Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(4):1212-1219. doi:10.7150/jca.54095 This issue Cite
Review
Chimeric antigen receptor T-cell immunotherapy in breast cancer: development and challenges
Sara Toulouie1, Gary Johanning2, Yihui Shi1 ![]()
1. California Northstate University, College of Medicine, Elk Grove CA, USA.
2. SunnyBay Biotech, Fremont, CA USA.
Received 2020-10-5; Accepted 2020-11-27; Published 2021-1-1
Abstract
Chimeric antigen receptor (CAR) T-cell therapy is an innovative form of immunotherapy wherein autologous T-cells are genetically modified to express chimeric receptors encoding an antigen-specific single-chain variable fragment and costimulatory molecules. Moreover, CAR T-cell therapy can only work successfully in patients who have an intact immune system. Therefore, patients receiving cytotoxic chemotherapy will be immunosuppressed making CAR-T therapy less effective. In adoptive CD8+ T-cell therapy (ACT), numerous tumor-specific, engineered T-cells are sourced from patients, expanded in vitro, and infused back expressing tumor-specific antigen receptors. The most successful ACT, anti-CD19 chimeric antigen receptor T-cell therapy directed against B-cell lymphoma, has proved to be efficacious. However, current efforts to utilize this approach for solid tumors, like breast cancer, have shown only modest improvement. Nevertheless, the potential efficacy of CAR-T therapy is promising in an era of immunological advances. By appropriately manipulating CAR T-cells to combat the immunosuppressive forces of the tumor microenvironment, significant eradication of the solid tumor may occur. This review discusses CAR T-cell therapy and its specificity and safety in adoptive cell transfers in breast cancer. We will highlight novel discoveries in CAR T-cell immunotherapy and the formidable barriers including suppression of T-cell function and localization at tumor sites.
Keywords: breast cancer, T-cell, immunotherapy, chimeric antigen receptor, TNBC
Introduction
Breast cancer has consistently been characterized as a leading cause of death in women. One-fourth of all women with cancer, or 1.5 million women, are diagnosed with breast cancer each year throughout the world [1].This illness is often diagnosed during a routine screening or incidentally and may spread to lymph nodes and metastasize to other organs, like the brain [2]. Due to its prominent hematologic spread, breast cancer screening is highly encouraged in women who exhibit risk factors such as age, family history, and exogenous hormone use [3]. Germ-line mutations in high-penetrance breast cancer susceptibility genes like BRCA1, BRCA2, p53 and PTEN have been seen in up to 10% of all breast cancers [4]. The pathogenesis of breast cancer includes neoplastic changes of myoepithelial cells that overcome growth suppressor genes and receive nutrient and oxygen supply by angiogenesis and using telomerase to engage in extensive replication of cells [5]. A comprehensive electronic search of PubMed/MEDLINE was conducted for studies published from the year 2000 through 2020 utilizing initially broad keywords pertaining to this study including “immunotherapy” and “CAR T-cell” that resulted in over three-thousand articles primarily highlighting the success of CAR T-cell therapy in hematologic diseases. Further narrowing of the search was conducted using additional keywords such as “solid tumor” and “breast cancer,” shortening the results down to 87 articles. This search further showcased the lack of extensive research on the utilization of CAR T-cell therapy in breast cancer and the potential limitations of immunotherapy in solid tumor applications like that of breast and lung.
In 2016, the American Joint Committee on Cancer updated breast cancer staging to include T, N, M, tumor grade, and expression of estrogen and progesterone receptors and HER2 to groups patients into risk categories that help define and guide treatment recommendation [6]. For early stage treatment, surgery remains the first treatment recommendation with neoadjuvant chemotherapy [7]. The current surgical treatment options include radical mastectomy of the entire breast tissue and axillary lymph nodes and a simple mastectomy including solely the breast removal and axillary procedures used in local control of the disease [8]. Unfortunately, many cancers develop resistance to traditional treatment modalities like chemotherapy and radiation. Nevertheless, novel advancements of immunotherapy including antibodies, vaccines, immune checkpoint inhibitors, and CAR T-cell therapy are promising [9]. One modality in particular termed CAR T-cell therapy involves the genetic modification of a patient's autologous T-cells to express a CAR specific to a tumor antigen of interest followed by mass proliferation and eventual infusion of these cells performed ex vivo [10].
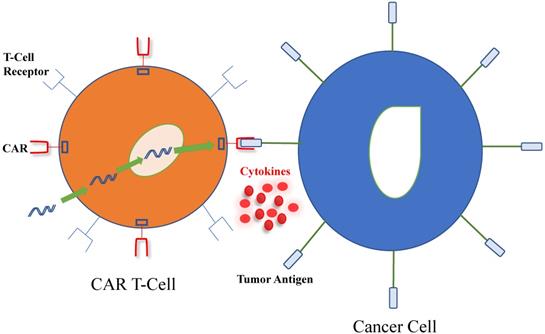
As described in Figure 1, a tumor-associated antigen (TAA) specific receptor known as CAR is introduced into T-cells, using a plasmid or viral vector, of which lentivirus is the most commonly used [11]. CAR combines the specificity of a monoclonal antibody with the inherent cytotoxic and memory capabilities of T-cells. Furthermore, its extracellular domain is derived from the antigen-binding site of a monoclonal antibody and defines CAR's high target affinity while its intracellular domain recapitulates the function of normal T-cells. The incorporation of costimulatory domains, such as CD28 or 4-1BB (CD137) for enhanced survival and proliferation render the CAR T-cells less susceptible than unmodified T cells to negative regulation from tumor cells. Additionally, CAR T-cells do not rely on dendritic cell antigen processing and presentation for its activation. The end effect is that the external targeting domain of CAR binds to the antigen, activating the CAR T- cell and its release of cytokines and other soluble mediators that may directly kill antigen-expressing target cells nearby [12].
Interaction between chimeric antigen receptor (CAR) T-cell and cancer cell. T-cells harvested from patients are genetically modified using viral or non-viral systems to produce the CAR, which includes an antibody-like surface domain, transmembrane domain and intracellular signaling domains. CAR T-cells interact with tumor antigens on cancer cells by using their extracellular surface domain, linked to intracellular costimulatory and signaling domains to amplify the immune response against tumor cells. Furthermore, pro-inflammatory cytokines and chemokines are produced and participate in the eradication of cancer cells.
Current literature highlights CAR T-cell therapy as a highly effective treatment modality in hematological cancers, like Acute Lymphocytic Leukemia (ALL), exhibiting a 92% full recovery [13]. However, in this review, we will explore current strategies and barriers of CAR T-cell therapy in the application to solid tumors, predominately breast cancer.
Discussion
Details of CAR-T Therapy
Clinical trials often focus on disseminated tumor cells that lay dormant for years but have metastatic potential in breast cancer patients. Many of these clinical trials involve the triple-negative breast cancer (TNBC) type, a very aggressive malignant type of tumor that is frequently resistant to standard breast cancer therapies due to the absence of estrogen receptors (ER), progesterone receptors, and epidermal growth factor receptors (EGFR), making novel therapeutic modalities such as CAR T-cells particularly attractive [14]. To effectively utilize CAR-T against metastatic breast cancer, the identification of an appropriate target antigen and consideration of additional genetic strategies to protect cells from the suppressive tumor microenvironment are critical [15].
As mentioned earlier, modified CAR T-cells contain hybrid receptors that include both an extracellular and intracellular antigen recognizing domain with extensive costimulatory regions that together allow for enhanced antigen affinity [16]. The length of the extracellular spacer domain affects tumor cell recognition and CAR T-cell function, likely because of the spatial interaction of CAR and tumor antigen, and the localization of the targeted epitope [17]. Additionally, CAR T-cells are major histocompatibility complex (MHC)-independent which excludes human leukocyte antigen (HLA) compatibility issues between donors and recipients. Another particular advantage of CAR T-cells includes their ability to cross the blood-brain barrier, a helpful tool when studying metastases to the central nervous system [18]. The activation of resilient T-cells requires three signals: T cell receptor (TCR) (signal 1), co-stimulation (signal 2), and cytokine (signal 3). To enhance activity, persistence, and efficacy, CAR-T therapy has been developed in stages that include first, second, and third generations [19]. Originally, first-generation CARs contained only a single signaling domain derived from CD3ζ which may explain the unpromising initial clinical results. Subsequently, second-generation CARs containing an additional co-stimulatory signaling molecule, such as 4-1BB, CD28, CD27, OX40 or ICOS have been developed to stimulate an endogenous immune response against tumor cells via epitope spreading [20]. Although second-generation CARs are relatively efficient in the immunosuppressive microenvironment, co-stimulation alone may not be sufficient [21]. The latest generations of CAR-T cells, also known as “armored CARs”, contain additional signaling domains from cytokine receptors and express inflammatory cytokines, such as interleukin-12 (IL-12) or IL-18 [11].
Current Targets and Chimeric Antigens
The various types of antigens recognized by CAR T-cells can be extended to carbohydrates and glycolipids that are often modified in cancer cell [22]. Additional engineering of CAR T-cells to include intracellular signaling motifs from costimulatory molecules such as CD28, 4-1BB, OX40, and ICOS serve to enhance their proliferation and ability to eradicate cancer cells and cytokine production [23]. A folate receptor alpha (FRα) specific chimeric antigen receptor was constructed and the gene encoding this receptor was inserted into T lymphocytes that killed TNBC cells upon amplification and purification [24]. One tumor associated antigen called mucin1 (MUC1) is associated with tumor invasiveness and metastatic potential in all cancer cells, including TNBC [25]. Immunotherapy was utilized to derive a CAR from the TAB004 monoclonal antibody to bind to an aberrant glycoform of MUC1, called tMUC1. It is important to note that tMUC1 is present in greater than 95% of all TNBC with no significant staining in normal breast epithelium [26]. Notably, the T-cells transfected with this specific CAR exhibit antigen-specific cytotoxicity against several TNBC cell lines [14].
Another tumor antigen known as integrin αvβ3 is expressed in several tumor entities including melanoma, glioblastoma, breast, pancreatic, and prostate cancer and promotes tumor cell survival and metastasis [27]. Data suggest that adoptive therapy with αvβ3-CAR T-cells has the potential to confer greater therapeutic efficacy compared to immunotherapy with anti-αvβ3 monoclonal antibodies (mABs) through direct cytolytic activity and modification of the tumor microenvironment. The use of immunotherapy in this setting limits tumor angiogenesis by destruction of αvβ3-expressing cancer associated fibroblasts and endothelial cells in tumor-associated blood vessels [28].
One study explored the cell-surface molecule c-Met, which is expressed in ~50% of breast tumors, prompting the construction of a CAR T-cell specific for c-Met in order to halt tumor growth in immune-incompetent mice with tumor xenografts [29]. Tumors treated with intertumoral injections of T-cells transduced with c-Met CAR mRNA were excised and analyzed by immunohistochemistry, revealing extensive tumor necrosis at the injection site, cellular debris, loss of c-Met immunoreactivity, all surrounded by macrophages at the leading edges and within necrotic zones. These findings conclude that intertumoral injections of mRNA c-Met-CAR T-cells are well tolerated and elicit an inflammatory response within tumors [30]. Similarly, a microphysiologic three-dimensional (3D) lung and breast cancer model that closely resembles the architectural and phenotypical features of primary tumors was used to evaluate the antitumor function of receptor tyrosine kinase-like orphan receptor 1-specific (ROR1-specific) CAR T-cells. Notably, ROR1-CAR T-cells penetrated deep into tumor tissue and eliminated multiple layers of tumor cells located above and below the basement membrane [31]. As seen in these studies, much consideration should be made into the use of tumor models to assure safety and efficacy before the clinical application of immunotherapy.
Several CAR T-cell trials are currently enrolling, including trials targeting CEA, HER2, mesothelin, MUC1, NKG2D ligands, ROR1, CD70, and CD133[32]. Among the many tested tumor associated antigens, HER2 is one antigen that is overexpressed in 20% to 30% of all breast cancers, influencing recurrence rates and ultimately survival. Despite the significant improvement in prognosis after therapy with the anti-HER2 antibodies trastuzumab and pertuzumab, a high proportion of patients will eventually experience recurrence even with the synergistic effects of both antibodies when given in the neoadjuvant setting [32,33]. Thus, alternative therapies or new combinations to overcome resistance to these antibodies are needed. It was found that dual-targeted T-cells co-expressing a HER2-and MUC-1-specific CAR effectively kill breast cancer cells that normally express both targets. Additionally, engineering T-cells to express a dominant-negative TGFβ receptor that restores T-cell effector function is being investigated in a clinical trial utilizing CAR T-cells that target the HER2 antigen [34].
Another potential target focuses on mesothelin, a cell-surface antigen overexpressed in 67% of TNBC samples [35]. Overexpression of mesothelin alone is sufficient to constitutively activate the NFκB, MAPK, and PI3K intracellular pathways promoting tumor cell proliferation and resistance to apoptosis [34]. Similarly, the CSPG4 tumor glycoprotein has been identified in 32 of 44 (72.7%) primary TNBC lesions and is inversely correlated with overall survival and time to recurrence. Mechanistically, interactions between chondroitin sulfate side chains of CSPG4 with P-selectin are thought to result in tumor cell activation and augment survival of circulating breast cancer cells [36]. CSPG4 presence was also detected in TNBC cancer stem cells, which are regarded as a major source for relapse and resistance [37]. Hence, CSPG4-CAR T-cells in TNBC can counteract this mechanism via down-regulating CSPG4 to impair metastasis and stunt breast cancer progression. In aggregate, CSPG4-CAR-T cells have the potential to mount a concerted attack against various targets, including primary TNBC cells, stromal cells, and cancer-associated fibroblasts, which assume a crucial role in maintaining the tumor microenvironment [14].
One study reported on the evaluation of disialoganglioside GD2 expressed in 35.5% of metastatic TNBC as a breast cancer stem cell specific target antigen for immunotherapy [38]. Importantly, GD2-CAR-T demonstrated excellent cytolytic activity against GD2 positive cell lines, independent of the tumor entity. This valuable therapeutic option may benefit high-risk breast cancer subtypes like TNBC once clinical trials have proven improved survival rates. Similarly, EGFR-targeted CAR-T cells of the third generation are potent and specific in suppression of TNBC cell growth. This capability was exhibited in vitro and in vivo in a xenograft mouse model, with minimal off-tumor cytotoxicity. The activation of the interferon γ, granzyme-perforin-PARP and Fas-FADD-caspase signaling pathways in TNBC cells further confirm that CAR-T as an immunotherapy tool may be used to treat TNBC in the clinical setting [39].
TEM8, a marker overexpressed on the vasculature of some solid tumors, has been proposed as a target of controversy [40]. A 2018 report stated that T-cells engineered to express a TEM8-specific CAR when injected into mouse models of TNBC, are both safe and effective in controlling tumor growth. This study used the L2 antibody and mouse models of TNBC and reported no toxic effects [41]. Conversely, a more recent study by Petrovic presented opposite findings using a panel of TEM8-specific CARs based on the same antibodies, however, causing significant toxicity in healthy mice through the targeting of TEM8 in healthy tissue. Reasons for these contradictory findings could lie in subtle changes in the design of the CAR. For example, different retroviral vectors may have led to different levels of CAR expression per cell [42]. This is of particular concern if strategies are employed to enhance the anti-tumor effects of the CAR, for example through dose escalation or increasing the levels of CAR expression and/or function in humans [43].
An intriguing antigen that is overexpressed in most cancers including breast cancer is the human endogenous retrovirus family K (HERV-K) [44]. We recently reported that HERV-K is expressed at especially high levels in the basal breast cancer subtype, which is similar to TNBC [45]. In contrast to most tumor-specific antigens, HERV-K is absent in nearly all normal human tissues [46]. We recently evaluated CAR T-cells targeting the HERV-K envelope protein (K-CAR T cells) for effectiveness in slowing tumor growth in a mouse model of breast cancer [47]. CTL assays were employed in in vitro studies to determine the cytotoxicity of K-CAR toward MDA-MB-231, a triple-negative breast cancer cell line, and significantly greater lysis was demonstrated for this cell line using K-CAR from breast cancer patients. In vivo studies with immunodeficient mice revealed that tumor growth, size, and weights were significantly decreased in mice bearing MDA-MB-231 or MDA-MB-435.eB1 transfectant cancer cell lines, which express a 258-fold increase in the cell growth protein c-erbB-2, were treated with K-CAR T-cells compared to control T-cells or no treatment [48]. The mechanism by which K-CAR T-cells repress tumor growth may not be a single mode, since reduced expression of HERV-K in tumor biopsies of the treated mice led to downregulated expression of HERV-K, and this was accompanied by upregulation of p53 and downregulation of its inhibitor MDM2, as well as decreased expression of p-ERK, compared with controls. Anti-HERV-K CAR T-cells have also been investigated for melanoma therapy in vivo [49].
Immunotherapy Combination Therapy
The future of antitumor therapeutics will most likely encompass cell-based therapy in addition to chemotherapy as it is unlikely that CAR T-cell treatment will successfully eradicate a tumor on its own [18]. Accurate patient selection and the use of immune checkpoint inhibitors in combination with modalities like photoimmunotherapy that activate the immune system may allow immunotherapy to advance the future of personalized care for breast cancer patients [50]. Multiple trials have examined chemotherapy in addition to PD-1/PD-L1 blockade with the goal of enhancing immune priming through antigen release and relieving immunosuppressive signals in the tumor microenvironment. Trastuzumab, a humanized mAb that binds to HER-2 homodimers, is often added to standard chemotherapy to prolong survival and decrease risk of relapse. The addition of Pertuzumab and Trastuzumab to chemotherapy drugs like Taxotere and Carboplatin gives the highest reported clinically partial response (cPR) rate reported to date [51]. One clinical trial in Phase 2 has suggested the combination of Pembrolizumab and radiotherapy in patients with metastatic TNBC to be both efficacious and safe for the 17 patients enrolled in the study. However, larger clinical trials of checkpoint blockade plus radiotherapy with predictive biomarkers are required to make a definitive claim [52].
Additionally, two recent reports have linked the gut microbiota composition to the promising clinical response of CAR T-cells in terms of efficacy and toxicity. Specifically, the immunostimulation by intestinal bacteria includes cross-reactions between microbial and tumor antigen stimulation of pattern-recognition receptors (PRRs) and the production of bacterial metabolites that might exert systemic modulatory effects [11]. In addition to genetically engineering T-cells to enhance an immune response to tumor infiltration, preconditioning to achieve host lymphodepletion by use of cyclophosphamide, fludarabine, or radiotherapy may promote engraftment of adoptively transferred T-cells. Likewise, CAR T-cell therapy may be used in conjunction with small-molecule inhibitors, monoclonal antibodies, and vaccines [36].
Challenges
In infant cases of ALL, CAR T-cells achieve up to 90% eradication of the tumor, while in solid tumors, the clinical efficacy is less rewarding due to toxic side effects and fewer clinical trial implementations [19]. The mechanism of toxicity includes the immediate release of large quantities of inflammatory cytokines in response to CAR T-cells binding to antigens on target tumor cells. The clinical side effect of such a manifestation is termed cytokine release syndrome (CRS), which is characterized by high fevers, myalgias, nausea, anorexia, and potential hemodynamic or respiratory instability [18]. The onset of CRS usually occurs several days after T-cell infusion at the peak of CAR T-cell expansions. The inflammatory cytokines may also activate endothelial cells of the blood-brain barrier, disrupting barrier integrity and driving the CAR-T therapy associated neurotoxicity [53]. As IL-6 serves as the primary mediator of CRS, such adverse reactions might respond to IL-6 receptor inhibitors like tocilizumab but this may require further supplementary treatment with corticosteroids to prevent any lethal consequences [54]. This so called “on-target/off-tumor effect” of CAR T-cells is mainly caused by T cells further attacking normal cells that express the target antigen causing severe “off-tumor” recognition. Nevertheless, this effect emphasizes the need for identification of tumor specific antigens for proper treatment of solid tumors with CAR T-cells [55]. Upon T-cell activation following tumor infiltration, multiple intracellular factors, such as diacylglycerol kinase (DGK), impair T-cell effector functions and promote T-cell anergy. Furthermore, the genetic deletion of DGKζ significantly increases the antitumor activity of mesothelin-targeted CAR T-cells [34].
Another possible concern with cell-based therapies is immunoediting of tumor antigens. As tumor cells are destroyed by CAR T-cells, which are formed from memory cells, they are directed against the tumor antigen they were originally engineered to recognize. In this setting, immunomodulation may prove to be a problem as antigenic shift may cause tumor cells to evolve in a way to create new tumor antigens that may not be recognized by the original CAR T-cells. Thus, this notable antigenic shift, or molecular alteration of an antigen due to recombination, may inadvertently cause CAR T-cells to act against the tumor antigen they were intended to recognize [18]. Abnormal vasculature, physical barriers from tumor fibroblasts in the surrounding stroma, and the multitude of immunosuppressive factors such as checkpoint pathways and cytokines prevent efficient solid tumor infiltration of CAR T-cells. The combination of solid tumors secreting chemokines, such as CXCL12 and CXCL5, and the lack of CAR T-cell expression of appropriate chemokine receptors hinder T-cell migration into the tumor site [55]. To combat these challenges, novel advancements in trafficking, penetration, and immunosuppressive barriers have been made to improve the efficacy of this mode of therapy. Enhanced expression of chemokine receptors CCR5, CCR2b, CCR4 improve the migration and homing of CAR T-cells to the tumor sites. In terms of penetration, overexpression of heparanase in CAR T-cells may enhance T-cell infiltration by the degradation of heparan sulfate proteoglycans, a key component of the extracellular matrix [56]. Likewise, targeting vasculature antigens such as vascular endothelial growth factor receptor (VEGFR-2), integrin alpha V beta 3 (αvβ3) or prostate-specific membrane antigen (PSMA) can also aid CAR T-cell infiltration into the tumor [57]. Lastly, secretion of anti-PD-L1 antibodies and IL-12 have shown to improve CAR T-cell function in an immunosuppressive environment. As mentioned earlier, checkpoint inhibitory proteins, such as PD-L1, are upregulated in tumors and the interaction of PD-L1 with its receptor, PD-1 may lead to T-cell exhaustion [19]. Under the selection pressure of adoptive cell therapy, cancer cells may evolve to escape the recognition by CD8 + T cells due to epitope mutation and tumor recurrence1. For instance, HER2 can undergo proteolysis to cleave the extracellular domain without compromising kinase activity. By using a dual-targeting CAR system, engineered T-cells coexpress two CARs that recognize two distinct antigens so that T-cells can be activated in the presence of either antigen to mitigate antigen-loss escape [20].
Metastasis to the brain from breast cancer poses a significant clinical challenge that may be treated with CAR-based immunotherapy [58]. One notable study utilized HER2-CARs containing the 4-1BB costimulatory domain and delivered them intracranially in orthotopic xenograft models to demonstrate robust antitumor efficacy for the treatment of multifocal brain metastases and leptomeningeal disease. This model suggests that regional delivery of CAR T-cells may circumvent systemic targeting of less restricted tumor antigens, like HER2 [59].
Conclusions
While the results of immunotherapy through CAR T-cells have been monumental in cancer therapy, further understanding of the inhibitory tumor microenvironment is needed. Combinatory use of multiple agents in a patient's treatment protocol is found to be advantageous in malignancies like breast cancer that express high immunogenicity and a heterogenous antigen profile. Collectively, studies comparing normal with benign breast tissue indicated early immune infiltration of B cells, T cells, and macrophages as the tumor progresses to ductal carcinoma in situ and on to invasive levels. Further exploration as to the direct role of these cells will be imperative in diagnosing breast cancer in its early stages through the identification of discreet cell populations expressing markers commonly associated with a pro-tumor phenotype [60].
The emergence of immuno-oncology has provided a unique opportunity for researchers and clinicians to concentrate on cancers like TNBC that lack targeted therapy universally and are recognized as the most complex and challenging breast cancer subtype to treat, with chemotherapy remaining the standard of care. Therefore, significant effort has been placed on the discovery of novel treatment strategies for subtype TNBC with the hope to broaden this therapeutic approach to other breast cancer subtypes in the future. One strategy in particular targets the use of immune checkpoints for immune escape, specifically the tumor's ability to suppress PD-L1 expression and decrease the activity of cytotoxic T-cells [61]. Typically, the binding of PD-L1 to PD-1 on CAR T-cells initiates an inhibitor signal that suppresses the function of CAR T-cells, leading to an exhausted phenotype. Nevertheless, the combination of CAR T-cells with PD-1 inhibitors can overcome tumor microenvironment immunosuppression as demonstrated by Lotfinejad et al. in a murine model of breast cancer. When PD-1 inhibitor antibodies were combined with anti-HER2 CAR T-cells, significant tumor volume reduction was seen and levels of IFN-γ and granzyme B increased, indicating improved immune response [62].
Healthy donor allogeneic CAR T-cells can be derived from an HLA-matched hematopoietic stem cell transplant donor. However, for the non-HLA-matched patient, current efforts to pursue gene-editing approaches using CRISPR/Cas9 have shown to be advantageous. Nevertheless, unwanted side effects of gene editing include off-target cleavage of genes and undesired translocations [63]. Ongoing research in the potentially rewarding fields of single-cell genomic analysis [64] and production of next-generation allogeneic CAR T-cells [65] will precisely define the mechanism of action of CAR-T in the breast tumor microenvironment and will provide a ready source of these agents for breast cancer patients. It is also important to consider the fiscal and mental effects of breast cancer. For example, it is expected that patients treated non-operatively are likely to require more intensive imaging follow-up and additional biopsies, potentially leading to increased costs and patient anxiety [66]. The potential efficacy of CAR-T therapy is promising and with the speed of technological advances, the hope is that CAR T-cells will be appropriately modified to combat the immunosuppressive forces of the tumor microenvironment and eradicate the solid tumor. The decreasing cost and increasing capacity of sequencing methods could significantly enhance the understanding of complex interactions while engineering the next generation of CAR-T cell therapy for solid malignancies [67].
Acknowledgements
Funding
This article is funded by NIH/NCI, grant No. 1R21CA238636-01 (Dr. Johanning PI, Dr. Shi Subaward PI).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sun YS, Zhao Z, Yang ZN, Xu F, Lu HJ, Zhu ZY. et al. Risk Factors and Preventions of Breast Cancer. Int J Biol Sci. 2017;13:1387-97
2. Seely JM, Alhassan T. Screening for breast cancer in 2018-what should we be doing today? Curr Oncol. 2018;25:S115-S24
3. Alkabban FM, Ferguson T. Breast Cancer. StatPearls. Treasure Island (FL). 2020
4. Abdulkareem IH. Aetio-pathogenesis of breast cancer. Niger Med J. 2013;54:371-5
5. Akram M, Iqbal M, Daniyal M, Khan AU. Awareness and current knowledge of breast cancer. Biol Res. 2017;50:33
6. Edge SB, Hortobagyi GN, Giuliano AE. New and important changes in breast cancer TNM: incorporation of biologic factors into staging. Expert Rev Anticancer Ther. 2019;19:309-18
7. Heilat GB, Brennan ME, French J. Update on the management of early-stage breast cancer. Aust J Gen Pract. 2019;48:604-8
8. Czajka ML, Pfeifer C. Breast Cancer Surgery. StatPearls. Treasure Island (FL). 2020
9. Cheng J, Zhao L, Zhang Y, Qin Y, Guan Y, Zhang T. et al. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front Oncol. 2019;9:1237
10. Almasbak H, Aarvak T, Vemuri MC. CAR T Cell Therapy: A Game Changer in Cancer Treatment. J Immunol Res. 2016;2016:5474602
11. Pilipow K, Darwich A, Losurdo A. T-cell-based breast cancer immunotherapy. Semin Cancer Biol. 2020
12. Maus MV, Levine BL. Chimeric Antigen Receptor T-Cell Therapy for the Community Oncologist. Oncologist. 2016;21:608-17
13. Miliotou AN, Papadopoulou LC. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr Pharm Biotechnol. 2018;19:5-18
14. Harrer DC, Dorrie J, Schaft N. CSPG4 as Target for CAR-T-Cell Therapy of Various Tumor Entities-Merits and Challenges. Int J Mol Sci. 2019 20
15. Bajgain P, Tawinwung S, D'Elia L, Sukumaran S, Watanabe N, Hoyos V. et al. CAR T cell therapy for breast cancer: harnessing the tumor milieu to drive T cell activation. J Immunother Cancer. 2018;6:34
16. Subklewe M, von Bergwelt-Baildon M, Humpe A. Chimeric Antigen Receptor T Cells: A Race to Revolutionize Cancer Therapy. Transfus Med Hemother. 2019;46:15-24
17. Stoiber S, Cadilha BL, Benmebarek MR, Lesch S, Endres S, Kobold S. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells. 2019 8
18. Nahas GR, Walker ND, Bryan M, Rameshwar P. A Perspective of Immunotherapy for Breast Cancer: Lessons Learned and Forward Directions for All Cancers. Breast Cancer (Auckl). 2015;9:35-43
19. Yong CSM, Dardalhon V, Devaud C, Taylor N, Darcy PK, Kershaw MH. CAR T-cell therapy of solid tumors. Immunol Cell Biol. 2017;95:356-63
20. Bayraktar S, Batoo S, Okuno S, Gluck S. Immunotherapy in breast cancer. J Carcinog. 2019;18:2
21. Jaspers JE, Brentjens RJ. Development of CAR T cells designed to improve antitumor efficacy and safety. Pharmacol Ther. 2017;178:83-91
22. Zhao L, Cao YJ. Engineered T Cell Therapy for Cancer in the Clinic. Front Immunol. 2019;10:2250
23. Guedan S, Posey AD Jr, Shaw C, Wing A, Da T, Patel PR. et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. 2018 3
24. Song DG, Ye Q, Poussin M, Chacon JA, Figini M, Powell DJ Jr. Effective adoptive immunotherapy of triple-negative breast cancer by folate receptor-alpha redirected CAR T cells is influenced by surface antigen expression level. J Hematol Oncol. 2016;9:56
25. Yazdanifar M, Zhou R, Mukherjee P. Emerging immunotherapeutics in adenocarcinomas: A focus on CAR-T cells. Curr Trends Immunol. 2016;17:95-115
26. Roy LD, Dillon LM, Zhou R, Moore LJ, Livasy C, El-Khoury JM. et al. A tumor specific antibody to aid breast cancer screening in women with dense breast tissue. Genes Cancer. 2017;8:536-49
27. Felding-Habermann B, O'Toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH. et al. Integrin activation controls metastasis in human breast cancer. Proc Natl Acad Sci U S A. 2001;98:1853-8
28. Wallstabe L, Mades A, Frenz S, Einsele H, Rader C, Hudecek M. CAR T cells targeting alphavbeta3 integrin are effective against advanced cancer in preclinical models. Adv Cell Gene Ther. 2018 1
29. Zhao X, Qu J, Hui Y, Zhang H, Sun Y, Liu X. et al. Clinicopathological and prognostic significance of c-Met overexpression in breast cancer. Oncotarget. 2017;8:56758-67
30. Tchou J, Wang LC, Selven B, Zhang H, Conejo-Garcia J, Borghaei H. et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat. 2012;133:799-804
31. Wallstabe L, Gottlich C, Nelke LC, Kuhnemundt J, Schwarz T, Nerreter T. et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight. 2019 4
32. Williams AD, Payne KK, Posey AD Jr, Hill C, Conejo-Garcia J, June CH. et al. Immunotherapy for Breast Cancer: Current and Future Strategies. Curr Surg Rep. 2017 5
33. Mitri Z, Constantine T, O'Regan R. The HER2 Receptor in Breast Cancer: Pathophysiology, Clinical Use, and New Advances in Therapy. Chemother Res Pract. 2012;2012:743193
34. Morello A, Sadelain M, Adusumilli PS. Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors. Cancer Discov. 2016;6:133-46
35. Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ. et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol Res. 2017;5:1152-61
36. Cooney CA, Jousheghany F, Yao-Borengasser A, Phanavanh B, Gomes T, Kieber-Emmons AM. et al. Chondroitin sulfates play a major role in breast cancer metastasis: a role for CSPG4 and CHST11 gene expression in forming surface P-selectin ligands in aggressive breast cancer cells. Breast Cancer Res. 2011;13:R58
37. Wang X, Osada T, Wang Y, Yu L, Sakakura K, Katayama A. et al. CSPG4 protein as a new target for the antibody-based immunotherapy of triple-negative breast cancer. J Natl Cancer Inst. 2010;102:1496-512
38. Seitz CM, Schroeder S, Knopf P, Krahl AC, Hau J, Schleicher S. et al. GD2-targeted chimeric antigen receptor T cells prevent metastasis formation by elimination of breast cancer stem-like cells. Oncoimmunology. 2020;9:1683345
39. Xia L, Zheng ZZ, Liu JY, Chen YJ, Ding JC, Xia NS. et al. EGFR-targeted CAR-T cells are potent and specific in suppressing triple-negative breast cancer both in vitro and in vivo. Clin Transl Immunology. 2020;9:e01135
40. Chaudhary A, Hilton MB, Seaman S, Haines DC, Stevenson S, Lemotte PK. et al. TEM8/ANTXR1 blockade inhibits pathological angiogenesis and potentiates tumoricidal responses against multiple cancer types. Cancer Cell. 2012;21:212-26
41. Byrd TT, Fousek K, Pignata A, Szot C, Samaha H, Seaman S. et al. TEM8/ANTXR1-Specific CAR T Cells as a Targeted Therapy for Triple-Negative Breast Cancer. Cancer Res. 2018;78:489-500
42. Petrovic K, Robinson J, Whitworth K, Jinks E, Shaaban A, Lee SP. TEM8/ANTXR1-specific CAR T cells mediate toxicity in vivo. PLoS One. 2019;14:e0224015
43. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257:107-26
44. Wang-Johanning F, Rycaj K, Plummer JB, Li M, Yin B, Frerich K. et al. Immunotherapeutic potential of anti-human endogenous retrovirus-K envelope protein antibodies in targeting breast tumors. J Natl Cancer Inst. 2012;104:189-210
45. Johanning GL, Malouf GG, Zheng X, Esteva FJ, Weinstein JN, Wang-Johanning F. et al. Expression of human endogenous retrovirus-K is strongly associated with the basal-like breast cancer phenotype. Sci Rep. 2017;7:41960
46. Zhao J, Rycaj K, Geng S, Li M, Plummer JB, Yin B. et al. Expression of Human Endogenous Retrovirus Type K Envelope Protein is a Novel Candidate Prognostic Marker for Human Breast Cancer. Genes Cancer. 2011;2:914-22
47. Zhou F, Li M, Wei Y, Lin K, Lu Y, Shen J. et al. Activation of HERV-K Env protein is essential for tumorigenesis and metastasis of breast cancer cells. Oncotarget. 2016;7:84093-117
48. Zhou F, Krishnamurthy J, Wei Y, Li M, Hunt K, Johanning GL. et al. Chimeric antigen receptor T cells targeting HERV-K inhibit breast cancer and its metastasis through downregulation of Ras. Oncoimmunology. 2015;4:e1047582
49. Krishnamurthy J, Rabinovich BA, Mi T, Switzer KC, Olivares S, Maiti SN. et al. Genetic Engineering of T Cells to Target HERV-K, an Ancient Retrovirus on Melanoma. Clin Cancer Res. 2015;21:3241-51
50. Tokumaru Y, Joyce D, Takabe K. Current status and limitations of immunotherapy for breast cancer. Surgery. 2020;167:628-30
51. Emens LA. Breast Cancer Immunotherapy: Facts and Hopes. Clin Cancer Res. 2018;24:511-20
52. Ho AY, Barker CA, Arnold BB, Powell SN, Hu ZI, Gucalp A. et al. A phase 2 clinical trialassessing theefficacy and safety of pembrolizumab and radiotherapy in patients with metastatic triple-negative breast cancer. Cancer. 2020;126:850-60
53. Jiang X, Xu J, Liu M, Xing H, Wang Z, Huang L. et al. Adoptive CD8(+) T cell therapy against cancer:Challenges and opportunities. Cancer Lett. 2019;462:23-32
54. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011
55. Ma S, Li X, Wang X, Cheng L, Li Z, Zhang C. et al. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int J Biol Sci. 2019;15:2548-60
56. Okolicsanyi RK, van Wijnen AJ, Cool SM, Stein GS, Griffiths LR, Haupt LM. Heparan sulfate proteoglycans and human breast cancer epithelial cell tumorigenicity. J Cell Biochem. 2014;115:967-76
57. Backer MV, Backer JM. Imaging key biomarkers of tumor angiogenesis. Theranostics. 2012;2:502-15
58. Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, Brown CE. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol Rev. 2019;290:60-84
59. Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK. et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2(+) Breast Cancer Metastasis to the Brain. Clin Cancer Res. 2018;24:95-105
60. Zhang C, Kurt RA. Indicators of a pro-tumor immune response are evident at early stages of breast cancer. Clin Transl Oncol. 2020
61. Zheng Y, Fang YC, Li J. PD-L1 expression levels on tumor cells affect their immunosuppressive activity. Oncol Lett. 2019;18:5399-407
62. Lotfinejad P, Kazemi T, Mokhtarzadeh A, Shanehbandi D, Jadidi Niaragh F, Safaei S. et al. PD-1/PD-L1 axis importance and tumor microenvironment immune cells. Life Sci. 2020;259:118297
63. Poorebrahim M, Sadeghi S, Fakhr E, Abazari MF, Poortahmasebi V, Kheirollahi A. et al. Production of CAR T-cells by GMP-grade lentiviral vectors: latest advances and future prospects. Crit Rev Clin Lab Sci. 2019;56:393-419
64. Chen GM, Azzam A, Ding YY, Barrett DM, Grupp SA, Tan K. Dissecting the Tumor-Immune Landscape in Chimeric Antigen Receptor T-cell Therapy: Key Challenges and Opportunities for a Systems Immunology Approach. Clin Cancer Res. 2020;26:3505-13
65. Depil S, Duchateau P, Grupp SA, Mufti G, Poirot L. 'Off-the-shelf' allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020;19:185-99
66. Monica Morrow MD. Will surgery be a part of breast cancer treatment in the future? Breast. 2019;48(Suppl 1):S110-S4
67. Hong M, Clubb JD, Chen YY. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell. 2020
Author contact
![]() Corresponding author: Yihui Shi, California Northstate University, College of Medicine. Phone No: 916-686-8362; E-mail: yihui.shiedu.
Corresponding author: Yihui Shi, California Northstate University, College of Medicine. Phone No: 916-686-8362; E-mail: yihui.shiedu.