacizumab plus paclitaxel versus placebo plus paclitaxel as first-line therapy for HER2-negative metastatic breast cancer (MERiDiAN): a double-blind placebo-controlled randomised phase III trial with prospective biomarker evaluation. Eur J Cancer. 2017;70:146-55
333. Oldenhuis CN, Oosting SF, Gietema JA, de Vries EG. Prognostic versus predictive value of biomarkers in oncology. Eur. J. Cancer. 2008;44:946-53
334. McShane LM, Altman DG, Sauerbrei W. et al. Statistics Subcommittee of NCI-EORTC Working Group on Cancer Diagnostics. Reporting recommendations for tumor MARKer prognostic studies (REMARK). Breast Cancer Res. Treat. 2006;100:229-35
335. Schneider BP, Li L, Shen F. et al. et al. Genetic variant predicts bevacizumab-induced hypertension in ECOG-5103 and ECOG-2100. Br J Cancer. 2014;111:1241-8
336. Jubb AM, Hurwitz HI, Bai W. et al. Impact of vascular endothelial growth factor-A expression, thrombospondin-2 expression, and microvessel density on the treatment effect of bevacizumab in metastatic colorectal cancer. J Clin Oncol. 2006;24:217-27
337. Gianni L, Romieu GH, Lichinitser M. et al. AVEREL: a randomized phase III Trial evaluating bevacizumab in combination with docetaxel and trastuzumab as first-line therapy for HER2-positive locally recurrent/metastatic breast cancer. J Clin Oncol. 2013;31:1719-25
338. Maru D, Venook AP, Ellis LM. Predictive biomarkers for bevacizumab: are we there yet? Clin Cancer Res. 2013;19:2824-7
339. Cao Y. Future options of anti-angiogenic cancer therapy. Chin J Cancer. 2016;35:21
340. Jain RK. Normalizing tumor microenvironment to treat cancer: Bench to bedside to biomarkers. J Clin Oncol. 2013;31:2205-18
341. Cao Y. Off-tumor target-beneficial site for antiangiogenic cancer therapy? Nat Rev Clin Oncol. 2010;7:604-8
342. Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7:987-9
343. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58-62
344. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417-27
345. Mancuso MR, Davis R, Norberg SM. et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest. 2006;116:2610-21
346. Griffioen AW, Mans LA, de Graaf AMA. et al. Rapid angiogenesis onset after discontinuation of sunitinib treatment of renal cell carcinoma patients. Clin Cancer Res. 2012;18:3961-71
347. Wolter P, Beuselinck B, Pans S, Schöffski P. Flare-up: An often unreported phenomenon nevertheless familiar to oncologists prescribing tyrosine kinase inhibitors. Acta Oncol. 2009;48:621-4
348. Schneider BP, Sledge GW Jr. Anti-vascular endothelial growth factor therapy for breast cancer: can we pick the winners? J Clin Oncol. 2011;29:2444-7
349. Couzin-Frankel J, Ogale Y. FDA. Once on 'fast track,' avastin now derailed. Science. 2011;333:143-4
350. Lupo G, Caporarello N, Olivieri M. et al. Anti-angiogenic therapy in cancer: downsides and new pivots for precision medicine. Front. Pharmacol. 2017;7:519
351. Perdrizet K, Leighl NB. The Role of Angiogenesis Inhibitors in the Era of Immune Checkpoint Inhibitors and Targeted Therapy in Metastatic Non-Small Cell Lung Cancer. Curr Treat Options Oncol. 2019;3:21
352. Abdalla A, Xiao L, Ullah MW. et al. Current Challenges of Cancer Anti-angiogenic Therapy and the Promise of Nanotherapeutics. Theranostics. 2018;8:533-48
353. Hironaka S1. Anti-angiogenic therapies for gastric cancer. Asia Pac J Clin Oncol. 2019;1:10
354. Hilmi M, Neuzilet C, Calderaro J. et al. Angiogenesis and immune checkpoint inhibitors as therapies for hepatocellular carcinoma: current knowledge and future research directions. J Immunother Cancer. 2019;7:333
355. Schmidt EV. Developing combination strategies using PD-1 checkpoint inhibitors to treat cancer. Semin Immunopathol. 2019;41:21-30
356. Manegold C, Dingemans AC, Gray JE. et al. The potential of combined immunotherapy and antiangiogenesis for the synergistic treatment of advanced NSCLC. J Thorac Oncol. 2017;12:194-207
357. Yi M, Jiao D, Qin S. et al. Synergestic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer. 2019;18:60
358. Fukumura D, Kloepper J, Amoozgar Z. et al. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol. 2018;15:325-40
Author contact
![]() Corresponding author: Yi Lu, Ph.D., Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Cancer Research Building, Room 258, 19 South Manassas Street, Memphis, TN 38163 (USA). Tel.: (901) 448-5436; Fax.: (901) 448-5496; E-mail: yluedu
Corresponding author: Yi Lu, Ph.D., Department of Pathology and Laboratory Medicine, University of Tennessee Health Science Center, Cancer Research Building, Room 258, 19 South Manassas Street, Memphis, TN 38163 (USA). Tel.: (901) 448-5436; Fax.: (901) 448-5496; E-mail: yluedu