Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2020; 11(19):5601-5611. doi:10.7150/jca.46173 This issue Cite
Research Paper
Involvement of the NF-κB signaling pathway in proliferation and invasion inhibited by Zwint-1 deficiency in Pancreatic Cancer Cells
Jae Hyeong Kim1, Yuna Youn1, Jong-chan Lee1,2, Jaihwan Kim1, Jin-Hyeok Hwang1,2 ![]()
1. Department of Internal Medicine, Seoul National University Bundang Hospital, Seongnam-si, Gyeonggi-do, 13620, Republic of Korea.
2. Department of Internal Medicine, Seoul National University College of Medicine, Seoul, 03080, Republic of Korea.
Received 2020-3-20; Accepted 2020-7-17; Published 2020-7-25
Abstract
Pancreatic cancer (PC) is an intractable cancer that is difficult to diagnose early and has a 5-year survival rate of less than 8%. ZW10-interacting kinetochore protein (ZWINT) is a crucial gene that contributes to chromosome instability and is essential for spindle assembly and kinetochore-microtubule attachment during meiosis and mitosis. However, the mechanism through which Zwint-1 promotes PC progression is yet to be elucidated. Here, we report that Zwint-1 is highly expressed in clinical PC specimens (based on analysis of the Gene Expression Profiling Interactive Analysis database) and various PC cell lines. Importantly, Zwint-1-deficient PC cells showed reduced nuclear factor-kappa B (NF-κB) (Ser536) phosphorylation along with inhibited proliferation and colony formation due to downregulation of NF-κB-regulated genes such as CCND1, cIAP1/2, and XIAP. In addition, Zwint-1-deficient PC cells showed reduced invasion and migration abilities, and decreased expression levels of the metalloproteinases MMP2 and MMP9. Furthermore, Zwint-1 deficiency arrested the PC cell cycle at the G2/M phase because the chromosomes failed to segregate properly, and the apoptosis rate in these cells gradually increased, accompanied by increased caspase-3 activation and anti-poly (ADP ribose) polymerase cleavage. Apoptosis caused by Zwint-1 deficiency was demonstrated to occur through caspase-dependent pathways based on experiments involving treatment with a pan-caspase inhibitor (Z-VAD-Fmk). Thus, Zwint-1 contributes to cell growth, invasion, and survival through NF-κB signaling pathways, suggesting that it could serve as a PC biomarker and new therapeutic target.
Keywords: Zwint-1, NF-κB, CCAN, KMN, RZZ, pancreatic cancer, cancer biology
Introduction
All eukaryotic cells must precisely replicate their genomes during mitosis and then distribute them to newly forming daughter cells. For proper chromosome segregation, sister chromosomes must bind to the microtubules extending from opposing spindle poles through the kinetochore, a dynamic multi-protein assembly on the centromere [1-4]. Kinetochores consist of hundreds of conserved proteins, which are divided into major structural sub-complexes such as the constitutive centromere-associated network (CCAN), KNL1-Mis12-Ndc80 (KMN) network, and ROD-Zwilch-ZW10 (RZZ) complexes [1-3, 5, 6]. The CCAN, which has several kinetochore subunits, is further assembled into a CENP-A nucleosome [7] and is constitutively localized to the centromere throughout the cell cycle [6, 8]. The CCAN acts as a foundation for kinetochore assembly and is a connector for centromeric chromatin, while the KMN network forms a complex with the microtubule-binding site of the kinetochore [8]. CENP-C binds to Ndc80 complexes through interactions with KNL1 and Mis12 complexes, and CENP-T is directly linked to Ndc80, contributing to KMN network localization [9]. Ndc80 complexes also directly bind microtubules [2]. RZZ is required for dynein/dynactin recruitment [10-12] and spindle assembly checkpoint activation at the kinetochore [13].
ZW10-interacting kinetochore protein 1 (Zwint-1) was initially identified as a protein that interacts with Zeste White 10 (ZW10) by a yeast two-hybrid screen, and was subsequently demonstrated to be a kinetochore component that plays an important role in spindle assembly and kinetochore-microtubule attachment during meiosis and mitosis [14-16]. Zwint-1 can directly interact with components of the KMN complex, specifically Ndc80 and Mis12, and acts as a bridge between the RZZ and KMN complexes required for kinetochore formation and spindle checkpoint activity [17]. Zwint-1 is also a mitotic checkpoint component required for the stable association of CENP-F and dynamitin with the kinetochore to ensure accurate chromosome segregation [15]. Recent studies have suggested that Zwint-1 could be a potential cancer biomarker [18, 19] given its high expression levels in several human malignancies such as prostate cancer, ovarian cancer, bladder cancer, lung cancer, hepatocellular carcinoma, and pulmonary adenocarcinoma [18, 20-25]. Moreover, knockdown of Zwint-1 has been shown to inhibit the proliferation, migration, invasion, and colony formation of lung cancer cell, and to enhance cell apoptosis [23].
However, little is known about the role of Zwint-1 in pancreatic cancer (PC). In this study, we investigated the expression of Zwint-1 in tissues from patients with PC in The Cancer Genome Atlas (TCGA) database and in PC cell lines, and evaluated the effects of Zwint-1 on PC cell tumorigenesis and progression using a Zwint-1 knockdown system in vitro.
Methods
Database analysis
Expression levels of ZWINT, CCAN (CENP-A, CENP-C, CENP-T), KMN (KNL1, Mis12, Ndc80), and RZZ (ROD, Zwilch, ZW10) complexes in 179 PC and 171 normal pancreas tissues were compared by Gene Expression Profiling Interactive Analysis (GEPIA) (http://gepia.cancer-pku.cn/) [26] based on patient data from TCGA [27] and normal tissue samples from Genotype-Tissue Expression (GTEx) [28, 29]. The TCGA dataset is based on RNA-sequencing performed on 179 PC tissues from 178 PC patients, including 178 primary tissues and one metastasis tissue. The expression was determined as transcripts per million (TPM), and gene expression levels were calculated using a log2 (TPM+1) scale for comparison. The cutoff values were |log2 fold change (FC)| of 1 and P value < 0.01. The overall survival (OS) of patients with PC was extracted from TCGA data and compared with the expression levels of the Zwint-1-related genes.
Cell culture and small interfering RNA (siRNA) knockdown
The human PC cell lines MIA PaCa-2 [American Type Culture Collection (ATCC) CRL-1420; ATCC, Manassas, VA, USA] and PANC-1 (ATCC CRL-1469) were grown in high-glucose Dulbecco's modified Eagle's medium (DMEM). The human pancreatic ductal adenocarcinoma (PDAC) cell lines AsPC-1 and Capan-1 were grown in RPMI medium. Noncancerous immortalized human pancreatic duct epithelial (HPDE) cells, obtained from Joo Kyung Park, MD (Samsung Medical Center, Seoul, South Korea), were grown in defined K-SFM medium. All cell culture media contained 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin (Gibco, Life Technologies, Grand Island, NY, USA). To block Zwint-1 expression, the cells were transfected with specific siRNA using Lipofectamine RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA, USA) and then tested for efficacy according to the manufacturer's instructions. Two different Zwint-1 siRNAs were prepared [30]. The two Zwint-1 siRNAs and control siRNA were purchased from Cosmo Bio Co., Ltd. (Tokyo, Japan).
Cell proliferation and viability assay
After adhesion for 24 h, the cells were transfected with Zwint-1 siRNA or control siRNA and incubated for 3 or 7 d. For cell proliferation and viability assays, MIA PaCa-2 and PANC-1 cells were seeded into 12-well plates at a density of 1 × 104 cells/well or 8 × 104 cells/well and tested using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution. For colony formation assays, the cells were seeded into 6-well plates at a density of 500-1000 cells/well and incubated for 14 d. The colonies were then fixed in 100% methanol, stained with 10% crystal violet, and counted. Each assay was performed in triplicate.
Wound-healing assay
The motility of Zwint-1 siRNA and control siRNA-transfected cells was examined by a wound-healing assay. The cells were seeded in 6-well plates at 5 × 105 cells per well. After 24 h, wounds were created in cell monolayers in each well using a P1000 pipette tip. The cells were then rinsed once with phosphate-buffered saline (PBS) and cultured for 24 h or 72 h. Cell motility was expressed as the cell migration rate.
Cell migration and invasion assays
Migration and invasion abilities under Zwint-1 deficiency were assessed using Transwell plates (Corning, Corning, NY, USA). For the cell migration assay, 5 × 104 cells in 200 μL of 1% serum DMEM were seeded directly into each well of the Transwell chambers with 8-μm pore membranes. For the cell invasion assay, the cells were seeded in the Transwell chambers coated with Matrigel (Corning), and medium containing 10% FBS was added to the lower chamber. After incubation for 24 h, the medium in the upper chamber was removed, and the cells were fixed and stained using the Differential Quik Stain Kit. The cells adhering to the upper surface of the membrane were removed using a cotton applicator. The number of cells on the lower side of the membrane was counted. Each experiment was performed in triplicate.
Western blotting
Proteins were extracted from whole cells using 1× RIPA buffer, and protein concentrations were determined using a BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Protein extracts were resuspended in 5× sample buffer [50 mM Tris (pH 6.8), 100 mM dithiothreitol, 2% sodium dodecyl sulfate (SDS), 0.1% bromophenol blue, and 10% glycerol], boiled for 5 min, and subjected to SDS-polyacrylamide gel electrophoresis on 8-15% gels. Separated proteins were transferred to transblot nitrocellulose membranes (Schleicher & Schuell, Keene, NH, USA), and membranes were blocked in 5% skim milk with TBST [10 mM Tris (pH 8.0), 150 mM NaCl, 0.05% Tween 20] and incubated with the following primary antibodies: anti-Zwint-1 (GeneTex), anti-p-NF-κB (p65) (Ser536) (Cell Signaling Technology, Danvers, MA, USA), anti-NF-κB (p65) (Cell Signaling Technology), anti-cyclin D1 (Cell Signaling Technology), anti- cellular inhibitor of apoptosis 1/2 (cIAP1) (Cell Signaling Technology), anti-cIAP2 (Cell Signaling Technology), anti-X-linked inhibitor of apoptosis (XIAP) (Cell Signaling Technology), anti-MMP2 (Cell Signaling Technology), anti-MMP9 (Cell Signaling Technology), anti-cyclin A2 (Cell Signaling Technology), anti-p-histone H3 pS10 (Abcam, Cambridge, MA, USA), anti-caspase-3 (Cell Signaling Technology), anti-poly (ADP ribose) polymerase (PARP; Cell Signaling Technology), and anti-β-actin (Cell Signaling Technology). Protein expression was detected based on chemiluminescent signals activated by SuperSignal West Pico Chemiluminescent Substrate (Pierce, Thermo Scientific, USA) after reaction with horseradish peroxidase-tagged secondary antibodies (Jackson Immunoresearch Laboratories, West Grove, PA, USA).
Fluorescence-activated cell sorting (FACS) analysis
For cell cycle and DNA content analysis, cultured cells were incubated in trypsin-ethylenediaminetetraacetic acid at 37°C in an atmosphere containing 5% CO2, collected by centrifugation, and washed once with 1× PBS. The cells were centrifuged, supernatants were removed, and then the cells were stained with 50 μg/mL propidium iodide (PI; Sigma-Aldrich, St. Louis, MO, USA), along with 100 U ribonuclease A from the bovine pancreas (Sigma-Aldrich). FACs analysis was performed with a FACSCalibur instrument (BD Biosciences, San Diego, CA, USA) according to standard protocols.
Apoptosis was also analyzed by FACS using fluorescein isothiocyanate (FITC)-conjugated annexin V (BD Biosciences, San Diego, CA, USA) and PI (Sigma-Aldrich) staining. The analysis was performed with a FACSCalibur (BD Biosciences) instrument according to the standard protocol. Apoptosis was blocked with the pan-caspase inhibitor Z-VAD-FMK (20 µM; R&D Systems, Minneapolis, MN, USA).
Immunofluorescence
Cells were grown on Thermo Scientific Nunc Lab-Tek II Chamber Slides, permeabilized with 0.5% Triton X-100 for 1 min, and fixed with 4% paraformaldehyde for 10 min. The fixed cells were incubated for 1 h at room temperature with blocking solution (1% bovine serum albumin) and then incubated overnight at 4°C with anti-CREST (Immuno Vision Technologies, Brisbane, CA) and anti-α-tubulin (Abcam, Cambridge, MA, USA) primary antibodies. The cells were then incubated with secondary antibodies and 100 ng/mL DAPI for 3 h. Samples were mounted in Prolong Gold Antifade reagent (Invitrogen) and viewed under a confocal microscope (Zeiss LSM710 with ZEN software).
Statistical analysis
All data are presented as the mean ± standard error of the mean (SEM) values from three independent experiments. Results were statistically analyzed using GraphPad Prism (version 5.0, GraphPad Software Inc., San Diego, CA, USA) using one-way or two-way analysis of variance followed by Bonferroni's multiple comparison tests.
Results
Zwint-1 expression is upregulated in PC patient tissues and PC cell lines
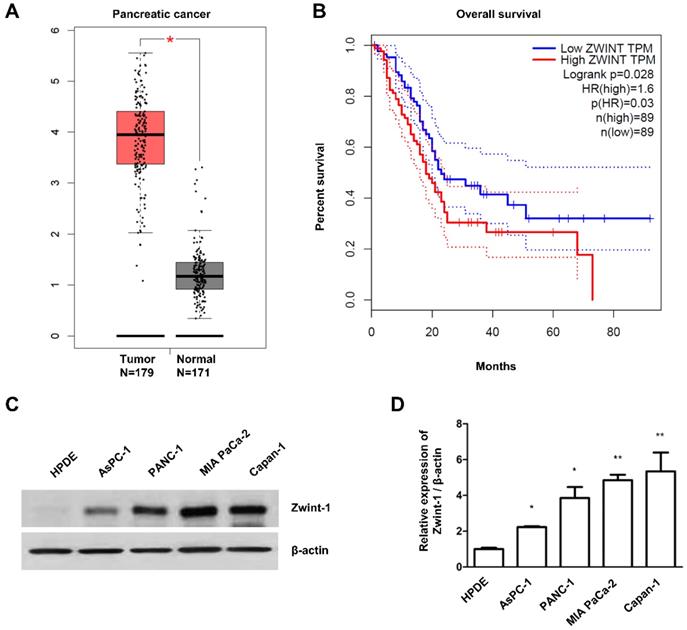
We first confirmed the mRNA expression of the CCAN (CENP-A, CENP-C, CENP-T), KMN (KNL1, Mis12, Ndc80), and RZZ (ROD, Zwilch, ZW10) complexes, as well as Zwint-1, in clinical PC specimens from TCGA (n = 179) and normal tissues from GTEx data (n = 171) using the publicly available GEPIA database. Zwint-1 expression was elevated in tumors, whereas expression of the genes involved in the CCAN, KMN, and RZZ complexes was not (Fig. 1A and Supplementary Fig. 1A). In addition, the OS of PC patients was inversely proportion to the Zwint-1 expression level (Fig. 1A and B). Other clinical and demographic information about the patients is shown in Supplementary Fig. 2A-I, which was obtained from the cBioportal database. We also measured the level of Zwint-1 protein in various PC lines, including AsPC-1, PANC-1, MIA PaCa-2, and Capan-1 cells, by western blotting, which demonstrated higher levels in AsPC-1, PANC-1, MIA-PaCa-2, and Capan-1 cells compared to those in normal HPDE cells (Fig. 1C and 1D).
Zwint-1 expression is elevated in PC tissues and cell lines. (A) Zwint-1 expression levels in PC tissues (n = 179) and normal tissues (n = 171). (B) OS based on the TCGA database. Each dot represents the Zwint-1 expression level in one sample. The OS of PC patients with low or high Zwint-1 expression levels was analyzed by the Kaplan-Meier method and a log rank test. The high dotted line demarcates samples with higher expression levels than the median value of TPM (the high expression cohort), whereas the low dotted line demarcates samples with lower expression levels than the median value of TPM (the low expression cohort). Median values are indicated by solid lines. *P < 0.005. TCGA, The Cancer Genome Atlas; TPM, transcripts per million; OS, overall survival; HR, hazard ratio. (C) Zwint-1 protein expression analyzed by western blotting in HPDE cells and different PC cell lines (AsPC-1, PANC-1, MIA PaCa-2, and Capan-1 cells). Cell lysates were immunoblotted with the indicated antibodies. (D) Levels of Zwint-1 protein expression in HPDE and PC cell lines. The Zwint-1/β-actin ratio was determined by densitometric analysis using ImageJ software. Error bars represent standard deviations of the means of three biological replicates. Values represent means ± SEMs. *P < 0.005, **P < 0.001. Results were analyzed by one-way analysis of variance followed by Bonferroni's multiple comparison tests.
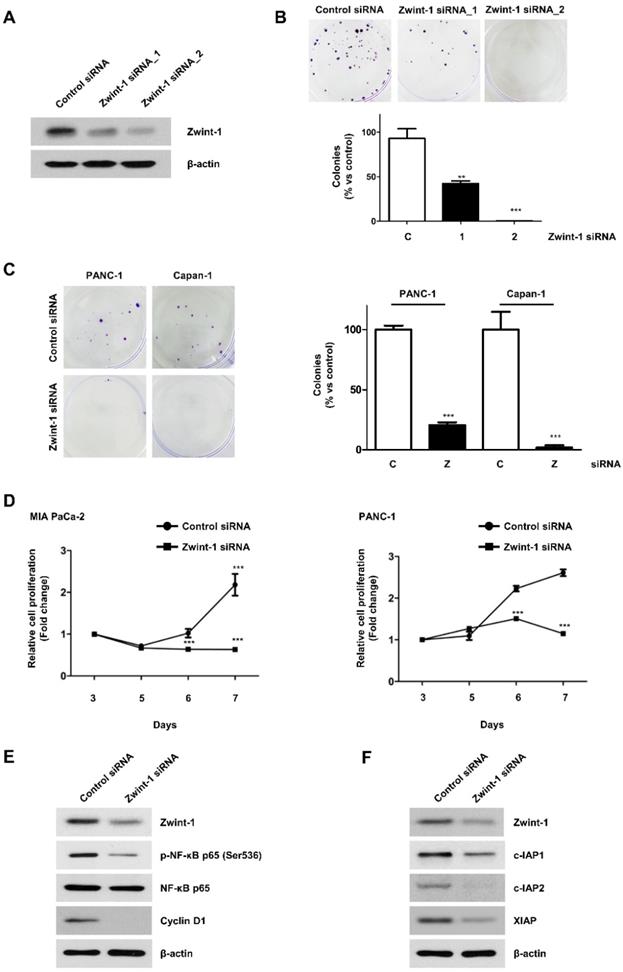
Zwint-1 deficiency inhibits the colony formation and proliferation of PC cells via the nuclear factor kappa B (NF-κB) signaling pathway
Between the two siRNAs tested, Zwint-1_2 had a greater effect on inhibiting Zwint-1 protein expression better (Fig. 2A), resulting in reduced colony formation in MIA PaCa-2 (Fig. 2B), PANC-1, and Capan-1 cells (Fig. 2C). Therefore, Zwint-1_2 siRNA was selected for use in subsequent experiments. The MTT assay showed that Zwint-1 deficiency significantly inhibited the proliferation of MIA PaCa-2 and PANC-1 cells for 3-7 d after siRNA transfection (Fig. 2D). Western blotting showed that the expression level of p-NF-κB (p65) (Ser536) was reduced due to Zwint-1 deficiency, whereas there was no difference in the expression level of NF-κB (p65) between Zwint-1-expressing and Zwint-1-deficient cells. In addition, the expression level of cyclin D1 (a target of NF-κB) was significantly reduced due to Zwint-1 deficiency in PC cells (Fig. 2E). Finally, the expression levels of cIAP1/2 and XIAP, targets of NF-κB involved in cell survival, were also markedly reduced in Zwint-1-deficient PC cells (Fig. 2F). These results indicated that Zwint-1 is involved in the proliferation and survival of PC cells at least partially through NF-κB signaling.
Zwint-1-deficient PC cells show reduced proliferation and colony formation mediated by NF-κB signaling. (A) MIA PaCa-2 cells were transfected with two different siRNA constructs to knock down Zwint-1 expression, and the expression levels of Zwint-1 protein were measured 48 h later by western blotting. (B) Colony formation assays on MIA PaCa-2 cells transfected with control or one of two Zwint-1 siRNAs (Zwint-1 siRNA_1 and siRNA_2). (C) Colony formation assays in PANC-1 and Capan-1 cells transfected with control siRNA and Zwint-1 siRNA_2. Values represent means ± SEMs. Results were analyzed using unpaired t-tests. ***P < 0.0001. (D) MTT assays to determine the proliferation of control and Zwint-1-deficient PC cells for 3 or 7 d. Curves were constructed based on biological triplicates with values expressed as means ± SEMs. ***P < 0.0001. Results were analyzed by two-way analysis of variance with Bonferroni's multiple comparison tests. (E) Western blotting to determine the levels of Zwint-1, p-NF-κB (Ser536), NF-κB p65, Cyclin D1, and β-actin in MIA PaCa-2 cells after transfection with control or Zwint-1 siRNA at 24 h. (F) Western blotting to determine the levels of Zwint-1, c-IAP1, c-IAP2, XIAP, and β-actin in MIA PaCa-2 cells after transfection with control or Zwint-1 siRNA at 48 h.
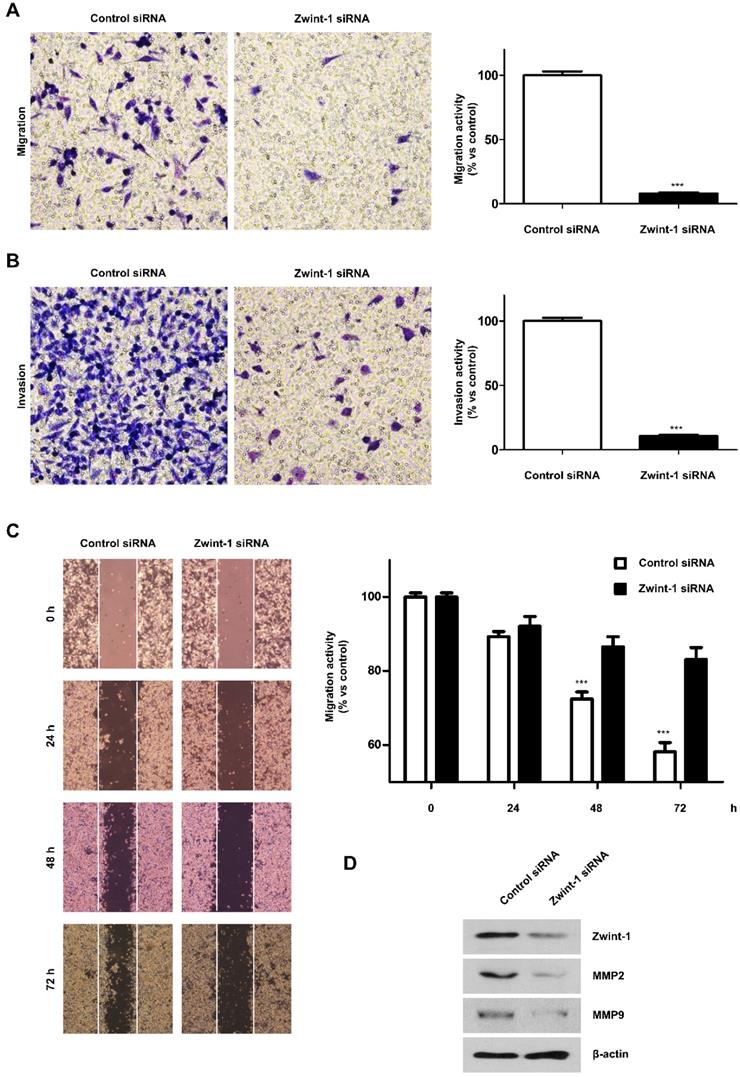
Zwint-1 deficiency reduces the migration and invasion capacity of PC cells
After inhibiting PC cell growth by culturing under low FBS conditions, the number of migrating Zwint-1-deficient MIA-PaCa-2 cells was markedly reduced compared to that of control cells (Fig. 3A). Transwell assays further showed that the number of invading Zwint-1-deficient MIA-PaCa-2 cells was reduced compared to that of the control cells (Fig. 3B), and the wound-healing ability was also decreased in Zwint-1-deficient PC cells (Fig. 3C) further confirming reduced migration potential, particularly after 48 h. These results were supported by the reduced expression levels of MMP-2 and MMP-9 under Zwint-1 deficiency (Fig. 3D), indicating that Zwint-1 plays essential roles in migration and invasion by activating MMP2 and MMP9 expression in PC cells.
Zwint-1-deficient PC cells show reduced migration, invasion, and wound healing abilities due to MMP2 and MMP9 inhibition. (A) Migration and (B) invasion assays of MIA PaCa-2 cells transfected with control or Zwint-1 siRNA at 24 h. (C) Wound-healing assays of MIA PaCa-2 cells transfected with control or Zwint-1 siRNA for 24-72 h. Values represent means ± SEMs. Results were analyzed using unpaired t-tests. ***P < 0.0001. (D) Western blotting to determine the levels of Zwint-1, MMP2, MMP9, and β-actin in MIA PaCa-2 cells after transfection with control or Zwint-1 siRNA at 48 h.
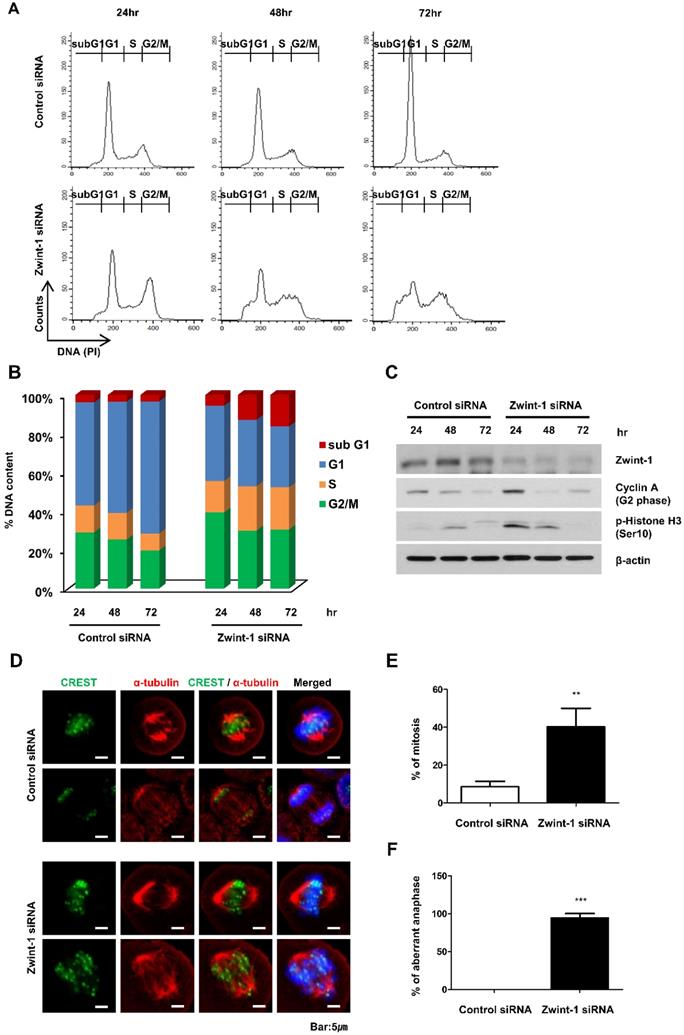
Zwint-1-deficient PC cells are arrested in the G2/M phase leading to high rates of apoptosis
Flow cytometry to determine the proportion of cells in the subG1, G1, S, and G2/M phases of the cycle demonstrated an increased proportion of Zwint-1-deficient PC cells in G2/M compared to that in controls. In addition, subG1 populations (apoptotic cells) increased compared to those in the controls at 24 to 72 h, whereas the G1 cell population was decreased in Zwint-1-deficient PC cells in a concentration-dependent manner (Fig. 4A, 4B). Using western blotting, we also measured the expression of cyclin A2 (a regulator of the G1 phase) and p-histone H3 (Ser10) (a regulator of the G2/M phase transition) to determine the molecular mechanisms altered by Zwint-1 deficiency. After 24 h of Zwint-1 siRNA transfection, cyclin A2 and p-histone H3 (Ser10) levels were significantly increased in the Zwint-1-deficient PC cells (Fig. 4C). Immunofluorescence observations of the centromere markers CREST and alpha-tubulin demonstrated well-aligned chromatids separated into two poles with a centromere in control cells. By contrast, although the tubulin of Zwint-1-deficient PC cells was attached to the centromere, no normally dividing cells were observed (Fig. 4D-4F). These results indicated that Zwint-1-deficient PC cells were arrested in the G2/M phase because they could not divide.
Zwint-1-deficient PC cells are arrested in the G2/M phase and undergo apoptosis. (A) MIA PaCa-2 cells were transfected with control or Zwint-1 siRNA and then subjected to flow cytometric analysis at 24, 48, and 72 h. The flow cytometry plots and data are representative of at least three separate experiments. (B) Percentages of cells in subG1 (apoptosis, red), G1 (blue), S (orange), and G2/M (green) phases. (C) Western blotting to determine the levels of Zwint-1, cyclin A2, p-histone H3, and β-actin in MIA PaCa-2 cells after transfection with control or Zwint-1 siRNA for 24, 48, and 72 h. (D) Spindle structures were visualized by immunofluorescent staining for CREST (green), a-tubulin (red), and DNA (blue). (E) Comparison of mitosis frequency in Zwint-1 siRNA-transfected cells versus control siRNA-transfected cells. (F) Comparison of aberrant anaphase frequencies in Zwint-1-trasnfected siRNA cells versus those in control siRNA-transfected cells. Values represent means ± SEMs. Results were analyzed using unpaired t-tests. **P < 0.001, ***P < 0.0001.
Apoptosis is increased in Zwint-1-deficient PC cells through caspase-dependent signaling
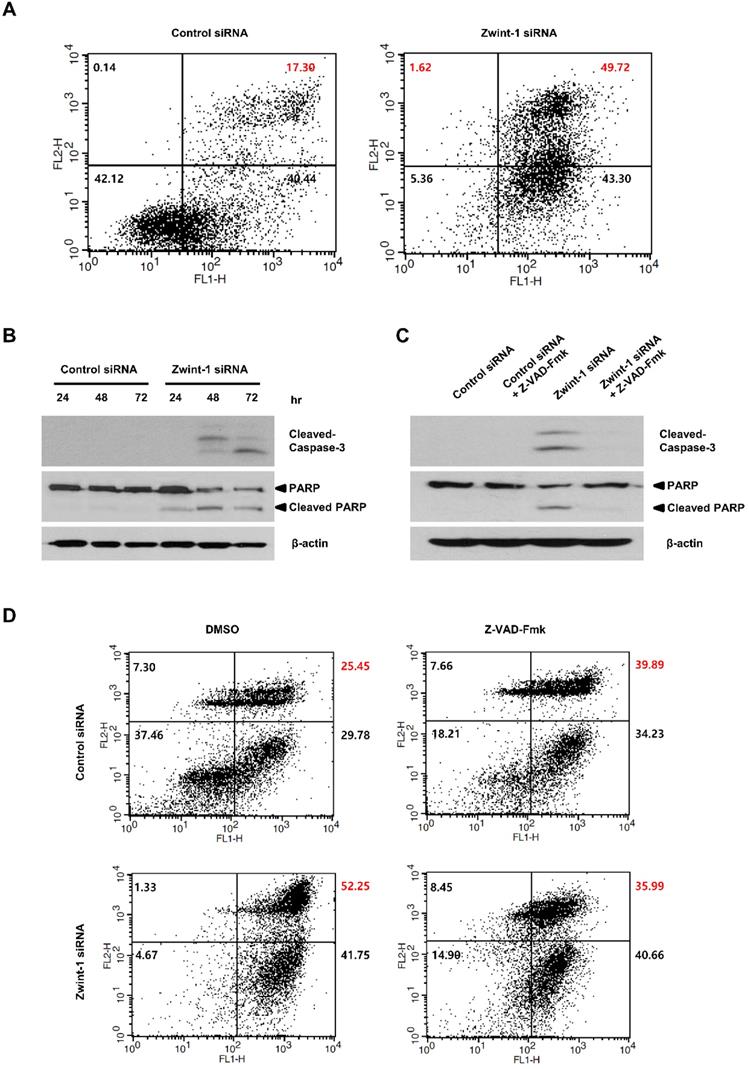
To determine whether Zwint-1-deficient PC cells were lost through apoptosis or necrosis, we double-stained the cells with annexin V-FITC and PI and quantified phenotypic changes in apoptotic cells. FACS analysis showed that the proportion of the late-apoptosis (annexin V-FITC-positive and Annexin V-FITC/PI-positive) population was significantly higher in Zwint-1-deficient cells compared to that in control cells at 72 h (Fig. 5A, upper right quadrant). The levels of caspase-3 and cleaved PARP proteins increased in Zwint-1-deficient PC cells compared to those of controls (Fig. 5B), further confirming a role of apoptosis in the observed cell death. These results are consistent with the increase in the subG1 population in Zwint-1-deficient PC cells. Treatment of Zwint-1-deficient PC cells with 20 μM of the pan-caspase inhibitor Z-VAD-Fmk reduced the rate of apoptosis (Fig. 5C), and also reduced the levels of caspase-3 activation and PARP cleavage increased underZwint-1 deficiency (Fig. 5D). These results indicated that increased apoptosis due to Zwint-1 deficiency occurs through caspase-dependent signaling pathways.
Zwint-1-deficient PC cells undergo apoptosis induced by a caspase-dependent pathway. (A) Apoptosis was measured by annexin V staining followed by flow cytometry. The lower left (LL) quadrant shows live cells, the lower right (LR) shows early-apoptotic cells, and the upper right (UR) shows late-stage apoptotic cells. (B) Western blotting to determine the levels of cleaved-caspase-3, PARP, and β-actin in MIA PaCa-2 cells after transfection with control or Zwint-1 siRNA for 24, 48, and 72 h. (C) Western blot analysis to determine the levels of caspase-3, PARP, and β-actin in MIA PaCa-2 cells after transfection with control or Zwint-1 siRNA with or without Z-VAD-Fmk (20 μM) treatment for 72 h. (D) Apoptosis measured by annexin V staining followed by flow cytometry. Cells were treated with Z-VAD-Fmk (20 µM) for 72 h.
Discussion
Zwint-1 is not only necessary for normal cell division during mitotic metaphase but is also highly expressed in various carcinomas. In this study, we showed that Zwint-1 is highly expressed in PC cells and tissues, and promotes the proliferation and invasion of PC cells through NF-κB signaling. In line with previous studies in patients with other cancer types [18, 20-25], Zwint-1 was found to be upregulated in PC tissues and cell lines, and patients with high Zwint-1 expression had lower OS than those with low Zwint-1 expression. Zwint-1-deficient PC cells exhibited suppressed colony formation, proliferation, migration, and invasion due to a reduction in NF-κB phosphorylation and the expression of related genes. These data support evidence that Zwint-1 knockdown inhibits lung cancer cell proliferation, migration, and invasion, as well as colony formation [23]. Furthermore, Zwint-1-deficient PC cells showed induced G2/M arrest due to abnormal cell division, which promoted apoptosis through caspase-dependent pathways. These findings suggest that Zwint-1 might play an important role in the promotion and progression of PC. To the best of our knowledge, this is the first report showing that Zwint-1 contributes to PC cell progression through NF-κB signaling.
The NF-κB family of transcription factors includes RelA (p65), RelB, c-Rel, NF-κB1 (p50/p105), and NF-κB2 (p52/p100), which play important roles in inflammation, cell proliferation and differentiation, immune responses, and cancer [31-33]. They share a Rel homology domain that regulates the development and progression of cancer by allowing the binding of NF-κB-specific DNA motifs [34]. Masking nuclear position signals prevents NF-κB transcription factors from being translocated into the cell nuclei by NF-κB inhibitor (IκB); thus, NF-κB remains dormant and inactive in the cytoplasm [35]. However, in most cancers, NF-κB is activated and is considered to be a major signal mediator that contributes to cancer development and progression by promoting cell proliferation, regulating apoptosis, stimulating angiogenesis, and increasing invasion and metastasis [36-38]. In particular, NF-κB signaling pathways play an important role in the development and progression of PC and drug resistance [39]. Indeed, we observed that Zwint-1-deficient PC cells inhibited temozolomide resistance (Supplementary Fig. 3A).
NF-κB also plays an essential role in controlling the transcription of genes such as cyclooxygenase-2 (COX2) and cyclin D1, which are important in the early and late stages of aggressive cancers; cIAP-1/2, XIAP, and cellular FLICE inhibitory protein (FLIP), which are important genes that encode apoptosis suppressor proteins; and MMP2, MMP9, and vascular endothelial growth factor (VEGF), which are important genes in invasion and angiogenesis [40-44]. In our study, various changes in proliferation, invasion, migration, and apoptosis were observed in Zwint-1-deficient PC cells, suggesting a role for NF-κB signaling. Zwint-1-deficient PC cells showed reduced phosphorylation of NF-κB and significantly reduced the protein expression levels of NF-κB target genes, including cyclin D1, cIAP1/2, XIAP, MMP2, and MMP9. Consistently, Zwint-1-deficient PC cells also showed reduced proliferation ability, invasion, and migration, as well as increased apoptosis.
Conclusion
Zwint-1 deficiency inhibits PC cell proliferation, invasion, and migration through the NF-κB signaling pathway, in addition to promoting cellular apoptosis through a caspase-dependent pathway.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by grant no's. NRF-2017R1C1B2009106 and NRF-2017R1A2B2012191 from the National Research Foundation of Korea (NRF). We would like to thank Editage (www.editage.co.kr) for English language editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fukagawa T, Earnshaw WC. The centromere: chromatin foundation for the kinetochore machinery. Developmental cell. 2014;30:496-508
2. McKinley KL, Cheeseman IM. The molecular basis for centromere identity and function. Nature reviews Molecular cell biology. 2016;17:16-29
3. Westhorpe FG, Straight AF. The centromere: epigenetic control of chromosome segregation during mitosis. Cold Spring Harbor perspectives in biology. 2014;7:a015818
4. Westhorpe FG, Straight AF. Chromosome Segregation: Reconstituting the Kinetochore. Current biology: CB. 2016;26:R1242-r5
5. Mosalaganti S, Keller J, Altenfeld A, Winzker M, Rombaut P, Saur M. et al. Structure of the RZZ complex and molecular basis of its interaction with Spindly. The Journal of cell biology. 2017;216:961-81
6. Pesenti ME, Weir JR, Musacchio A. Progress in the structural and functional characterization of kinetochores. Current opinion in structural biology. 2016;37:152-63
7. Yan K, Yang J, Zhang Z, McLaughlin SH, Chang L, Fasci D. et al. Structure of the inner kinetochore CCAN complex assembled onto a centromeric nucleosome. Nature. 2019;574:278-82
8. Hara M, Fukagawa T. Critical Foundation of the Kinetochore: The Constitutive Centromere-Associated Network (CCAN). Progress in molecular and subcellular biology. 2017;56:29-57
9. Rago F, Gascoigne KE, Cheeseman IM. Distinct organization and regulation of the outer kinetochore KMN network downstream of CENP-C and CENP-T. Current biology: CB. 2015;25:671-7
10. Cheerambathur DK, Gassmann R, Cook B, Oegema K, Desai A. Crosstalk between microtubule attachment complexes ensures accurate chromosome segregation. Science (New York, NY). 2013;342:1239-42
11. Gassmann R, Essex A, Hu JS, Maddox PS, Motegi F, Sugimoto A. et al. A new mechanism controlling kinetochore-microtubule interactions revealed by comparison of two dynein-targeting components: SPDL-1 and the Rod/Zwilch/Zw10 complex. Genes & development. 2008;22:2385-99
12. Gassmann R, Holland AJ, Varma D, Wan X, Civril F, Cleveland DW. et al. Removal of Spindly from microtubule-attached kinetochores controls spindle checkpoint silencing in human cells. Genes & development. 2010;24:957-71
13. Rodriguez-Rodriguez JA, Lewis C, McKinley KL, Sikirzhytski V, Corona J, Maciejowski J. et al. Distinct Roles of RZZ and Bub1-KNL1 in Mitotic Checkpoint Signaling and Kinetochore Expansion. Current biology: CB. 2018;28:3422-9.e5
14. Starr DA, Saffery R, Li Z, Simpson AE, Choo KH, Yen TJ. et al. HZwint-1, a novel human kinetochore component that interacts with HZW10. Journal of cell science. 2000;113( Pt 11):1939-50
15. Wang H, Hu X, Ding X, Dou Z, Yang Z, Shaw AW. et al. Human Zwint-1 specifies localization of Zeste White 10 to kinetochores and is essential for mitotic checkpoint signaling. The Journal of biological chemistry. 2004;279:54590-8
16. Woo Seo D, Yeop You S, Chung WJ, Cho DH, Kim JS, Su Oh J. Zwint-1 is required for spindle assembly checkpoint function and kinetochore-microtubule attachment during oocyte meiosis. Scientific reports. 2015;5:15431
17. Obuse C, Iwasaki O, Kiyomitsu T, Goshima G, Toyoda Y, Yanagida M. A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nature cell biology. 2004;6:1135-41
18. Xu Z, Zhou Y, Cao Y, Dinh TL, Wan J, Zhao M. Identification of candidate biomarkers and analysis of prognostic values in ovarian cancer by integrated bioinformatics analysis. Medical oncology (Northwood, London, England). 2016;33:130
19. Liu J, Meng H, Li S, Shen Y, Wang H, Shan W. et al. Identification of Potential Biomarkers in Association With Progression and Prognosis in Epithelial Ovarian Cancer by Integrated Bioinformatics Analysis. Frontiers in genetics. 2019;10:1031
20. Bhattacharjee A, Richards WG, Staunton J, Li C, Monti S, Vasa P. et al. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:13790-5
21. Endoh H, Tomida S, Yatabe Y, Konishi H, Osada H, Tajima K. et al. Prognostic model of pulmonary adenocarcinoma by expression profiling of eight genes as determined by quantitative real-time reverse transcriptase polymerase chain reaction. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2004;22:811-9
22. Ho JR, Chapeaublanc E, Kirkwood L, Nicolle R, Benhamou S, Lebret T. et al. Deregulation of Rab and Rab effector genes in bladder cancer. PloS one. 2012;7:e39469
23. Peng F, Li Q, Niu SQ, Shen GP, Luo Y, Chen M. et al. ZWINT is the next potential target for lung cancer therapy. Journal of cancer research and clinical oncology. 2019;145:661-73
24. Urbanucci A, Sahu B, Seppala J, Larjo A, Latonen LM, Waltering KK. et al. Overexpression of androgen receptor enhances the binding of the receptor to the chromatin in prostate cancer. Oncogene. 2012;31:2153-63
25. Ying H, Xu Z, Chen M, Zhou S, Liang X, Cai X. Overexpression of Zwint predicts poor prognosis and promotes the proliferation of hepatocellular carcinoma by regulating cell-cycle-related proteins. OncoTargets and therapy. 2018;11:689-702
26. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic acids research. 2017;45:W98-w102
27. Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K. et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nature genetics. 2013;45:1113-20
28. The Genotype-Tissue Expression (GTEx) project. Nature genetics. 2013;45:580-5.
29. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science (New York, NY). 2015;348:648-60
30. Georges SA, Biery MC, Kim SY, Schelter JM, Guo J, Chang AN. et al. Coordinated regulation of cell cycle transcripts by p53-Inducible microRNAs, miR-192 and miR-215. Cancer research. 2008;68:10105-12
31. Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Molecular cancer. 2013;12:86
32. Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680-4
33. Ryseck RP, Weih F, Carrasco D, Bravo R. RelB, a member of the Rel/NF-kappa B family of transcription factors. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas. 1996;29:895-903
34. Huguet C, Crepieux P, Laudet V. Rel/NF-kappa B transcription factors and I kappa B inhibitors: evolution from a unique common ancestor. Oncogene. 1997;15:2965-74
35. Lenardo M, Pierce JW, Baltimore D. Protein-binding sites in Ig gene enhancers determine transcriptional activity and inducibility. Science (New York, NY). 1987;236:1573-7
36. Prabhu L, Mundade R, Korc M, Loehrer PJ, Lu T. Critical role of NF-kappaB in pancreatic cancer. Oncotarget. 2014;5:10969-75
37. Lu T, Stark GR. Cytokine overexpression and constitutive NFkappaB in cancer. Cell cycle (Georgetown, Tex). 2004;3:1114-7
38. Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochemical pharmacology. 2006;71:1397-421
39. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431-6
40. Shi H, Ma J, Mi C, Li J, Wang F, Lee JJ. et al. Amorfrutin A inhibits TNF-alpha-induced NF-kappaB activation and NF-kappaB-regulated target gene products. International immunopharmacology. 2014;21:56-62
41. Li J, Ma J, Wang KS, Mi C, Wang Z, Piao LX. et al. Baicalein inhibits TNF-alpha-induced NF-kappaB activation and expression of NF-kappaB-regulated target gene products. Oncology reports. 2016;36:2771-6
42. Cao LP, Song JL, Yi XP, Li YX. Double inhibition of NF-kappaB and XIAP via RNAi enhances the sensitivity of pancreatic cancer cells to gemcitabine. Oncology reports. 2013;29:1659-65
43. Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochimica et biophysica acta. 2010;1799:775-87
44. Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S. et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461-6
Author contact
![]() Corresponding author: Jin-Hyeok Hwang, MD, PhD. Department of Internal Medicine, Seoul National University Bundang Hospital 82, Gumi-ro 173 Beon-gil, Bundang-gu, Seongnam-si, Gyeonggi-do 13620, Republic of Korea. Tel.: + 82-31-787-7017, Fax: + 82-31-787-4051, E-mail: woltoongac.kr.
Corresponding author: Jin-Hyeok Hwang, MD, PhD. Department of Internal Medicine, Seoul National University Bundang Hospital 82, Gumi-ro 173 Beon-gil, Bundang-gu, Seongnam-si, Gyeonggi-do 13620, Republic of Korea. Tel.: + 82-31-787-7017, Fax: + 82-31-787-4051, E-mail: woltoongac.kr.