Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(4):954-964. doi:10.7150/jca.50137 This issue Cite
Review
Autophagy-mediated tumor cell survival and progression of breast cancer metastasis to the brain
Aparna Maiti ![]() , Nitai C. Hait
, Nitai C. Hait ![]()
Division of Breast Surgery and Department of Surgical Oncology, Department of Molecular & Cellular Biology, Roswell Park Comprehensive Cancer Center, Buffalo, New York, 14263, USA.
Received 2020-6-30; Accepted 2020-10-7; Published 2021-1-1
Abstract
Brain metastases represent a substantial amount of morbidity and mortality in breast cancer (BC). Metastatic breast tumor cells committed to brain metastases are unique because they escape immune surveillance, can penetrate the blood-brain barrier, and also adapt to the brain tissue microenvironment (TME) for colonization and outgrowth. In addition, dynamic intracellular interactions between metastatic cancer cells and neighboring astrocytes in the brain are thought to play essential roles in brain tumor progression. A better understanding of the above mechanisms will lead to developing more effective therapies for brain metastases. Growing literature suggests autophagy, a conserved lysosomal degradation pathway involved in cellular homeostasis under stressful conditions, plays essential roles in breast tumor metastatic transformation and brain metastases. Cancer cells must adapt under various microenvironmental stresses, such as hypoxia, and nutrient (glucose) deprivation, in order to survive and progress. Clinical studies reveal that tumoral expression of autophagy-related proteins is higher in brain metastasis compared to primary breast tumors. In this review, we outline the molecular mechanisms underlying autophagy-mediated BC cell survival and metastasis to the brain.
Keywords: breast cancer, brain metastases, astrocytes, autophagy, cell survival
Introduction
Metastasis is a complex interlinked multi-step process and accounts for 90% of BC mortality [1, 2]. Breast cancer brain metastasis (BCBM) occurs in approximately 15-30% of women with late-stage BC, and it is often seen in women with more aggressive cancers such as triple-negative or HER2+ [3].
Available therapies for brain metastasis include whole-brain radiation therapy (WBRT) and stereotactic radiosurgery (SRS) [4]. Although SRS offers the additional ability to treat tumors in more of a normal tissue sparing manner compared to WBRT, however, combining the two techniques have been shown to improve survival and local tumor control in select patients. Current clinical management of brain metastases has limitations in controlling the disease and can impair neurocognitive function, with patient's survival less than 12 months after diagnosis [5]. Thus, this underscores the need to understand this brain-specific metastatic process to better identify women at risk of developing brain metastasis, and finding ways their occurrence altogether. Therefore, an urgent need exists to identify new genetic signatures and underlying mechanisms of BCBM and to discover novel molecular targets to prevent this process.
In BC, it may take years or decades for cancer cells to precede distant relapse [6, 7], suggesting BC cells initially lack the full competence for outgrowth in distant organs until they slowly they get modified to survive in secondary sites. Metastatic tumor cells must first overcome the blood-brain barrier (BBB), which acts as a selective interface between the peripheral circulation and the central nervous system [8, 9]. Metastatic tumor cells must adapt themselves to a different microenvironment from that of the primary site [10, 11]. How breast tumor metastatic cells adapt a specific niche in the brain for their self-renewal is an intriguing question for therapeutic and prognostic purposes. Although this complex topic requires further investigations, several pieces of evidence report on the role of autophagy in different stages of breast tumor metastasis [12-14].
In this review, we discuss that autophagy may be the mechanism responsible for metastatic cancer cells undergoing adaptation to escape all adverse environments. With the help of astrocytes, metastatic cancer cells colonize in the brain.
Autophagy helps cancer cells to be resistant to stressors
Macroautophagy (autophagy) is a well-established conserved mechanism that targets intracellular components for degradation and recycles protein and organelles to generate amino acids, nucleotides, fatty acids, sugars, and ATP to survive under various stresses, such as hypoxia and nutrient (glucose) deprivation [15, 16]. Growing literature suggests autophagy plays a complex role in tumorigenesis [17]. Autophagy has been implicated and plays a crucial role in preserving physiological tissue homeostasis by multiple mechanisms, including enhancing metabolic/redox homeostasis, and maintaining stemness by preventing senescence, with these effects suppressing malignant transformation [18-20]. Most early disseminated cancer cells are detected in the bone marrow of BC patients and have a putative BC stem cell phenotype [21]. Autophagy is shown to act as both pro-metastatic and anti-metastatic factors [13, 22-24]. In the early stages of tumor metastasis, autophagy has been shown to be linked with anti-metastasis via the limitation of cancer necrosis and inflammatory responses [25, 26].
However, recent studies have suggested that autophagy promotes multiple steps in tumor progression, survival, and colonization in distal organs [13, 22, 27, 28]. Autophagy is activated in the central part of solid tumors, where cells are exposed to extremely stressful conditions, including hypoxia and nutrient deprivation. Autophagy helps cancer cells to overcome these stresses [29-31]. During nutrient deprivation, autophagy is enhanced and acts to promote tumor survival and growth in advanced cancers [32, 33]. Autophagy manages the high metabolic demands of proliferating tumors by recycling intracellular components to supply metabolic substrates [34, 35]. Therefore, autophagy contributes to tumor-cell survival by enhancing the nutrient supply to meet metabolic demands affording them stress tolerance. Oxidative stress responses involving PKR-like endoplasmic reticulum kinase (PERK)-autophagy survival signaling has been shown to contribute to mammary gland development and tumorigenesis (reviewed in [36]). Hyperactivation of the mammalian target of rapamycin complex 1 (mTORC1) promotes BC progression through enhancing glucose starvation-induced autophagy and AKT signaling [37]. One of the crucial regulators of autophagy is mTORC1, linked with tumor progression, metastasis, and metabolism [38]. Published results have implicated mitochondrial complex I activity modulates AKT/mTORC1-mediated autophagy and BC progression and metastasis to the distant organs, including the brain [39].
Other published papers show that autophagy markers are easily detected in ductal carcinoma in situ lesions from human patients [40, 41]. Further observations suggest that autophagy regulation and biology in BC could be more complicated. Published results have also implicated defective autophagy in breast tumors could also enhance tumorigenesis [18, 42, 43]. On the other hand, the inhibition of autophagy or knockdown of autophagy genes can also result in tumor-cell death [44], suggesting a very promising target in BC treatment [43]. Several studies have revealed that anticancer drug resistance increases via the upregulation of autophagy, which functions as a protective mechanism in cancer cells undergoing anticancer therapy [45, 46]. Inflammation is also induced by autophagy in the cancer microenvironment and cancer adjacent cells, leading to cancer progression. Inflammatory regulators are increased in the tumor microenvironment, which contributes to tumorigenesis [47]. Some studies show that inflammation triggers high levels of reactive oxygen species (ROS) in cancer cells, macrophages, and other immune cells. These cells are in terms of secrete chemokines and cytokines, including interleukin-6 (IL-6), tumor necrosis factor-α (TNFα), interleukin-10 (IL-10), and transforming growth factor-β (TGFβ) into the tumor microenvironment (TME) [48]. These cytokines play crucial roles in maintaining chronic inflammation, which contributes to cancer progression via inflammation [49]. The multifaceted roles of autophagy have been implicated in modulating breast tumor cell motility and invasion [50-54], cancer stem cell viability and differentiation [41, 55, 56], tumor cell dormancy [20], and escape from immune surveillance [57], with emerging functions in establishing the pre-metastatic niche (reviewed in [13]). One recent study suggested disseminated dormant BC cells survive and metastasize to distant organs via the autophagy process. Furthermore, results have implicated that inhibition of autophagic flux in the dormant BC cells enhances the accumulation of damaged mitochondria and ROS, which results in programmed cell death [58]. Earlier studies using the mammosphere formation by BC cells have suggested that the autophagy-related gene ATG4A promotes a BC stem-like phenotype [59]. Autophagy proteins LC3A and LC3B expressions are a common feature of solid tumors and are associated with proliferation, metastasis, and poor outcome [54, 60]. The presence of LC3B puncta and HMGB1 gene expression in the malignant cells is found to be correlated with immune infiltration associated with BC progression and predicted residual risk of relapse after adjuvant chemotherapy [57, 61].
Another study suggests that induction of autophagy genes, ATG7 and conversion of autophagy marker (LC3A to LC3B), and tetraspanin proteins (CD9, CD63, CD81), are useful biomarkers and provide novel therapeutic targets for the treatment and prevention of cancer [62]. In agreement, one recent study suggested upregulated expression of ATG7 in BC tissue is significantly associated with reduced overall survival (OS), relapse-free survival (RFS), and distant metastasis-free survival (DMFS) in BC patients [63]. Another recent study was performed to assess the significance of the expression of autophagy markers, including beclin-1 (BECN1), LC3A, LC3B, and p62 in the molecular subtypes of TNBC patient samples using tissue microarray analysis. Data suggested that among the subtypes of TNBC, the molecular apocrine TNBC subtype showed a higher expression of nuclear p62 and beclin-1 than other markers, which indicated higher autophagy activity [64]. Further follow-up studies by another group have suggested that autophagy-related markers are overexpressed in the TNBC patient samples using immunohistochemical analysis [65]. In agreement, these pieces of evidence suggest LC3B may play a critical function in the progression of TNBC and could be a useful marker in prognostic evaluation for patients with TNBC [66]. Gene expression omnibus database (http://www.ncbi.nlm.nih.gov/geo/) analysis has identified a set of eight autophagy-related genes (BCL2, BIRC5, EIF4EBP1, ERO1L, FOS, GAPDH, ITPR1 and VEGFA) associated with a prognostic signature of BC patients [67]. One recent study suggested that LC3B and ATG17 elevated expression in brain metastases are correlated with significantly shorter survival time in TNBC patients [68]. These results are in line with a strategy to target autophagy in the treatment of brain metastases in BC patients. Some essential genes and gene products participating in the process of autophagy and BCBM, and their functions, are summarized in Table 1. Collectively, these observations suggest that the autophagic signaling network is involved in BC cell survival under stressful conditions and tumor metastasis.
Pivotal genes/proteins participating in the process of autophagy and BCBM
| Genes/Proteins | Autophagy in BC | BC | BCBM | Functions | References |
|---|---|---|---|---|---|
| Beclin 1 | Lethal autophagy | Tumorigenesis | Not known | Heterozygous disruption of the beclin 1 is found in human BC, a tumor suppressor. It also enhances BC stem-like/progenitor cells. | [69, 70] |
| ATG4A | Survival autophagy | Tumorigenesis | Not known | ATG4A promotes BC stem-like phenotype. | [59] |
| ATG5 | Lethal autophagy | Inhibition of tumorigenesis | Reduced BCBM | Downregulation of ATG5-dependent macroautophagy linked with BC growth and metastasis to the brain. | [39, 71] |
| ATG3, ATG7 | Survival autophagy | Tumorigenesis | Not known | Promotes the survival of dormant BC cells and metastatic tumor recurrence. | [58, 72] |
| LC3 | Survival autophagy | Tumorigenesis | Not known | TNBC cell proliferation, survival, migration, and invasion, and may contribute to tumor growth and progression. | [73] |
| LC3B | Survival autophagy | Tumorigenesis | Elevated in BCBM | Elevated levels are linked with shorter survival in TNBC patients. | [68] |
| P62/SQSTM1 | Lethal autophagy | Tumorigenesis | Not known | Interacts with vimentin to enhance BC metastasis. Metastatic dormancy of BC. | [72, 74] |
| ULK1 | Lethal autophagy | Inhibition of tumorigenesis | Not known | ULK1 phosphorylates Exo70 to suppress BC metastasis. Prognostic marker of BC patients. | [75, 76] |
| BIRC5 | Lethal autophagy | Inhibition of tumorigenesis | Not known | Levels of BIRC5 and ATG7 were inversely correlated. A novel ATG12-ATG5 conjugate interactor. It induces lethal autophagy in BC cells. | [77] |
| MTA1 | Survival autophagy | Tumorigenesis | Not known | A novel regulator of autophagy induces tamoxifen resistance in BC cells. | [78] |
| CEMIP and BiP | Survival autophagy | Tumorigenesis | Not known | Overexpressed, protective autophagy is observed in response to hypoxia, associated with tumor progression. | [79] |
| KISS1 | Lethal autophagy | Inhibition of metastases development | Reduced expression of KISS1, enhances BCBM | Astrocytes promote BCBM via KISS1-mediated autophagy | [80, 81] |
| GRP94 | Survival autophagy | Tumorigenesis | Induced BCBM | It is overexpressed in BCBM, involved in autophagy-mediated survival in endoplasmic reticulum stress. | [82] |
| FIP200/ATG17 | Survival autophagy | Tumorigenesis | Protein expression higher in BCBM | A significant correlation is observed between higher FIP200/ATG17 expression in primary-BC tumors and shorter disease-free survival. | [68] |
| ST6GALNAC5 | Not known | Tumorigenesis | Induced BCBM | It enhances BC cell adhesion to brain endothelial cells and their passage through the BBB. | [83] |
| Semaphorin 4D (SEMA4D) | Not known | Tumorigenesis | Induced BCBM | Semaphorin 4D/plexin B1 interactions mediate BCBM by promoting CTC transmigration through the BBB. | [84] |
| PTEN | Lethal autophagy | Inhibition of metastases development | Downregulated in BCBM | PTEN loss and elevated PI3K/AKT signaling are found in BCBM patients. | [85] |
Crossing the blood-brain barrier
The metastatic cascade can be divided into two major phases (i) physical translocation of a cancer cell from the primary tumor to the microenvironment of a distant tissue and (ii) colonization. During physical translocation, cancer cells acquire invasive phenotypes, invade the surrounding matrix, and entering the lymph or blood circulation system [1]. The bone marrow of patients with BC correlates with increased metastatic burden, aggressiveness, and reduced survival [86]. Animal studies have shown that only a tiny percentage of tumor cells are capable of completing the intricate steps of metastasis, the most limiting of which is the colonization of tumor cells at distant sites [87]. Autophagy is also a pro-metastatic cancer cell survival factor [13]. In order to undergo metastasis, cancer cells must be able to survive and proliferate in the circulatory system and disseminate to secondary sites as well. Therefore, a subset of invasive circulating tumor cells (CTC) that survive the circulation is capable of extravasation through the BBB for metastatic colonization in the brain [88, 89]. Cancer cell death undergoes apoptosis due to loss of extracellular matrix (ECM) attachment, called anoikis. Substantial evidence indicates that autophagy enables ECM-detached cancer cells to avoid anoikis and survive [90].
The ability of tumor cells to initiate growth in the brain is dependent mostly on cross-talk between tumor and brain resident cells. A clear understanding of the biological mechanism of the brain microenvironment and how cancer cells change themselves to survive in that environment is critical to tackling the clinical challenges of brain metastasis. As metastatic cancer cells arrive in the brain's vasculature, these cells are challenged to cross the BBB that separates the brain from the general circulation [91]. The BBB is surrounded by a thick basement membrane supported by pericytes, which are fibroblast-like cells that are wrapped around endothelial cells connected by tight junctions. Finally, astrocytes form the outer layer of the BBB [92]. The BBB's tight junctions are composed of a variety of proteins, including claudins and occludins [93], and possess high electrical resistance [94]. The primary function of this barrier is the maintenance of the homeostatic environment for neuronal function [93].
It has been suggested that cancer cell-derived marker proteins are the prognostic factors for organotropic metastasis to the brain [95]. In agreement with this concept, recent studies have suggested that brain-specific gene expressions, such as SEMA4D, and mechanisms in the BC CTCs can transmigrate through BBB [84]. Other studies have identified genes from BC clinical samples, such as cyclooxygenase COX2 (also known as PTGS2), the epidermal growth factor receptor (EGFR) ligand HBEGF, and the brain-specific 2,6-sialyltransferase ST6GALNAC5 as mediators of BC cell passage through the BBB [83]. Cancer cells need to secrete matrix metalloproteinases (MMPs) and other proteases to disrupt the basement membrane and tight junctions as occurs in metastasis to other distant sites [96-98]. It has been demonstrated that secretion of MMP2, MMP3, and MMP9 by metastatic BC cells is playing a crucial role in BCBM [99]. Cancer cells and tumor-associated cells, such as macrophages, produce a variety of mediators, which facilitate endothelial cell junction openings [100]. It has been shown that secreted vascular endothelial growth factor (VEGF) and TGF-β induce opening the endothelial junctions by disrupting the VE‑cadherin-β‑catenin complex [101-103].
A variety of cytokines, chemokines can also destabilize these junctions and inflammatory mediators frequently expressed by cancer cells and tumor-associated cells, including interleukin-1β (IL-1β), TNF-α, interferon-γ (IFN-γ), CCL2, and CXCL8 (reviewed in [104]). The interaction between surviving cancer cells and the brain microenvironment plays a crucial role in cancer cell progression to the brain (reviewed in [105]). Cancer-derived extracellular vesicles (EVs), including exosomes, are known to mediate cell-cell communications via delivery of their contents, including proteins, mRNAs, and microRNAs (miRNAs) [106], and are reported capable of breaking down the BBB through modulating actin dynamics and promote brain metastasis [107]. One such important recent study suggested that brain metastasis of BC cells or lung tumor cell-derived exosomes enriched with a Wnt-related protein, CEMIP, are shown to promote brain metastasis. Exosomal CEMIP upregulated pro-inflammatory mediators such as TNF, PTGS2, and CCL/CXCL in microglia, which promote BBB dysfunction and brain metastasis [108]. As indicated [95, 109], brain metastasis is a complex process that is only recently beginning to be understood, that may require activation of multiple pathways to cross the BBB and adapt to the brain environment, that has potential metastatic cancer cells crossing the BBB through mediator signaling and cell-cell interaction in the brain microenvironment to survive and colonize the brain.
Astrocytes cross-talk with the cancer cells to pass the barrier
Host cells in the microenvironment influence metastasis of tumor cells. Breast tumor metastasis represents a complex interaction between tumor cells and brain endothelial cells, pericytes, microglia, M2 macrophages, and astrocytes. These different cells are capable of tipping tumor biology toward cell extravasation and influencing the biologic behavior of metastatic cell growth and progression by exhibiting and favoring elevated autophagy. Brain metastasis of BC cells is highly adaptable in the brain by expressing brain-specific gene products and mediators that mediate survival in the brain microenvironment [83]. There is an urgent need to identify brain-specific new targets for developing novel therapeutics to prevent BCBM. The tumor microenvironment in the brain is represented predominantly by normal astrocytes in the brain tissues [110, 111]. Astrocytes are one of the subtypes of glial cells that become activated in response to central nervous system (CNS) stresses. In the brain environment, the homeostatic function of astrocytes includes supporting the BBB [111, 112], delivering nutrients to neurons [113], regulating the brain response to inflammation [114], and protecting neurons from hypoxia [115].
Astrocytes express glucose transporters for allowing glucose to enter into the brain and glycoproteins for pumping drugs and other compounds out of the brain. They also possess the capability to modify the tightness of the BBB in response to stresses like hypoxia [116]. After escaping elimination by astrocytes, cancer cells can take advantage of the protective benefits that astrocytes usually support neurons by a variety of mechanisms. Brain metastatic cancer cells can activate the pro-growth and survival effects of astrocytes by secretion of IL-1β [117] or CCL2 [118], and have been shown to enhance the differentiation of neural progenitor cells into astrocytes [119]. Reactive astrocytes have altered gene expression patterns, including the up-regulation of Glial Fibrillary Acidic Protein (GFAP), which have been frequently observed in metastatic brain tumors in different animal models and human patients ([8, 120], and reviewed in [110, 121]. The reciprocal cross-talk between brain metastatic cancer cells and astrocytes promote outgrowth and stimulate the therapeutic resistance of cancer cells in the brain [122]. The report by Kim et al. [123] showed the first evidence on the role of the interaction of BC cells with astrocytes in promoting brain metastatic transformation (through the up-regulation of different pro-survival genes, including those stimulating autophagy). These cell-cell interactions and cancer cell survival are associated with increased resistance to various chemotherapeutics.
A better understanding of therapeutic resistance and brain metastasis lies in the assimilated function of astrocytes. Numerous studies have demonstrated that a variety of factors produced and secreted by reactive astrocytes can support metastatic cancer cell growth and progression in the brain. These include neurotrophic factors such as TGF-α, ATP, endothelin-1 (ET-1), glutamate, IL-1β, IL-6, TNF-α, macrophage inflammatory protein MIP-2, CCL2, CXCL12, glial cell-derived neurotrophic factor (GDNF), NO, and bioactive lysophospholipid sphingosine-1-phosphate (S1P) [80, 112, 124], and reviewed in [110, 125]. The lipid mediator S1P is produced inside cells by the sphingosine kinases (SphK1 and SphK2) and involved in inflammation and cancer progression [126, 127]. However, S1P has complex roles in the tumor microenvironment. The brain has the highest levels of S1P, which is predominantly produced by the sphingosine kinase 2 (SphK2) [128, 129]. It has been shown cerebellar astrocytes produce S1P in response to bFGF and induces proliferation of astrocytes via extracellular signaling through G-protein coupled receptors (S1PRs) [130].
One recent study suggests that extracellular S1P signaling through S1P receptor 3 (S1PR3) mediates its effect through astrocytic secretion of IL-6 and CCL2, and relaxes endothelial cell adhesion. Astrocytic neuroinflammatory signaling through S1PR3 has been documented to modulate BBB permeability that enhances BCBM [109, 131]. This study is important because it suggests that abnormal angiogenesis-related signaling through S1PR3 in cancer cells or astrocytes could impact the permeability of the BBB and provides a potential therapeutic target of BCBM. Another study demonstrated that astrocytes promote brain metastasis of BC by upregulating autophagy signaling pathways via the CXCL12-MIR345-KISS1 axis [80, 132]. BCBM progression exhibits low levels of KISS1 expression. Recently published data suggests that KISS1 expression downregulated ATG5/7-mediated survival autophagy and BCBM [80]. One published study suggested that KISS1 expression is significantly higher in primary BC compared with brain metastases [81]. Together, these studies have suggested that KISS1-mediated survival autophagy is a potential diagnostic marker for BCBM, as well as a new anticancer therapeutics. Cancer cell and astrocyte gap junction communications have shown influencing barrier permeability associated with promoting cancer cell metastasis to the brain [133]. Cancer cells express protocadherin 7 (PCDH7), promote carcinoma-astrocyte gap junctions, which then induces IFNα and TNF production and activation of STAT1 and NF-κB pathways in cancer cells to support brain metastasis [133].
Metabolic adaptation of cancer cells to the brain microenvironment
Metastatic BC cells must overcome the metabolic adaptation challenges to survive and colonize in the brain microenvironment. It has been proposed that tumor cells acquire a metabolic signature adapted for survival at particular metastatic sites, which dictate where they are able to form distal colonies. This hypothesis is consistent with the growing body of evidence showing that the more metabolically flexible the primary tumor cells are, the more likely they are to survive the metastatic process and thrive in distant organs. The mammalian brain utilizes glucose as the primary energy source for its physiological functions, where astrocytes consume glucose, and neurons preferentially take up and oxidize lactate over glucose-derived pyruvate [134]. It has been postulated that primary breast tumors that undergo the Warburg effect [135] of metabolic reprogramming become suited to growth in the brain microenvironment [136]. Brain metastatic cancer cells exhibit altered gluconeogenesis and oxidation of glutamine/branched-chain amino acids as alternative energy sources for their adaptation in the foreign low-glucose environment [137, 138]. They possess an enhanced ability to utilize glutamine as an energy source and as a precursor to cellular building blocks, including purines, pyrimidines, and non-essential amino acids [139, 140]. However, it is unknown whether metastatic brain cells possess an intrinsic ability to utilize glutamine better or is a modified phenotype for the adaptation to the brain environment. A mechanistic understanding of the regulation and cause of this increased glutamine utilization can serve as a therapeutic target for BCBM. One recent study discovered glucose-regulated protein GRP94 over-expression is critical for BCBM, which relives metabolic stress in low-glucose conditions by coordinating the unfolded protein response and pro-survival autophagy [82]. These studies suggest autophagy-mediated mechanisms could be a potential therapeutic target in BCBM for designing new therapies.
Targeting autophagy in BC metastasis
Autophagy and regulatory machinery signaling in cancer are explained in many recent reviews [12, 13, 141]. As mentioned earlier, the dual roles of autophagy have been documented in BC as well. The loss of autophagy molecule BECN1 is a frequent event in many human tumors, including breast [69], and is associated with the promotion of tumorigenesis [142]. Conversely, overexpression of BECN1 in the MCF-7 BC cell line decreases tumorigenesis [42]. In contrast, autophagy has been shown to enhance breast tumor progression and metastasis. For example, published results suggest that mammary epithelial conditional knockout of the essential autophagy protein FIP200 has reduced tumorigenesis and metastasis in the MMTV-PyMT mouse model [44, 143]. Another study suggests that DNA damage and oxidative stress-mediated BC cell survival occur through autophagy and the involvement of p53 [144]. Recent studies suggest that the effector of the Hippo signaling pathway yes-associated protein (YAP) is involved in promoting autophagy-mediated TNBC cell survival and metastatic properties [145, 146]. Altering autophagic flux [141] by manipulating autophagy signaling has been recognized as an attractive cancer therapy strategy.
Autophagy is induced by almost all conventional BC treatments and is considered a potential pharmacological target in the clinic. Autophagy inhibitors chloroquine (CQ) or hydroxychloroquine (HCQ) is in clinical trials for solid cancer therapy. One phase II trial suggested that the combined treatment of mTOR inhibitor everolimus and HCQ increased six-month median progression-free survival (PFS) in patients with BC [147]. It has been suggested that anti-inflammatory polyphenolic stilbenoid resveratrol [148] induces autophagy, inhibits cancer stem cell proliferation, and suppresses the Wnt/beta-catenin pathway in BC cells [149]. Resveratrol has also been shown to induce BECN1 independent autophagy-mediated cell death in MCF-7 cells, suggesting a novel mechanism of cell death in BC cells [150].
The phosphatidylinositol 3-kinase (PI3K)/AKT pathway, which is one of the significant survival pathways, is highly active in BC patients due to frequent mutation in the PI3K gene [151]. Autophagy activators such as PI3K/mTOR inhibitors can also enhance cell cycle arrest and potential therapeutic strategies for treating metastatic BC patients [141]. Everolimus (mTOR inhibitor) was the first rapamycin analog approved in combination with endocrine therapy for patients with hormone receptor (HR)-positive metastatic BC [151]. Everolimus and aromatase inhibitor exemestane have significantly enhanced OS in HR-positive metastatic BC patients [152, 153]. The Food and Drug Administration (FDA) has already approved these strategies for treating advanced metastatic BC patients. Other autophagy activators such as inhibitors of mammalian target of rapamycin complex (mTORC)-1 and mTORC2 provide promising results for treating metastatic breast tumors [151, 154, 155].
Cyclin-dependent kinase (CDK)-4/6 inhibitors interrupt cell division, and cell growth is the FDA approved drug for treating patients with metastatic BC. CDK4/6 inhibition also induces senescence and autophagy in BC cells, and blocking autophagy sensitizes BC cells to CDK4/6 inhibition, and enhances programmed cell death. These data suggest potential strategies to overcome the limitation of CDK4/6 inhibition in the clinic for metastatic BC patients [156, 157]. The recent preclinical study also suggested that CDK4/6 inhibition in combination with mTOR inhibition effectively reduces metastatic triple-negative breast tumor growth in a mouse model [158], suggesting these novel strategies are essential for further investigation in the clinical setting. Metastatic colonization of the aggressive TNBC or HER2+ BC cells in the brain includes amplifying the epidermal growth factor receptor (EGFR) in the BC cells [159]. Amplification of EGFR is often associated with loss of phosphatase and tensin homolog (PTEN), a positive regulator of downstream EGFR effectors, including PI3K/AKT pathways, which not only promotes cell survival, but also regulates c-Myc to facilitate metabolic reprogramming [160, 161]. PTEN loss and elevated PI3K/AKT signaling were found in brain metastasis formation in BC patients [85, 87], suggesting that targeting autophagy and PI3K could be a potential strategy to treat BCBM patients.
Conclusion
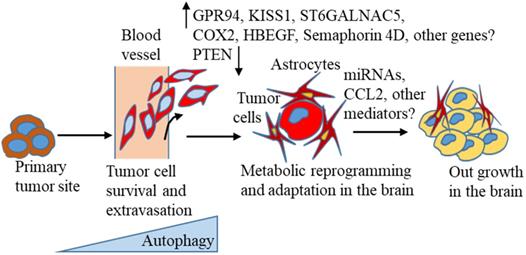
A large body of recent evidence suggests that autophagy is activated in tumor progression and different stages of the metastasis process [141, 162]. Thus, targeting this pathway could be useful in treating BC distant metastasis, including BCBM. However, in reality, autophagy-targeting drugs have yielded contradictory or limited results, as summarised in this recent review [141], suggesting a precise understanding of the autophagic pathway related to BC progression and distant metastasis, including BCBM is extremely necessary. Despite the increasing incidence of brain metastasis, knowledge about the molecular mechanisms of brain metastasis development, and optimal treatment strategies is limited and less investigated than other metastasis formation. New markers for predicting BCBM occurrence in the primary tumors are urgently needed for early detection of the possibility of a high risk of brain lesion formation. To turn brain metastasis into a treatable disease, researchers need to understand the molecular changes that occur, as proposed in the context of autophagy (Figure 1), based on the current literature that suggests breast tumors escape from the primary site, evade immune surveillance, and finally, colonize successfully in the brain environment.
Autophagy-mediated cell survival under stress is involved in the invasiveness of BC cells. This contributes to cancer cell intravasation, survival in the circulation, escape from immune surveillance, and extravasation of cells [13, 14]. Organotropic gene expression, gene signatures, and metabolic advantages are shown that mediate BC metastasis to the brain [82-84, 132, 136, 163]. Autophagy-mediated metabolic adaptation, interaction with astrocytes, and mediator signaling [109, 132, 136, 138, 163, 164] are also proposed to modify cancer cells, which are able to establish and grow in the brain environment.
Abbreviations
ATP: adenosine triphosphate; ATGs: autophagy-related genes; BC: breast cancer; BBB: blood-brain barrier; BCBM: breast cancer brain metastasis; BEN1: beclin-1; BCL2: B-cell lymphoma 2; BIRC5: baculoviral IAP repeat-containing protein 5; BiP: binding immunoglobulin protein; bFGF: basic fibroblast growth factor; CTC: circulating tumor cells; CCL2: C-C motif chemokine ligand 2; CXC: C-X-C motif chemokine ligand 8; CD: cluster of differentiation; CNS: central nervous system; CEMIP: cell migration-inducing protein; CQ: chloroquine; CDK: cyclin-dependent kinase; DMFS: distant metastasis free survival; EGFR: epidermal growth factor receptor; ET-1: endothelin-1; ECM: extracellular matrix; EIF4EBP1: eukaryotic translation initiation factor 4E binding protein 1; ERO1L: endoplasmic reticulum oxidoreductase 1 alpha; EVs: extracellular vehicles; FIP200: FAK family-interacting protein of 200 kDa; FOS: Fos proto-oncogene; FDA: Food and Drug Administration; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; GRP94: glucose-regulated protein 94; GFAP: glial fibrillar acidic protein; GDNF: glial cell-derived neurotrophic factor; HR: hormone receptor; HER2: human epidermal growth factor receptor 2; HMGB1: high mobility group box 1; HCQ: hydroxychloroquine; IL-6: interleukin-6; IL-10: interleukin-10; IL-1β: interleukin-1β; IFN: interferon; ITPR1: inositol 1,4,5-trisphosphate receptor type 1; KISS1: kisspeptin 1; LC3A: light-chain 3A; LC3B: light-chain 3B; mTOR: mammalian target of rapamycin; MTA1: metastasis-associated 1; mTORC1: mammalian target of rapamycin complex 1; miRNAs: MicroRNAs; MIR345: MicroRNA 345; MMPs: matrix metalloproteinases; MIP-2: macrophage inflammatory protein 2; MMTV-PyMT: mouse mammary tumor virus-polyoma virus middle T antigen; NO: nitric oxide; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; OS: overall survival; PFS: progression-free survival; PERK: PKR-like endoplasmic reticulum kinase; PTEN: phosphatase and tensin homolog; PI3K: phosphatidylinositol 3-kinase; PCDH7: protocadherin 7; PTGS2: prostaglandin-endoperoxide synthase 2; ROS: reactive oxygen species; RFS: relapse free survival; RNA: ribonucleic acid; ST6GALNAC5: ST6 N-acetylgalactosaminide alpha-2,6-sialyltransferase 5; S1P: sphingosine-1-phosphate; SphK2: sphingosine kinase 2; S1PRs: S1P receptors; S1PR3: S1P receptor 3; STAT1: signal transducer and activator of transcription 1; SRS: stereotactic radiosurgery; TNBC: triple-negative breast cancer; TNFα: tumor necrosis factor-α; TGF: transforming growth factor; TME: tumor microenvironment; ULK1: Unc-51 like autophagy activating kinase 1; VEGFA: vascular endothelial growth factor A; WBRT: whole-brain radiation therapy; Wnt: wingless-type MMTV integration site family member; YAP: Yes-associated protein.
Acknowledgements
Supported by the Roswell Park Health Research Incorporated (HRI) Start Up Funds #714084-01 (NCH). The authors would like to thank Dr. Suzanne Hess (Roswell Park Comprehensive Cancer Center) for critical review of the manuscript.
Author contributions
Conceptualization, writing-review, and editing, AM and NCH. All the authors have reviewed and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science (New York, NY). 2011;331:1559-64
2. Jin X, Mu P. Targeting Breast Cancer Metastasis. Breast Cancer (Auckl). 2015;9:23-34
3. Bachmann C, Schmidt S, Staebler A, Fehm T, Fend F, Schittenhelm J. et al. CNS metastases in breast cancer patients: prognostic implications of tumor subtype. Med Oncol. 2015;32:400
4. Kaal EC, Niel CG, Vecht CJ. Therapeutic management of brain metastasis. Lancet Neurol. 2005;4:289-98
5. Chargari C, Campana F, Pierga JY, Vedrine L, Ricard D, Le Moulec S. et al. Whole-brain radiation therapy in breast cancer patients with brain metastases. Nat Rev Clin Oncol. 2010;7:632-40
6. Schmidt-Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ. et al. From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci U S A. 2003;100:7737-42
7. Karrison TG, Ferguson DJ, Meier P. Dormancy of mammary carcinoma after mastectomy. J Natl Cancer Inst. 1999;91:80-5
8. Lorger M, Felding-Habermann B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am J Pathol. 2010;176:2958-71
9. Wrobel JK, Toborek M. Blood-brain Barrier Remodeling during Brain Metastasis Formation. Mol Med. 2016;22:32-40
10. Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239-52
11. Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends in cell biology. 2015;25:198-213
12. Mowers EE, Sharifi MN, Macleod KF. Functions of autophagy in the tumor microenvironment and cancer metastasis. FEBS J. 2018;285:1751-66
13. Mowers EE, Sharifi MN, Macleod KF. Autophagy in cancer metastasis. Oncogene. 2017;36:1619-30
14. Dower CM, Wills CA, Frisch SM, Wang HG. Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy. 2018;14:1110-28
15. Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms. Autophagy. 2018;14:207-15
16. Cheon SY, Kim H, Rubinsztein DC, Lee JE. Autophagy, Cellular Aging and Age-related Human Diseases. Exp Neurobiol. 2019;28:643-57
17. Rybstein MD, Bravo-San Pedro JM, Kroemer G, Galluzzi L. The autophagic network and cancer. Nat Cell Biol. 2018;20:243-51
18. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY. et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062-75
19. Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S. et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621-35
20. Garcia-Prat L, Martinez-Vicente M, Perdiguero E, Ortet L, Rodriguez-Ubreva J, Rebollo E. et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37-42
21. Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G. et al. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin Cancer Res. 2006;12:5615-21
22. Kenific CM, Thorburn A, Debnath J. Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol. 2010;22:241-5
23. Debnath J. The multifaceted roles of autophagy in tumors-implications for breast cancer. J Mammary Gland Biol Neoplasia. 2011;16:173-87
24. Avivar-Valderas A, Bobrovnikova-Marjon E, Alan Diehl J, Bardeesy N, Debnath J, Aguirre-Ghiso JA. Regulation of autophagy during ECM detachment is linked to a selective inhibition of mTORC1 by PERK. Oncogene. 2013;32:4932-40
25. Salah FS, Ebbinghaus M, Muley VY, Zhou Z, Al-Saadi KR, Pacyna-Gengelbach M. et al. Tumor suppression in mice lacking GABARAP, an Atg8/LC3 family member implicated in autophagy, is associated with alterations in cytokine secretion and cell death. Cell Death Dis. 2016;7:e2205
26. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P. et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011;334:1573-7
27. Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14:611-22
28. Langley RR, Fidler IJ. The seed and soil hypothesis revisited-the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer. 2011;128:2527-35
29. Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res. 2014;24:42-57
30. Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823-30
31. He L, Zhang J, Zhao J, Ma N, Kim SW, Qiao S. et al. Autophagy: The Last Defense against Cellular Nutritional Stress. Adv Nutr. 2018;9:493-504
32. Luo T, Fu J, Xu A, Su B, Ren Y, Li N. et al. PSMD10/gankyrin induces autophagy to promote tumor progression through cytoplasmic interaction with ATG7 and nuclear transactivation of ATG7 expression. Autophagy. 2016;12:1355-71
33. Liu M, Jiang L, Fu X, Wang W, Ma J, Tian T. et al. Cytoplasmic liver kinase B1 promotes the growth of human lung adenocarcinoma by enhancing autophagy. Cancer Sci. 2018;109:3055-67
34. Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344-8
35. Liu EY, Ryan KM. Autophagy and cancer-issues we need to digest. J Cell Sci. 2012;125:2349-58
36. Avivar-Valderas A, Wen HC, Aguirre-Ghiso JA. Stress signaling and the shaping of the mammary tissue in development and cancer. Oncogene. 2014;33:5483-90
37. Chen Y, Wei H, Liu F, Guan JL. Hyperactivation of mammalian target of rapamycin complex 1 (mTORC1) promotes breast cancer progression through enhancing glucose starvation-induced autophagy and Akt signaling. J Biol Chem. 2014;289:1164-73
38. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21-35
39. Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ. et al. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013;123:1068-81
40. Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J. et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol. 2011;31:3616-29
41. Espina V, Mariani BD, Gallagher RI, Tran K, Banks S, Wiedemann J. et al. Malignant precursor cells pre-exist in human breast DCIS and require autophagy for survival. PLoS One. 2010;5:e10240
42. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H. et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672-6
43. Zhou Y, Rucker EB 3rd, Zhou BP. Autophagy regulation in the development and treatment of breast cancer. Acta Biochim Biophys Sin (Shanghai). 2016;48:60-74
44. Liu F, Lee JY, Wei H, Tanabe O, Engel JD, Morrison SJ. et al. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood. 2010;116:4806-14
45. Xiao X, Wang W, Li Y, Yang D, Li X, Shen C. et al. HSP90AA1-mediated autophagy promotes drug resistance in osteosarcoma. J Exp Clin Cancer Res. 2018;37:201
46. Chen Z, Jiang Q, Zhu P, Chen Y, Xie X, Du Z. et al. NPRL2 enhances autophagy and the resistance to Everolimus in castration-resistant prostate cancer. Prostate. 2019;79:44-53
47. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K. et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921-5
48. Vaupel P, Mayer A. Hypoxia and anemia: effects on tumor biology and treatment resistance. Transfus Clin Biol. 2005;12:5-10
49. Lin WW, Karin M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175-83
50. Sharifi MN, Mowers EE, Drake LE, Collier C, Chen H, Zamora M. et al. Autophagy Promotes Focal Adhesion Disassembly and Cell Motility of Metastatic Tumor Cells through the Direct Interaction of Paxillin with LC3. Cell Rep. 2016;15:1660-72
51. Mowers EE, Sharifi MN, Macleod KF. Novel insights into how autophagy regulates tumor cell motility. Autophagy. 2016;12:1679-80
52. Lock R, Kenific CM, Leidal AM, Salas E, Debnath J. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014;4:466-79
53. Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S. et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 2014;4:914-27
54. Lazova R, Camp RL, Klump V, Siddiqui SF, Amaravadi RK, Pawelek JM. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res. 2012;18:370-9
55. Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Vellon L, Menendez JA. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle. 2011;10:3871-85
56. Han Y, Fan S, Qin T, Yang J, Sun Y, Lu Y. et al. Role of autophagy in breast cancer and breast cancer stem cells (Review). Int J Oncol. 2018;52:1057-70
57. Ladoire S, Enot D, Senovilla L, Ghiringhelli F, Poirier-Colame V, Chaba K. et al. The presence of LC3B puncta and HMGB1 expression in malignant cells correlate with the immune infiltrate in breast cancer. Autophagy. 2016;12:864-75
58. Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat Commun. 2018;9:1944
59. Wolf J, Dewi DL, Fredebohm J, Muller-Decker K, Flechtenmacher C, Hoheisel JD. et al. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013;15:R109
60. Sivridis E, Koukourakis MI, Zois CE, Ledaki I, Ferguson DJ, Harris AL. et al. LC3A-positive light microscopy detected patterns of autophagy and prognosis in operable breast carcinomas. Am J Pathol. 2010;176:2477-89
61. Ladoire S, Penault-Llorca F, Senovilla L, Dalban C, Enot D, Locher C. et al. Combined evaluation of LC3B puncta and HMGB1 expression predicts residual risk of relapse after adjuvant chemotherapy in breast cancer. Autophagy. 2015;11:1878-90
62. Kumar D, Gupta D, Shankar S, Srivastava RK. Biomolecular characterization of exosomes released from cancer stem cells: Possible implications for biomarker and treatment of cancer. Oncotarget. 2015;6:3280-91
63. Zhou J, Hang D, Jiang Y, Chen J, Han J, Zhou W. et al. Evaluation of genetic variants in autophagy pathway genes as prognostic biomarkers for breast cancer. Gene. 2017;627:549-55
64. Kim S, Jung WH, Koo JS. Differences in autophagy-related activity by molecular subtype in triple-negative breast cancer. Tumour Biol. 2012;33:1681-94
65. Choi J, Jung W, Koo JS. Expression of autophagy-related markers beclin-1, light chain 3A, light chain 3B and p62 according to the molecular subtype of breast cancer. Histopathology. 2013;62:275-86
66. Zhao H, Yang M, Zhao J, Wang J, Zhang Y, Zhang Q. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med Oncol. 2013;30:475
67. Gu Y, Li P, Peng F, Zhang M, Zhang Y, Liang H. et al. Autophagy-related prognostic signature for breast cancer. Mol Carcinog. 2016;55:292-9
68. Hashemi-Sadraei N, Muller-Greven GM, Abdul-Karim FW, Ulasov I, Downs-Kelly E, Burgett ME. et al. Expression of LC3B and FIP200/Atg17 in brain metastases of breast cancer. J Neurooncol. 2018;140:237-48
69. Negri T, Tarantino E, Orsenigo M, Reid JF, Gariboldi M, Zambetti M. et al. Chromosome band 17q21 in breast cancer: significant association between beclin 1 loss and HER2/NEU amplification. Genes Chromosomes Cancer. 2010;49:901-9
70. Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V. et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2013;32:2261-72 72e 1-11
71. Han Q, Deng Y, Chen S, Chen R, Yang M, Zhang Z. et al. Downregulation of ATG5-dependent macroautophagy by chaperone-mediated autophagy promotes breast cancer cell metastasis. Sci Rep. 2017;7:4759
72. La Belle Flynn A, Calhoun BC, Sharma A, Chang JC, Almasan A, Schiemann WP. Autophagy inhibition elicits emergence from metastatic dormancy by inducing and stabilizing Pfkfb3 expression. Nat Commun. 2019;10:3668
73. Hamurcu Z, Delibasi N, Gecene S, Sener EF, Donmez-Altuntas H, Ozkul Y. et al. Targeting LC3 and Beclin-1 autophagy genes suppresses proliferation, survival, migration and invasion by inhibition of Cyclin-D1 and uPAR/Integrin beta1/ Src signaling in triple negative breast cancer cells. J Cancer Res Clin Oncol. 2018;144:415-30
74. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1-222
75. Mao L, Zhan YY, Wu B, Yu Q, Xu L, Hong X. et al. ULK1 phosphorylates Exo70 to suppress breast cancer metastasis. Nat Commun. 2020;11:117
76. Tang J, Deng R, Luo RZ, Shen GP, Cai MY, Du ZM. et al. Low expression of ULK1 is associated with operable breast cancer progression and is an adverse prognostic marker of survival for patients. Breast Cancer Res Treat. 2012;134:549-60
77. Lin TY, Chan HH, Chen SH, Sarvagalla S, Chen PS, Coumar MS. et al. BIRC5/Survivin is a novel ATG12-ATG5 conjugate interactor and an autophagy-induced DNA damage suppressor in human cancer and mouse embryonic fibroblast cells. Autophagy. 2019 p: 1-18
78. Lee MH, Koh D, Na H, Ka NL, Kim S, Kim HJ. et al. MTA1 is a novel regulator of autophagy that induces tamoxifen resistance in breast cancer cells. Autophagy. 2018;14:812-24
79. Banach A, Jiang YP, Roth E, Kuscu C, Cao J, Lin RZ. CEMIP upregulates BiP to promote breast cancer cell survival in hypoxia. Oncotarget. 2019;10:4307-20
80. Kaverina N, Borovjagin AV, Kadagidze Z, Baryshnikov A, Baryshnikova M, Malin D. et al. Astrocytes promote progression of breast cancer metastases to the brain via a KISS1-mediated autophagy. Autophagy. 2017;13:1905-23
81. Ulasov IV, Kaverina NV, Pytel P, Thaci B, Liu F, Hurst DR. et al. Clinical significance of KISS1 protein expression for brain invasion and metastasis. Cancer. 2012;118:2096-105
82. Santana-Codina N, Muixi L, Foj R, Sanz-Pamplona R, Badia-Villanueva M, Abramowicz A. et al. GRP94 promotes brain metastasis by engaging pro-survival autophagy. Neuro Oncol. 2019
83. Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX. et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459:1005-9
84. Klotz R, Thomas A, Teng T, Han SM, Iriondo O, Li L. et al. Circulating Tumor Cells Exhibit Metastatic Tropism and Reveal Brain Metastasis Drivers. Cancer Discov. 2020;10:86-103
85. Wikman H, Lamszus K, Detels N, Uslar L, Wrage M, Benner C. et al. Relevance of PTEN loss in brain metastasis formation in breast cancer patients. Breast Cancer Res. 2012;14:R49
86. Braun S, Vogl FD, Naume B, Janni W, Osborne MP, Coombes RC. et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N Engl J Med. 2005;353:793-802
87. Witzel I, Oliveira-Ferrer L, Pantel K, Muller V, Wikman H. Breast cancer brain metastases: biology and new clinical perspectives. Breast Cancer Res. 2016;18:8
88. Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298-306
89. Zhang L, Ridgway LD, Wetzel MD, Ngo J, Yin W, Kumar D. et al. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci Transl Med. 2013;5:180ra48
90. Guadamillas MC, Cerezo A, Del Pozo MA. Overcoming anoikis-pathways to anchorage-independent growth in cancer. J Cell Sci. 2011;124:3189-97
91. Abbott NJ. Dynamics of CNS barriers: evolution, differentiation, and modulation. Cell Mol Neurobiol. 2005;25:5-23
92. Brown LS, Foster CG, Courtney JM, King NE, Howells DW, Sutherland BA. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front Cell Neurosci. 2019;13:282
93. Wolburg H, Lippoldt A. Tight junctions of the blood-brain barrier: development, composition and regulation. Vascul Pharmacol. 2002;38:323-37
94. Butt AM, Jones HC, Abbott NJ. Electrical resistance across the blood-brain barrier in anaesthetized rats: a developmental study. J Physiol. 1990;429:47-62
95. Yuzhalin AE, Yu D. Brain Metastasis Organotropism. Cold Spring Harb Perspect Med. 2019
96. Feng S, Cen J, Huang Y, Shen H, Yao L, Wang Y. et al. Matrix metalloproteinase-2 and -9 secreted by leukemic cells increase the permeability of blood-brain barrier by disrupting tight junction proteins. PLoS One. 2011;6:e20599
97. Boecke A, Carstens AC, Neacsu CD, Baschuk N, Haubert D, Kashkar H. et al. TNF-receptor-1 adaptor protein FAN mediates TNF-induced B16 melanoma motility and invasion. Br J Cancer. 2013;109:422-32
98. Sevenich L, Joyce JA. Pericellular proteolysis in cancer. Genes Dev. 2014;28:2331-47
99. Mendes O, Kim HT, Stoica G. Expression of MMP2, MMP9 and MMP3 in breast cancer brain metastasis in a rat model. Clin Exp Metastasis. 2005;22:237-46
100. Reymond N, d'Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858-70
101. Miles FL, Pruitt FL, van Golen KL, Cooper CR. Stepping out of the flow: capillary extravasation in cancer metastasis. Clin Exp Metastasis. 2008;25:305-24
102. Drabsch Y, ten Dijke P. TGF-beta signaling in breast cancer cell invasion and bone metastasis. J Mammary Gland Biol Neoplasia. 2011;16:97-108
103. Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56:549-80
104. Stamatovic SM, Keep RF, Andjelkovic AV. Brain endothelial cell-cell junctions: how to "open" the blood brain barrier. Curr Neuropharmacol. 2008;6:179-92
105. Preusser M, Winkler F, Valiente M, Manegold C, Moyal E, Widhalm G. et al. Recent advances in the biology and treatment of brain metastases of non-small cell lung cancer: summary of a multidisciplinary roundtable discussion. ESMO Open. 2018;3:e000262
106. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007;9:654-9
107. Tominaga N, Kosaka N, Ono M, Katsuda T, Yoshioka Y, Tamura K. et al. Brain metastatic cancer cells release microRNA-181c-containing extracellular vesicles capable of destructing blood-brain barrier. Nat Commun. 2015;6:6716
108. Rodrigues G, Hoshino A, Kenific CM, Matei IR, Steiner L, Freitas D. et al. Tumour exosomal CEMIP protein promotes cancer cell colonization in brain metastasis. Nat Cell Biol. 2019;21:1403-12
109. Achrol AS, Rennert RC, Anders C, Soffietti R, Ahluwalia MS, Nayak L. et al. Brain metastases. Nat Rev Dis Primers. 2019;5:5
110. Lorger M. Tumor microenvironment in the brain. Cancers (Basel). 2012;4:218-43
111. Gee JR, Keller JN. Astrocytes: regulation of brain homeostasis via apolipoprotein E. Int J Biochem Cell Biol. 2005;37:1145-50
112. Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200:629-38
113. Pellerin L, Pellegri G, Bittar PG, Charnay Y, Bouras C, Martin JL. et al. Evidence supporting the existence of an activity-dependent astrocyte-neuron lactate shuttle. Dev Neurosci. 1998;20:291-9
114. Kang YC, Kim KM, Lee KS, Namkoong S, Lee SJ, Han JA. et al. Serum bioactive lysophospholipids prevent TRAIL-induced apoptosis via PI3K/Akt-dependent cFLIP expression and Bad phosphorylation. Cell death and differentiation. 2004;11:1287-98
115. Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24:2143-55
116. Cabezas R, Avila M, Gonzalez J, El-Bacha RS, Baez E, Garcia-Segura LM. et al. Astrocytic modulation of blood brain barrier: perspectives on Parkinson's disease. Front Cell Neurosci. 2014;8:211
117. Xing F, Kobayashi A, Okuda H, Watabe M, Pai SK, Pandey PR. et al. Reactive astrocytes promote the metastatic growth of breast cancer stem-like cells by activating Notch signalling in brain. EMBO Mol Med. 2013;5:384-96
118. Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang WC. et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature. 2015;527:100-4
119. Neman J, Choy C, Kowolik CM, Anderson A, Duenas VJ, Waliany S. et al. Co-evolution of breast-to-brain metastasis and neural progenitor cells. Clin Exp Metastasis. 2013;30:753-68
120. Fitzgerald DP, Palmieri D, Hua E, Hargrave E, Herring JM, Qian Y. et al. Reactive glia are recruited by highly proliferative brain metastases of breast cancer and promote tumor cell colonization. Clin Exp Metastasis. 2008;25:799-810
121. Zhang M, Olsson Y. Hematogenous metastases of the human brain-characteristics of peritumoral brain changes: a review. J Neurooncol. 1997;35:81-9
122. Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XH, Lee DJ. et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156:1002-16
123. Kim SJ, Kim JS, Park ES, Lee JS, Lin Q, Langley RR. et al. Astrocytes upregulate survival genes in tumor cells and induce protection from chemotherapy. Neoplasia. 2011;13:286-98
124. Sarvaiya PJ, Guo D, Ulasov I, Gabikian P, Lesniak MS. Chemokines in tumor progression and metastasis. Oncotarget. 2013;4:2171-85
125. Hoelzinger DB, Demuth T, Berens ME. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst. 2007;99:1583-93
126. Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nature reviewsMolecular cell biology. 2003;4:397-407
127. Hait NC, Maiti A. The Role of Sphingosine-1-Phosphate and Ceramide-1-Phosphate in Inflammation and Cancer. Mediators of inflammation. 2017;2017:4806541
128. Edsall LC, Spiegel S. Enzymatic measurement of sphingosine 1-phosphate. Anal Biochem. 1999;272:80-6
129. Hait NC, Wise LE, Allegood JC, O'Brien M, Avni D, Reeves TM. et al. Active, phosphorylated fingolimod inhibits histone deacetylases and facilitates fear extinction memory. Nature neuroscience. 2014;17:971-80
130. Bassi R, Anelli V, Giussani P, Tettamanti G, Viani P, Riboni L. Sphingosine-1-phosphate is released by cerebellar astrocytes in response to bFGF and induces astrocyte proliferation through Gi-protein-coupled receptors. Glia. 2006;53:621-30
131. Gril B, Paranjape AN, Woditschka S, Hua E, Dolan EL, Hanson J. et al. Reactive astrocytic S1P3 signaling modulates the blood-tumor barrier in brain metastases. Nat Commun. 2018;9:2705
132. Ulasov IV, Borovjagin AV, Timashev P, Cristofanili M, Welch DR. KISS1 in breast cancer progression and autophagy. Cancer Metastasis Rev. 2019;38:493-506
133. Chen Q, Boire A, Jin X, Valiente M, Er EE, Lopez-Soto A. et al. Carcinoma-astrocyte gap junctions promote brain metastasis by cGAMP transfer. Nature. 2016;533:493-8
134. Pellerin L. Brain energetics (thought needs food). Curr Opin Clin Nutr Metab Care. 2008;11:701-5
135. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-33
136. Ciminera AK, Jandial R, Termini J. Metabolic advantages and vulnerabilities in brain metastases. Clin Exp Metastasis. 2017;34:401-10
137. Venneti S, Dunphy MP, Zhang H, Pitter KL, Zanzonico P, Campos C. et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med. 2015;7:274ra17
138. Chen EI, Hewel J, Krueger JS, Tiraby C, Weber MR, Kralli A. et al. Adaptation of energy metabolism in breast cancer brain metastases. Cancer Res. 2007;67:1472-86
139. Chen J, Lee HJ, Wu X, Huo L, Kim SJ, Xu L. et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. 2015;75:554-65
140. Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427-33
141. Cuomo F, Altucci L, Cobellis G. Autophagy Function and Dysfunction: Potential Drugs as Anti-Cancer Therapy. Cancers (Basel). 2019 11
142. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A. et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809-20
143. Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25:1510-27
144. Huo Y, Cai H, Teplova I, Bowman-Colin C, Chen G, Price S. et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov. 2013;3:894-907
145. Chen W, Bai Y, Patel C, Geng F. Autophagy promotes triple negative breast cancer metastasis via YAP nuclear localization. Biochem Biophys Res Commun. 2019;520:263-8
146. Pavel M, Renna M, Park SJ, Menzies FM, Ricketts T, Fullgrabe J. et al. Contact inhibition controls cell survival and proliferation via YAP/TAZ-autophagy axis. Nat Commun. 2018;9:2961
147. Xu R, Ji Z, Xu C, Zhu J. The clinical value of using chloroquine or hydroxychloroquine as autophagy inhibitors in the treatment of cancers: A systematic review and meta-analysis. Medicine (Baltimore). 2018;97:e12912
148. Ko JH, Sethi G, Um JY, Shanmugam MK, Arfuso F, Kumar AP. et al. The Role of Resveratrol in Cancer Therapy. Int J Mol Sci. 2017 18
149. Fu Y, Chang H, Peng X, Bai Q, Yi L, Zhou Y. et al. Resveratrol inhibits breast cancer stem-like cells and induces autophagy via suppressing Wnt/beta-catenin signaling pathway. PLoS One. 2014;9:e102535
150. Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ. 2008;15:1318-29
151. McKenna M, McGarrigle S, Pidgeon GP. The next generation of PI3K-Akt-mTOR pathway inhibitors in breast cancer cohorts. Biochim Biophys Acta Rev Cancer. 2018;1870:185-97
152. Hurvitz SA, Andre F, Jiang Z, Shao Z, Mano MS, Neciosup SP. et al. Combination of everolimus with trastuzumab plus paclitaxel as first-line treatment for patients with HER2-positive advanced breast cancer (BOLERO-1): a phase 3, randomised, double-blind, multicentre trial. Lancet Oncol. 2015;16:816-29
153. Piccart M, Hortobagyi GN, Campone M, Pritchard KI, Lebrun F, Ito Y. et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2dagger. Ann Oncol. 2014;25:2357-62
154. Petrossian K, Nguyen D, Lo C, Kanaya N, Somlo G, Cui YX. et al. Use of dual mTOR inhibitor MLN0128 against everolimus-resistant breast cancer. Breast Cancer Res Treat. 2018;170:499-506
155. Guichard SM, Curwen J, Bihani T, D'Cruz CM, Yates JW, Grondine M. et al. AZD2014, an Inhibitor of mTORC1 and mTORC2, Is Highly Effective in ER+ Breast Cancer When Administered Using Intermittent or Continuous Schedules. Mol Cancer Ther. 2015;14:2508-18
156. Vijayaraghavan S, Karakas C, Doostan I, Chen X, Bui T, Yi M. et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun. 2017;8:15916
157. Lambert LA, Qiao N, Hunt KK, Lambert DH, Mills GB, Meijer L. et al. Autophagy: a novel mechanism of synergistic cytotoxicity between doxorubicin and roscovitine in a sarcoma model. Cancer Res. 2008;68:7966-74
158. Yamamoto T, Kanaya N, Somlo G, Chen S. Synergistic anti-cancer activity of CDK4/6 inhibitor palbociclib and dual mTOR kinase inhibitor MLN0128 in pRb-expressing ER-negative breast cancer. Breast Cancer Res Treat. 2019;174:615-25
159. Hohensee I, Lamszus K, Riethdorf S, Meyer-Staeckling S, Glatzel M, Matschke J. et al. Frequent genetic alterations in EGFR- and HER2-driven pathways in breast cancer brain metastases. Am J Pathol. 2013;183:83-95
160. Masui K, Cavenee WK, Mischel PS. mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol Metab. 2014;25:364-73
161. Masui K, Cavenee WK, Mischel PS. mTORC2 dictates Warburg effect and drug resistance. Cell Cycle. 2014;13:1053-4
162. Huang F, Wang BR, Wang YG. Role of autophagy in tumorigenesis, metastasis, targeted therapy and drug resistance of hepatocellular carcinoma. World J Gastroenterol. 2018;24:4643-51
163. Palmieri D, Bronder JL, Herring JM, Yoneda T, Weil RJ, Stark AM. et al. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2007;67:4190-8
164. Santana-Codina N, Marce-Grau A, Muixi L, Nieva C, Marro M, Sebastian D. et al. GRP94 Is Involved in the Lipid Phenotype of Brain Metastatic Cells. Int J Mol Sci. 2019 20
Author contact
![]() Corresponding authors: Nitai C. Hait, Ph.D., Department of Surgical Oncology, Department of Molecular & Cellular Biology, Roswell Park Comprehensive Cancer Center, Medical Research Complex (MRC), Elm and Carlton Streets, Buffalo, NY 14263 (USA). Tel: (716) 845-8527; Fax: (716) 845-1668; E-mail: nitai.haitorg; or Aparna Maiti, Ph.D., Department of Surgical Oncology, Department of Molecular & Cellular Biology, Roswell Park Comprehensive Cancer Center, Medical Research Complex (MRC), Elm and Carlton Streets, Buffalo, NY 14263 (USA). Tel: (716) 845-3505; E-mail: aparna.maitiorg.
Corresponding authors: Nitai C. Hait, Ph.D., Department of Surgical Oncology, Department of Molecular & Cellular Biology, Roswell Park Comprehensive Cancer Center, Medical Research Complex (MRC), Elm and Carlton Streets, Buffalo, NY 14263 (USA). Tel: (716) 845-8527; Fax: (716) 845-1668; E-mail: nitai.haitorg; or Aparna Maiti, Ph.D., Department of Surgical Oncology, Department of Molecular & Cellular Biology, Roswell Park Comprehensive Cancer Center, Medical Research Complex (MRC), Elm and Carlton Streets, Buffalo, NY 14263 (USA). Tel: (716) 845-3505; E-mail: aparna.maitiorg.