Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(5):1386-1397. doi:10.7150/jca.52418 This issue Cite
Research Paper
GTW inhibits the Epithelial to Mesenchymal Transition of Epithelial Ovarian Cancer via ILK/AKT/GSK3β/Slug Signalling Pathway
Ying Feng1, Fuyin Le1, Puyuan Tian2, Yanying Zhong1, Fuliang Zhan1, Genhua Huang1, Hui Hu1, Tingtao Chen1,2 ![]() , Buzhen Tan1
, Buzhen Tan1 ![]()
1. Department of Obstetrics & Gynecology, The Second Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, PR China.
2. Institute of Translational Medicine, Nanchang University, Nanchang, Jiangxi 330031, PR China.
Received 2020-8-25; Accepted 2020-12-9; Published 2021-1-1
Abstract

Background: Epithelial ovarian cancer (EOC) accounts for the most lethal of all gynaecological cancers which is attributed to metastasis, invasiveness and drug resistance. A crucial link has been found between epithelial-mesenchymal transition (EMT) and cancer metastasis and chemo-resistance. Previous studies have confirmed that one of the main components of tripterygium glycosides (GTW)-triptolide (TPL) has anticancer effects.
Methods: The purpose of this study is to determine whether GTW could inhibit EMT in A2780/DPP cells in vitro and in vivo, and explore the underlying mechanism.
Results: In vitro results showed that GTW inhibited cell proliferation, invasion and migration, and intensified the sensitivity of A2780/DDP cells to cisplatin (DDP). GTW, especially GTW+DDP, significantly inhibited the expression of N-cadherin, integrin-linked kinase (ILK), phospho-protein kinase B/AKT (PKB/p-AKT), phospho-glycogen synthase kinase (p-GSK3β) and Slug, while it increased E-cadherin levels by inhibiting EMT via the ILK/AKT/GSK3β/Slug signalling pathway. Animal results indicated that GTW, especially GTW+DDP, significantly reduced tumour burden, prolonged the life span of mice, and down-regulated the levels of tumour markers CA125 and HE4 by regulating EMT through the ILK/AKT/GSK3β/Slug signalling pathway.
Conclusion: Our results highlighted the significance of EMT in EOC metastasis, invasiveness and resistance to DDP and investigated the potential role of GTW as an adjuvant therapeutic agent in chemo-resistant EOC.
Keywords: epithelial ovarian cancer (EOC), epithelial-mesenchymal transition (EMT), A2780/DDP cells, Cisplatin (DDP) resistance, Tripterygium glycosides (GTW), ILK/AKT/GSK3β/Slug pathway
Introduction
Ovarian cancer is the deadliest gynaecological malignancy and the main cause of cancer-related deaths in women, with epithelial ovarian cancer (EOC) being the most common type of malignant ovarian tumour, occurring in 90% of cases [1]. The treatment of ovarian cancer broadly encompasses surgical treatment, radiotherapy and chemotherapy, molecular targeted therapy, immunotherapy and photodynamic therapy, depending on cancer stage, the histopathological type, the age of the patient, the desire of the patient to have children and general health [2, 3]. EOC patients often presents with advanced stage, often received cytoreductive surgery followed by platinum-based chemotherapy, and 80% of these women will develop platinum-resistant and relapse within 12-18 months of their primary treatment [4, 5]. Therefore, there is an urgent need to develop new therapies to improve patient prognosis.
Drug resistance is a multi-factorial phenomenon involving various mechanisms, characterised by decreasing cisplatin uptake, enhancing DNA damage tolerance and repair, increasing the efflux of cisplatin caused by cellular glutathione or metallothionein cause [6-8]. Moreover, recent reports have indicated that cancer progression, migration, invasion, and chemotherapeutic resistance may occur as a result of epithelial-mesenchymal transition (EMT) [9]. The acquisition of an EMT phenotype has been demonstrated in paclitaxel-resistant ovarian cancer cells [10], tamoxifen/adriamycin-resistant breast cancer cells [11], gemcitabine/5-FU-resistant pancreatic cancer cells [12], gefitinib-resistant non-small cell lung cancer and oxaliplatin-resistant colorectal cancer cells [13], and blockade of the EMT pathways is critical for preventing cancer cell migration and invasion as well as for restoring drug sensitivity [14, 15].
Several key signalling pathways, including the phosphatidylinositol 3-kinase (PI3K) pathway, are known to be involved in EMT [16]. As a component of the PI3K pathway located upstream of protein kinase B/AKT (PKB/AKT), integrin-linked kinase (ILK) is a serine/threonine protein kinase, which was originally identified by interacting with β1 and β3 integrin ligands [17]. ILK has been shown to play a fundamental role in regulating the survival, proliferation, migration, invasion, and angiogenesis of cells by mediating the integrin signal in various cells [18-20]. It also plays an important role in EMT for activated ILK via phosphorylating its downstream target kinases, PKB/AKT at Ser 473 and glycogen synthase kinase (GSK3β) at Ser 9 [21], which in turn affects cell survival, cell cycle, and changes extracellular matrix (ECM) modifications and cell adhesion [22]. Thus, EOC patients may benefit from targeted therapies that inhibit EMT.
Tripterygium glycosides (GTW) is a fat-soluble mixture obtained from the roots of the genus Tripterygium wilfordii. It has been approved by the China State Food and Drug Administration (Z32021007) for the conventional treatment of inflammatory bowel disease (IBD) (Crohn's disease (CD) and ulcerative colitis (UC)) [23, 25] and rheumatoid arthritis (RA) [24]. In addition GTW possess other biological activities for the treatment of lupus, cancer, and nephritic syndrome [26, 27]. Recent studies have shown that triptolide (TPL), one of the main components of GTW, could inhibit EMT through the PI3K/AKT/mTOR pathway to exert anti-tumour effect [28]. In the previous research by our team, we verified the effect of TPL on drug-resistant EOC cells in vitro and in vivo [29-31]. Our group indicated that TPL had synergistically enhanced the cytotoxicity of cisplatin (DDP), and promoted the apoptosis of the SKOV3PT cells through the inhibition of nuclear factor kappa-B (NF-κB) in a p53-independent pathway inducing mitochondria-derived reactive oxygen species (ROS) accumulation [29]. However, whether GTW has an inhibitory effect like TPL on the proliferation, invasion and metastasis of drug-resistant EOC remains unclear.
Our study aimed to investigate whether GTW could inhibit metastasis and enhance the sensitivity of drug-resistant human EOC cells (A2780/DDP) to DDP by suppressing EMT via targeting the ILK/AKT/GSK3β/Slug signalling pathway, and explore the underlying mechanism.
Materials and methods
Cell lines and culture
The cisplatin-resistant A2780/DDP cell line (human epithelial ovarian carcinoma-derived) was purchased from Cell Bank of Chinese Academy of Sciences (Shangha, China). A2780/DDP was cultured in RPMI-1640 medium (Gibco Life Technologies, Grand Island, NY, USA) containing 10% foetal bovine serum (Biological Industries, Israel), 100 U/mL penicillin/streptomycin (Solarbio, China) in a 37 °C incubator containing 5% CO2.
Cell viability assay
A2780/DDP cells at logarithmic growth stage were seeded in 96-well plates with 1000 cells/well with five replicates for each test condition, and allowed to attach overnight in a humidified atmosphere containing 5% CO2 at 37 °C. Then, the cells were exposed to 100 µL/well various concentrations of GTW (Preferred, Chengdu, China) (0, 50, 200, 800, 1600, 3200 µg/mL) and DDP (Hansoh Pharma, China) (0.5625, 1.25, 2.5, 5, 10 and 20 µg/mL) for 24 h. After incubation, 20 µL of fresh medium containing 10 µL of CCK-8 solution (Beijing Zoman Biothchnology Co.,Ltd, China) was added to each well and incubated for 1.5 h at 37 °C. The absorbance at 450 nm was measured by a micoplate reader (Bio-Rad, CA, USA).
Cellular migration and invasion assays
A 24-well Boyden chamber (8 μm pore size; Corning Costar, USA) was used for transwell migration assay. Here, 2 × 104 or 4 × 104 A2780/DDP cells were loaded onto the top of the 24-well migration chamber in 100 µL serum-free media, and filled 600 µL medium containing 20% FBS into the lower chamber. Cells after incubating with a range concentrations of GTW (0, 50, 200, 800, 1600 and 3200 µg/mL) and a various concentrations of DDP (0.5625, 1.25, 2.5, 5, 10 and 20 µg/mL) in the incubator for 24 h, the cells has migrated into the lower surface of the filter were fixed with 4% paraformaldehyde for 15 minutes, and then stained with 0.1% crystal violet solution for 30 minutes. Pictures (100×) were taken with an Olympus IX51 (Olympus Optical, Melville, NY, USA) inverted microscope and 5 visual fields were counted. The sterile Borden chamber was also used for invasion measurements. BD Matrigel (BD Biosciences, USA) was placed in a -4 °C refrigerator overnight before commencing the experiment, at which point the BD matrix will become liquid. It was diluted with serum-free medium in an 1:8 ratio. When the upper chamber has been pre-coated with Matrigel, operations similar to migration analysis were performed.
Small interfering RNA (siRNA) transfection
Slug (U) and ILK (I) specific siRNA were purchased from RIBO Company (Shanghai, China). The logarithmic growth phase A2780/DDP cells were divided into 8 groups: (1) blank control group; (2) DDP group (10 µg/mL DDP for 24 h); (3) GTW group (800 µg/mL GTW for 24 h); (4) DDP+GTW group (800 µg/mL GTW and 10 µg/mL DDP for 24 h); (5) U or I (10 μM for 24 h); (6) DDP+U or DDP+I group; (7) GTW+U or GTW+I group; and (8) DDP+GTW+U or DDP+GTW+I group. For siRNA transfection, A2780/DDP cells were planted in 6-well plates without streptomycin treated with 50-60% confluence before transfection. Cell transfection was carried out using GeneTran III Transfection Reagent (Biomiga, Inc., USA). After incubation for 6 h, the culture medium was changed into fresh RPMI-1640 medium containing 10% FBS. After 48 h of transfection, cells were harvested for the following cell experiments. For A2780/DDP the sequence for Slug specific siRNA is 5'-CCCAUUCUGAUGUAAAGAATT-3', 5'-UUCUUUACAUCAGAAUGGGTT-3', respectively. Also, the sequence for ILK specific siRNA is 5'-GACCCAAAUUUGACAUGAUTT-3', 5'-AUCAUGUCAAAUUUGGGUCTT-3', respectively, in A2780/DDP cells.
Detection of AKT and GSK3β activity using their inhibitor
The AKT inhibitor (A) and GSK3β inhibitor (S) were purchased from APExBIO (Houston, USA). The logarithmic growth phase A2780/DDP cells were divided into 8 groups: (1) blank control group; (2) DDP group (10 µg/mL DDP for 24 h); (3) GTW group (800 µg/mL GTW for 24 h); (4) DDP+GTW group (800 µg/mL GTW and 10 µg/mL DDP for 24 h); (5) A or S (10 µM for 24 h); (6) DDP+A or DDP+S group; (7) GTW+A or GTW+S group; and (8) DDP+GTW+A or DDP+GTW+S group. A2780/DDP cells (5×105 per well) were plated in six-well plates overnight and the experimental conditions were set in triplicate. Then MK2206 (AKT inhibitor, 10 μM) or AR-A014418 (GSK3β inhibitor, 10 μM) was added to prevent AKT or GSK3β activity. After 24 h of incubation, cells were collected for subsequent experiments.
Western blotting
The protein of cells were lysed in RIPA lysis buffer (Solarbio, China) on ice, and the tumour tissues were homogenised in RIPA lysis buffer (Solarbio, China), before samples were centrifuged at 10000 rpm for 30 min at 4 °C. The protein concentration of the samples was measured using BCA kit (Thermo scientific, USA), subsequently, they were boiled for 5-10 min in 1× protein loading buffer. The proteins were resolved using polyacrylamide gel electrophoresis, electrotransferred to the polyvinylidene difluoride membranes, and then nonspecific binding sites were blocked using 5% bovine serum albumin (BSA) (Servicebio, China) in Tris buffered saline with Tween 20 (TBST) (Solarbio, China) for 2 h at room temperature. Polyvinylidene fluoride (PVDF) membranes were then incubated with the following primary antibodies at 4 °C overnight: anti-β-actin (1:5000; Proteintech, Cat# 60008-1-lg; RRID: not registered; having reactivity with mouse), anti-AKT (1:1000; Proteintech, Cat# 10176-2-AP; RRID: AB_2224574; having reactivity with mouse), anti-p-AKT (1:2000; Proteintech, Cat# 66444-1-lg; RRID: not registered; having reactivity with mouse), anti-GSK3β (1:1000; Proteintech, Cat# 22104-1-AP; RRID: not registered; having reactivity with mouse), anti-p-GSK3β (1:1000; Proteintech, Cat# 14850-1-AP; RRID: not registered; having reactivity with mouse), anti-ILK (1:1000; Proteintech, Cat# 12955-1-AP; RRID: AB_2127053; having reactivity with mouse), anti-Slug (1:1000; abcam, Cat# ab180714; RRID: AB_2728773; having reactivity with mouse), anti-N-cadherin (1:2000; Proteintech, Cat# 22018-1-AP; RRID: AB_2813891; having reactivity with mouse), and anti-E-cadherin (1:5000; Proteintech, Cat# 20874-1-AP; RRID: AB_10697811; having reactivity with mouse). Cells were washed with TBST buffer, and then appropriate amounts of secondary antibody, e.g. goat anti-rabbit secondary antibody (1:1000; Abcam, Cat# 205718; RRID: not registered), or goat anti-mouse secondary antibody (1:1000; Abcam, Cat# 97265; RRID: not registered), conjugated with horseradish peroxidase (HRP) was added for another 1.5 h at room temperature.
Mouse model of ovarian cancer
Female nude mice, aged 6 weeks, were purchased from Hunan SJA Laboratory Animal Co., Ltd. (Changsha, Hunan, China; RRID: MGI_5656552) randomly divided into four groups (N = 12 per group), and acclimatised for one week. Mice received 1×106 A2780/DDP cells subcutaneously suspended in PBS. When the tumour reaches 50 mm3, mice were treated as follows: (1) Model (50 mL/kg/day PBS was intraperitoneally injected for a total of 14 days); (2) DDP (4 mg/kg/day DPP was intraperitoneally injected for the first and eighth day, 2 days in total); and (3) GTW (0.15 mg/kg/day GTW gavage, 0.15 mg/kg/day for a total of 14 days), DDP + GTW (4 mg/kg/day DPP was intraperitoneally injected for the first and eighth day, 2 days in total and 0.15 mg/kg/day GTW gavage, 0.15 mg/kg/day for a total of 14 days). The tumour volume was measured twice a week and weighed the mice. At day 22, three mice from each group were euthanized; their tumours were removed and frozen at -80°C for further study. The remaining mice were used to assess survival curves. Tumour volume was determined as follows:
Tumour volume = length × width2 × 0.5 (repeated three times)
ELISA
Blood was collected from mice of each group (day 22) via retro-orbital bleeding. Fresh blood was allowed to stand at room temperature for 10 minutes, and then spun in a centrifuge (3000 rpm, 10 min). After centrifugation, the serum fraction was immediately transferred to a new centrifuge tube and stored at -80 °C until use. The levels of tumour markers (CA125 and HE4) in serum samples were determined through the mouse ELISA kits (both from eBioscience, USA).
Statistical analysis
Statistical analysis was performed by one-way ANOVA or two-way ANOVA and expressed as mean ± SD. P < 0.05 indicates a statistically significant difference.
Results
GTW prohibits the proliferation, migration and invasion characteristics of A2780/DDP cells
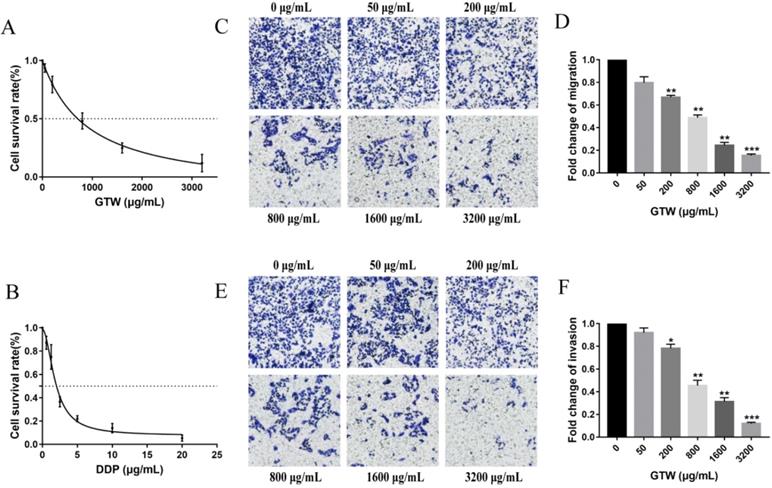
To detect the effect of GTW on the proliferation of epithelial drug-resistant EOC cells, A2780/DDP cells were treated with a range of concentrations of GTW (0, 50, 200, 800, 1,600 and 3,200 µg/mL) for 24 hours. As shown in Figure 1A, GTW had the effect of inhibiting the proliferation of A2780/DDP cells in a dose-dependent manner, and the IC50 of GTW on A2780/DDP cells was 882.1 µg/mL. Furthermore, we treated A2780/DDP cells with DDP (0.5625, 1.25, 2.5, 5, 10 and 20 µg/mL) (Figure 1B); similar results were obtained showing that the cell proliferation inhibition of DDP was dose-dependent, as the IC50 of DDP on A2780/DDP cells was 1.891 µg/mL. Transwell migration and invasion tests were used to determine the influence of GTW on the migration and invasion abilities of A2780/DDP cells. As expected, the migration and invasion abilities of A2780/DDP cells were significantly inhibited after 24 hours of GTW treatment (Figure 1C-F) (P < 0.05). All of the results showed that GTW had a significant inhibitory effect on proliferation, migration and invasion of A2780/DDP cells in vitro.
GTW inhibited the proliferation, migration, and invasion capabilities of A2780/DDP cells in vitro. (A) A2780/DDP cells were treated with a range of concentrations of GTW for 24 hours to examine cell survival; (B) A2780/DDP cells were treated with a range of concentrations of DDP for 24 hours to examine cell survival; (C-F) Transwell migration and invasion assay of A2780/DDP cells after treatment with increased concentrations of GTW (×100 magnification). ∗ P < 0.05, ∗∗ P < 0.005, and ∗∗∗ P < 0.001.
GTW inhibited EMT in A2780/DDP cells by targeting the Slug
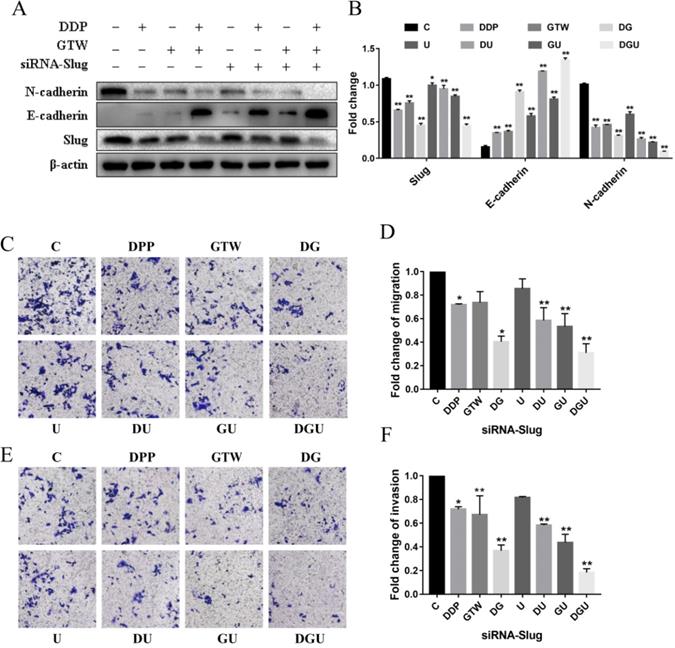
In view of the fact that GTW can effectively suppress the migration and invasion of A2780/DDP cells, we used western blot to detect the expression of EMT-related factors to assess whether GTW could affect EMT and sensitise the drug sensitivity of drug-resistant of EOC cells to DDP. As shown in Figure 2A-B, supplementation with GTW or DDP up-regulated the expression of epithelial marker E-cadherin and down-regulated the mesenchymal marker N-cadherin to a certain extent compared with the control group (P < 0.005). In addition, the expression of the transcription factor Slug was also inhibited after exposure to GTW or DDP. Furthermore, GTW combined with DDP played a synergistic effect on the up- and down-regulation of the above proteins (P < 0.005). To confirm the role of Slug channel in EMT, specific siRNA was performed to interfere with the production of Slug in the A2780/DDP cell line. After transfection of siRNA-Slug, the expression of Slug and N-cadherin was reduced while the expression of E-cadherin was increased in A2780/DDP cells, and siRNA-Slug intensified the synergistic effect of GTW and DDP combination (P < 0.005). Additionally, we also studied the effects of Slug silencing on cell migration and invasion followed by GTW, DDP and GTW+DDP treatment (Figure 2C-F). Unsurprisingly, the migration and invasion abilities of cells were suppressed most effectively in siRNA-Slug treatment followed by treatment with the combination of GTW+DDP (P < 0.005).
GTW inhibited EMT in A2780/DDP cells through targeting Slug. (A-B) A2780/DDP cells were transfected with siRNA-Slug and cultured for an additional 48 h, then incubated with GTW (800 µg/mL) or DDP (10 µg/mL) for 24 hours. Western blot analysis for the expression of Slug, E-cadherin, and N-cadherin protein; (C-F) Transwell migration and invasion assay of A2780/DDP cells after transfection with siRNA-Slug combined with GTW, DDP or GTW+DDP treatment. *P < 0.005, **P < 0.001.
GTW inhibited EMT by targeting ILK in A2780/DDP cells
Next, we detected the levels of ILK and its downstream targets (p-AKT and p-GSK3β) in A2780/DDP cells after GTW treatment. ILK expression was significantly reduced, and the expression of p-AKT and p-GSK3β was also decreased after 24 hours of GTW or DDP incubation (Figure 3A-B). In addition, the level of Slug was also down-regulated, consistent with the results of our former research. When associating GTW with DDP, the expression of these proteins was further suppressed (P < 0.005). To confirm whether GTW inhibited the EMT of drug-resistant EOC cells by targeting ILK, we silenced the ILK in A2780/DPP cells by transfecting them with siRNA-ILK. After siRNA-ILK transfection, we found that the levels of ILK, p-AKT, p-GSK3β and Slug were all down-regulated in A2780/DDP cells, and the siRNA-ILK reinforced the synergistic effect of GTW and DDP combination (P < 0.005).
GTW repressed EMT by targeting ILK in A2780/DDP cells. (A-B) A2780/DDP cells were transfected with siRNA-ILK and treated with GTW (800 µg/mL) or DDP (10 µg/mL) for 24 hours. The expression of ILK, AKT, p-AKT, GSK-3β, p-GSK-3β, Slug, E-cadherin, and N-cadherin protein; (C-F) Transwell migration and invasion assay of A2780/DDP cells after transfection with siRNA-ILK combined with GTW, DDP or GTW+DDP treatments. *P < 0.005, **P < 0.001.
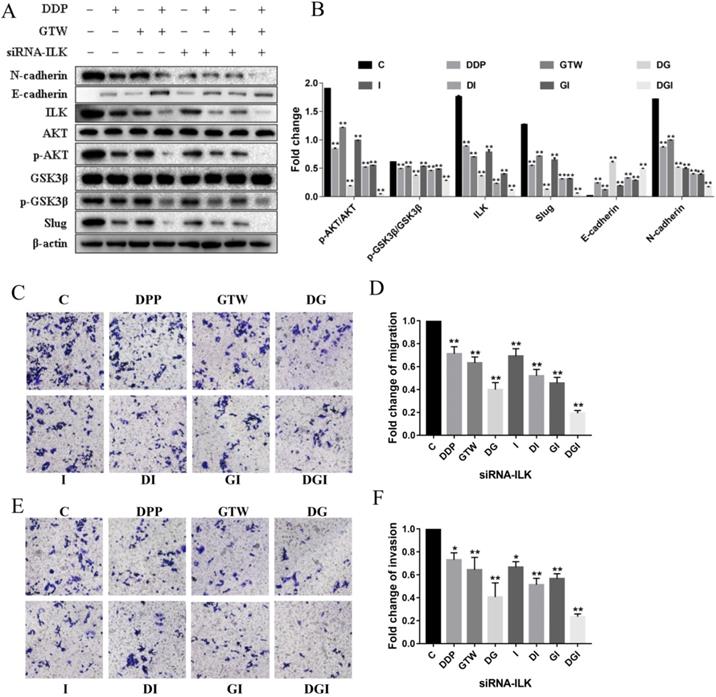
We also detected EMT related markers in A2780/DDP cells. ILK knockdown can further increase the expression of E-cadherin and suppress the level of N-cadherin in A2780/DDP cells after GTW, DDP and GTW+DDP treatment; in particular, GTW+DPP has the most obvious effect (P < 0.005). Furthermore, we also studied the effects of ILK silencing on cell migration and invasion followed by GTW, DDP and GTW+DDP treatment (Figure 3C-F). After siRNA-ILK transfection, the migration and invasion abilities of A2780/DDP cells were weakened, while the combination of GTW+DDP showed the most obvious inhibitory effect (P < 0.005). Based on the above results, we concluded that GTW inhibited EMT by targeting ILK in A2780/DDP cells.
GTW inhibited EMT of A2780/DDP cells via the ILK/AKT signalling pathway
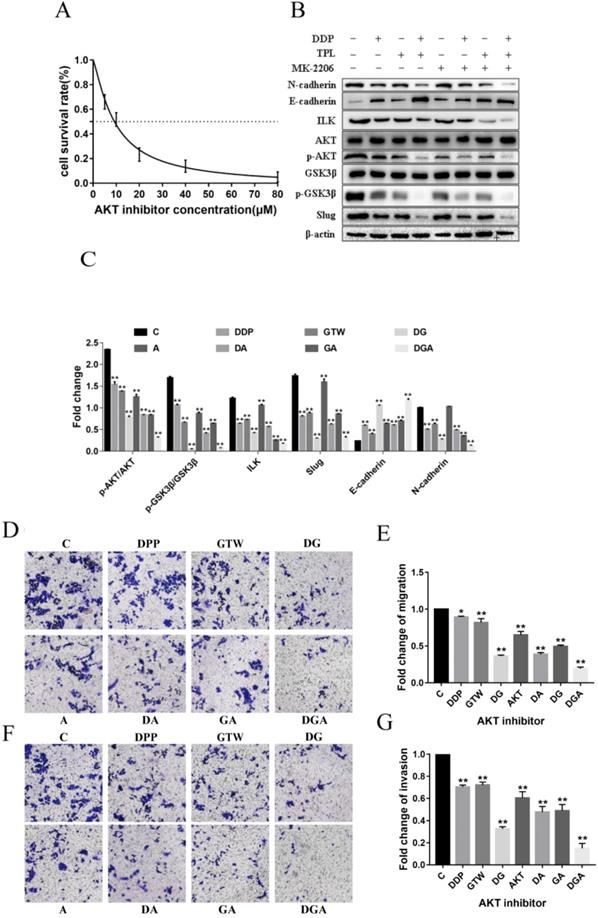
To prove whether GTW inhibited EMT of A2780/DDP cells by down-regulating AKT, we incubated the cells with an AKT inhibitor (MK2206) for 6 h. As shown in Figure 4A, MK2206 inhibited the growth of A2780/DDP cells in a dose-dependent manner. MK2206 restored the GTW, DDP and GTW+DDP-induced down-regulation of p-AKT, p-GSK3β, except for ILK. Additionally, MK2206 also restored the GTW, DDP and GTW+DDP-mediated up-regulation of E-cadherin levels, and the down-regulation of N-cadherin levels in A2780/DDP cells (Figure 4B-C) (P < 0.005). Similarly, we conducted transwell experiments on A2780/DDP cells after MK2206 inhibitor incubation. The AKT inhibitor restored the effects of GTW, DDP and GTW DDP on migration and invasion abilities (Figure 4D-G) (P < 0.005). Therefore, GTW suppressed EMT of A2780/DDP via the ILK/AKT signalling pathway.
GTW suppressed the EMT of A2780/DDP cells via the ILK/AKT signalling pathway. (A) (A) A2780/DDP cells were treated with a range of concentrations of AKT inhibitor (MK2206) for 24 hours to examine cell survival; (B-C) A2780/DDP cells were treated with 10 µM MK2206 and exposed with GTW (800 µg/mL) or DDP (10 µg/mL) for 24 hours. Protein levels of ILK, AKT, p-AKT, GSK3β, p-GSK3β, Slug, E-cadherin and N-cadherin were shown; (D-G) Transwell migration and invasion assay of A2780/DDP cells after incubation with AKT inhibitor combined with GTW, DDP or GTW+DDP treatment. *P < 0.005, **P < 0.001.
GTW inhibits EMT of A2780/DDP cells through the ILK/AKT/GSK3β signalling pathway
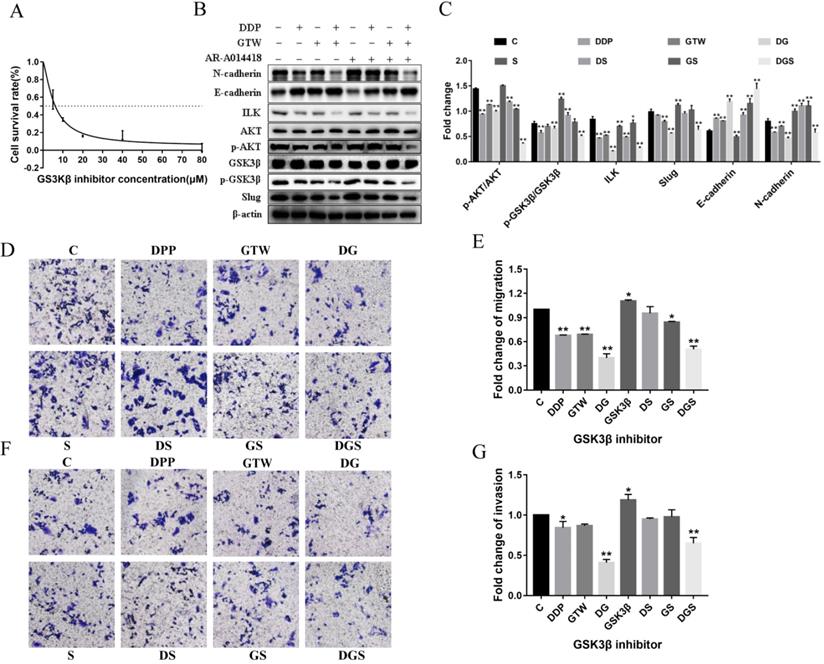
To determine whether ILK acts by targeting GSK3β to suppress the EMT of A2780/DDP cells, we incubated the cells with GSK3β inhibitor (AR-A014418) at different concentrations for 6 h. As shown in Figure 5A, AR-A014418 inhibited the growth of A2780/DDP cells in a dose-dependent manner. AR-A014418 reversed the GTW, DDP and GTW+DDP-induced down-regulation of p-GSK3β and Slug expression except for ILK and AKT (P < 0.05). In addition, AR-A014418 also reversed the GTW, DDP and GTW+DDP-mediated up-regulation of E-cadherin levels, and the down-regulation of N-cadherin levels in A2780/DDP cells (Figure 5B-C) (P < 0.05). Similarly, we conducted transwell experiments on A2780/DDP cells after GSK3β inhibitor treatment (Figure 5D-G). As expected, GSK3β inhibitor reversed the effects of GTW, DDP and GTW DDP on cell migration and invasion abilities (P < 0.05). The above results indicated that GTW inhibited the EMT of A2780/DDP cells through the ILK/AKT/GSK3β signalling pathway.
GTW suppressed the EMT of A2780/DDP cells via the ILK/AKT/GSK3β signalling pathway. (A) A2780/DDP cells were treated with a range of concentrations of GSK3β inhibitor (AR-A014418) for 24 hours to examine cell survival; (B-C) A2780/DDP cells were treated with 10 µM AR-A014418 and exposed with GTW (800 µg/mL) or DDP (10 µg/mL) for 24 hours. The expression of ILK, AKT, p-AKT, GSK3β, p-GSK3β, Slug, E-cadherin and N-cadherin protein; (D-G) Transwell migration and invasion assay of A2780/DDP cells after incubation with GSK3β inhibitor combined with GTW, DDP or GTW+DDP treatment. *P < 0.05, **P < 0.001.
GTW suppressed the proliferation of human tumour cells in vivo
On the basis of the in vitro findings, we moved on to determine the role of GTW in vivo. A2780/DDP cells were injected hypodermically into the armpits of female nude mice. The anti-tumour effects were assessed after treatment with GTW alone, DDP alone and a combination of GTW+DDP. As shown in Figure 6A-B, DDP, GTW and GTW+DDP considerably delayed tumour progression, of which GTW+DDP effectively reduced the tumour volumes compared with the control group (P < 0.05). Also, we found that GTW, DDP and DDP+GTW can significantly prolong the survival time of mice, and DDP+GTW enhanced the survival rate up to day 50 (Figure 6C).
EMT repressed the proliferation, migration, and invasion abilities of A2780/DDP cells in vivo. (A-B) Weights (N = 3) and volumes (N = 4) of tumours in different treatment groups; (C) Kaplan-Meier survival curves of mice bearing melanoma models after the intraperitoneal administration of GTW or DDP (N = 5); (D-E) CA125 and HE4 levels in serum were measured by ELISA; (F-G) Relative expressions of ILK, AKT, p-AKT, GSK3β, p-GSK3β, Slug, E-cadherin and N-cadherin in tumour tissues. *P < 0.05, **P < 0.005, ***P < 0.001.
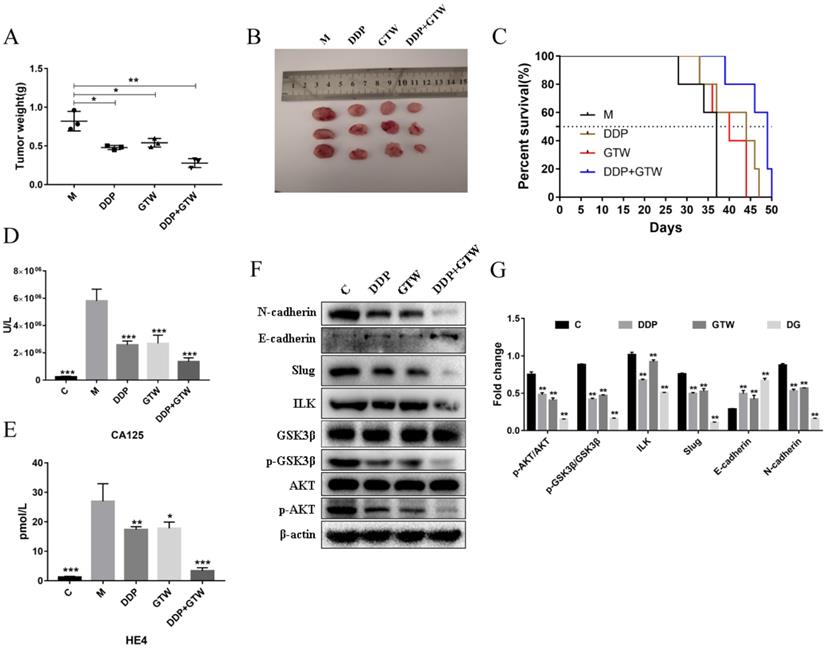
Furthermore, we studied the expression of serum tumour markers CA125 and HE4. As shown in Figure 6D-E, both GTW and DDP reduced the levels of CA125 and HE4 in nude mice serum, while DDP+GTW showed a stronger inhibitory effect (P < 0.005). Therefore, we believed that GTW combined with DDP could improve the prognosis of mice by reducing the serum levels of CA125 and HE4.
GTW inhibited the EMT of tumour cells through the ILK/AKT/GSK3β/Slug pathway in vivo
The tumours removed from nude mice were subjected to western blot assays. It was found that both GTW and DDP up-regulated the level of E-cadherin and down-regulated the levels of N-cadherin, ILK, p-AKT p-GSK3β, and Slug. DDP+GTW had a more obvious regulatory effect on the above related-protein expressions in tumour tissues (Figure 6F-G) (P < 0.05). The above data showed that GTW suppressed the EMT of A2780/DDP cells by targeting ILK/AKT/GSK3β/Slug in vivo.
Discussion
Among cancers in women, EOC has the highest fatality-to-incidence ratio. Most patients are diagnosed at an advanced stage with large-scale metastasis and development of chemo-resistant properties [32]. EMT is a process whereby cancer cells achieved migratory and invasive properties as a result of alterations in cell-cell adhesion, cell polarity and cell-extracellular matrix interaction [33]. Therefore, it is related to cancer metastasis and the acquisition of chemotherapy resistance properties of other cancers such as colorectal cancer [34], lung cancer [35], and pancreatic cancer [36]. Previous studies showed that TPL inhibited the migration and invasion abilities of drug-resistant EOC cells in vitro and in vivo [29-31]. However, whether GTW has the ability to modulate EMT and drug resistance of DDP-resistant EOC cells and the potential mechanisms are still not fully understood.
The absence or decreased expression of E-cadherin is regarded as a major characteristic of EMT that is a consequence of the actions of a number of TFs (Snail, Slug, Twist, ZEB1/2 and SIP1) that target the E-box sequences within the promoter region of E-cadherin [37-39]. Slug is closely related to tumour metastasis of ovarian cancer [40]. In this study, we found that the proliferation, migration and invasion of A2780/DDP cells were decreased in a dose-dependent manner after GTW treatment (Figure 1). GTW, especially GTW+DDP, could up-regulated E-cadherin expression and mediated N-cadherin down-regulation. We found that GTW suppressed the migration and invasion abilities of A2780/DDP cells and strengthened cell-sensitivity to DDP by inhibiting EMT (Figure 2). Slug was also suppressed after GTW incubation. Additionally, Slug silence suppressed EMT, which was verified by molecular and functional experiments. Based on this, we firmly believed that GTW inhibited EMT of A2780/DDP cells by targeting Slug (Figure 2).
Accumulating genetic and cancer biology evidence demonstrate that the PI3K/AKT signalling pathway is a central mechanism controlling EMT features, for its definite effects on cancer cell growth and survival [16, 41]. As the upstream regulatory molecule of AKT, ILK is a key factor that regulates the interaction between extracellular matrix and cells by phosphorylating of its downstream substrates AKT and GSK3β [42]. ILK has significant effects on the development and progression of human carcinoma [20, 43, 44], including promoting the metastatic behaviour of EOC cells [45]. It has been reported that ILK facilitates cancer cell migration, invasion and drug-resistance by regulating EMT processes [46]. What is more, emodin restrained EMT through the ILK/AKT/mTOR signalling pathway in breast cancer [47]. However, whether GTW can suppress ILK level in DDP-resistant EOC cells is unclear. In our study, we observed that GTW, especially GTW+DDP could significantly inhibit the expression of ILK in A2780/DDP cells. In order to clarify how ILK regulate EMT, we transfected A2780/DDP cells with siRNA-ILK to knock down the ILK expression. GTW, especially GTW+DDP, strengthened the influence of siRNA-ILK on the expression of E-cadherin and N-cadherin. In addition, siRNA-ILK inhibited the migration and invasion abilities of A2780/DDP cells. The existing research results showed that GTW weakened the migration and invasion abilities of A2780/DDP cells and strengthened cell-sensitivity to DDP by inhibiting EMT via targeting ILK (Figure 3).
ILK acts as a vital regulator in the PI3K/AKT pathway to phosphorylate its downstream targets such as AKT and GSK3β to regulate their activities [44]. ILK interacts with AKT and stimulates phosphorylation of AKT, leading to its activation. ILK can also direct and/or indirect stimulate phosphorylation of GSK3β. In addition, AKT participates in regulating the wingless/integrated (Wnt)/β-catenin signalling pathway by phosphorylating GSK3β, leading to the inactivation of GSK3β [48, 49]. In hepatocellular carcinoma (HCC), transfection with an ILK expression vector was able to recover the decreased expression of its downstream genes AKT and GSK3β phosphorylation, and affected cell proliferation and apoptosis [44]. In oral squamous cell carcinoma cells (OSCC), the knockdown of ILK level inhibited EMT by suppressing the phosphorylation of downstream signalling targets AKT and GSK3β both in vitro and in vivo [50]. In the present study, we confirmed that GTW, especially GTW+DDP, inhibited EMT by targeting ILK and the expression of p-AKT and p-GSK3β was down-regulated, consistent with ILK (Figure 3). Moreover, siRNA-ILK significantly decreased the levels of ILK, p-AKT, p-GSK3β, and Slug in A2780/DDP cells, which were reinforced by GTW, especially GTW+DDP (Figure 3). The AKT inhibitor reversed the GTW and GTW+DDP-induced effects of E-cadherin, N-cadherin, p-AKT, p-GSK3β, and Slug, except ILK (Figure 4). Similarly, in addition to ILK and p-AKT, the GSK3β inhibitor restored the effects of E-cadherin, N-cadherin, p-GSK3β and Slug induced by GTW and GTW+DDP (Figure 5). In terms of xenografts in nude mice, GTW, especially GTW+DDP, could decelerate tumour progression, prolong the survival rate of mice and down-regulate tumour-related protein expression (Figure 6). To sum up, our data confirmed that GTW inhibited EMT related metastasis and chemo-resistance (DDP-resistance) both in vitro and in vivo through the ILK/AKT/GSK3β/Slug pathway.
Cancer antigen 125 (CA125) is a commonly and initially used tumour biomarker that is enhanced in 80% of females with advanced-stage ovarian cancer [51], and higher level of CA125 usually indicates poor treatment outcomes and prognosis [52]. Human epididymis protein 4 (HE4) is considered a new biomarker for diagnosing ovarian cancer either at the early or late stages. In addition, HE4 levels also had good sensitivity in ovarian cancer patients when monitoring the prognosis condition postoperatively [53]. Overall, the combination of the two markers is more reliable and will increase the predictive value [54, 55]. In our research, we showed that GTW, especially GTW+DDP could significantly reduce the levels of CA125 and HE4 in the serum of nude mice (Figure 6). The above results indicated that GTW can improve the prognosis of drug-resistant EOC and strengthened cancer cell-sensitivity to DDP.
Conclusion
Overall, our study indicated that GTW can inhibit the proliferation, migration and invasion abilities as well as intensify sensitivity to DDP of A2780/DDP cells by suppressing EMT in vitro and in vivo. Furthermore, we confirmed that the role of GTW in EMT was regulated via the ILK/AKT/GSK3β/Slug signalling pathway. All of these results indicate that GTW is used as an adjuvant therapy for drug-resistant EOC in clinic. However, the lack of normal and other cancer cells as internal and external controls makes it a limitation in the present study.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 81760729, 82060638) and the Jiangxi Government (20202BABL206102, 20202BAB216005), “double 10-thousand plan” of Jiangxi Province (innovation and technology professionals as the highend talent).
Authors' contributions
Buzhen Tan and Tingtao Chen designed the current experiment; Fuyin Le, Ying Feng, Puyuan Tian, Yanying Zhong, Fuliang Zhan, Genhua Huang, Hui Hu performed the experiments; Fuyin Le, Tingtao Chen, Puyuan Tian, Buzhen Tan and Ying Feng analysed all data and wrote this article. All authors read and approved the final manuscript.
Ethical approval
This study was approved by the Ethics Committee of the Nanchang Royo Biotech Co,. Ltd (RYE2019062701) and all studies were conducted according to approved guidelines.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Coburn SB, Bray F, Sherman ME, Trabert B. International patterns and trends in ovarian cancer incidence, overall and by histologic subtype. Int J Cancer. 2017;140:2451-60
2. Stephen A, Cannistra M D. Cancer of the ovary. New Engl J Med. 2004;329:1550-59
3. Marcus CS, Maxwell GL, Darcy KM, Hamilton CA, McGuire WP. Current approaches and challenges in managing and monitoring treatment response in ovarian cancer. J Cancer. 2014;5:25-30
4. Morgan RD, Clamp AR, Evans DGR, Edmondson RJ, Jayson GC. PARP inhibitors in platinum-sensitive high-grade serous ovarian cancer. Cancer Chemoth Pharm. 2018;81:647-58
5. Jayson GC, Kohn EC, Kitchener HC, Ledermann JA. Ovarian cancer. Lancet. 2014;384:1376-88
6. Kartalou M, Essigmann JM. Mechanisms of resistance to cisplatin. Mutat Res. 2001;478:23-43
7. Wernyj RP, Morin PJ. Molecular mechanisms of platinum resistance: still searching for the Achilles' heel. Drug Resist Updat. 2004;7:227-32
8. Ohmichi M, Hayakawa J, Tasaka K, Kurachi H, Murata Y. Mechanisms of platinum drug resistance. Trends Pharmacol Sci. 2005;26:113-6
9. Savagner P. The epithelial-mesenchymal transition (EMT) phenomenon. Ann Oncol. 2010;21(Suppl 7):vii89-92
10. Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A. et al. Chemoresistance to paclitaxel induces epithelialmesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol. 2007;31:277-83
11. Hiscox S, Jiang WG, Obermeier K, Taylor K, Morgan L, Burmi R. et al. Tamoxifen resistance in MCF7 cells promotes EMT-like behaviour and involves modulation of beta-catenin phosphorylation. Int J Cancer. 2006;118:290-301
12. Shah AN, Summy JM, Zhang J, Park SI, Parikh NU, Gallick GE. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann Surg Oncol. 2007;14:3629-37
13. Rho JK, Choi YJ, Lee JK, Ryoo BY, Na IIl, Yang SH. et al. Epithelial to mesenchymal transition derived from repeated exposure to gefitinib determines the sensitivity to EGFR inhibitors in A549, a non-small cell lung cancer cell line. Lung Cancer. 2009;63:219-26
14. Rosano L, Cianfrocca R, Spinella F, Castro VD, Nicotra MR, Lucidi A. et al. Acquisition of chemoresistance and EMT phenotype is linked with activation of the endothelin A receptor pathway in ovarian carcinoma cells. Clin Cancer Res. 2011;17:2350-60
15. Du F, Wu X, Liu Y, Wang T, Qi X, Mao Y. et al. Acquisition of paclitaxel resistance via PI3Kdependent epithelialmesenchymal transition in A2780 human ovarian cancer cells. Oncol Rep. 2013;30:1113-18
16. Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFbeta in cancer. FEBS Lett. 2012;586:1959-70
17. Zervas CG, Psarra E, Williams V, Solomon E, Vakaloglou KM, Brown NH. A central multifunctional role of integrin-linked kinase at muscle attachment sites. J Cell Sci. 2011;124:1316-27
18. Yamaji S, Suzuki A, Kanamori H, Mishima W, Yoshimi R, Takasaki H. et al. Affixin interacts with alpha-actinin and mediates integrin signaling for reorganization of F-actin induced by initial cell-substrate interaction. J Cell Biol. 2004;165:539-51
19. Oloumi A, McPhee T, Dedhar S. Regulation of E-cadherin expression and beta-catenin/Tcf transcriptional activity by the integrin-linked kinase. BBA. 2004;1691:1-15
20. Tan C, Hennequart SC, Troussard A, Fazli L, Costello P, Sutton K. et al. Regulation of tumor angiogenesis by integrin-linked kinase (ILK). Cancer Cell. 2004;5:79-90
21. Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B-AKT by the integrin-linked kinase. Proc Natl Acad Sci. 1998;95:11211-6
22. Wu C, Dedhar S. Integrin-linked kinase (ILK) and its interactors: a new paradigm for the coupling of extracellular matrix to actin cytoskeleton and signaling complexes. J Cell Biol. 2001;55:505-10
23. Ren J, Wu X, Liao N, Wang G, Fan C, Liu S. et al. Prevention of postoperative recurrence of Crohn's disease: Tripterygium wilfordii polyglycoside versus mesalazine. J Int Med Res. 2013;41:176-87
24. Goldbach-Mansky R, Wilson M, Fleischmann R, Olsen N, Silverfield J, Kempf P. et al. Comparison of Tripterygium wilfordii Hook F Versus Sulfasalazine in the Treatment of Rheumatoid Arthritis. Ann Intern Med. 2009;151:229-40
25. Zhang H, Chen W. nterleukin 6 inhibition by triptolide prevents inflammation in a mouse model of ulcerative colitis. Exp Ther Med. 2017;14:2271-6
26. Li J, Shen F, Guan CW, Wang WW, Sun XZ, Fu XL. et al. Activation of Nrf2 Protects Against Triptolide-Induced Hepatotoxicity. Plos One. 2014;9:1-10
27. Zhou J, Xi C, Wang W, Fu X, Liang JQ, Qiu Y. et al. Triptolide-induced oxidative stress involved with Nrf2 contribute to cardiomyocyte apoptosis through mitochondrial dependent pathways. Toxicol Lett. 2014;230:454-66
28. Huang Y, Wu S, Zhang Y, Wang L, Guo Y. et al. Antitumor effect of triptolide in T-cell lymphoblastic lymphoma by inhibiting cell viability, invasion, and epithelial-mesenchymal transition via regulating the PI3K/AKT/mTOR pathway. Onco Targets Ther. 2018;11:769-79
29. Zhong YY, Chen HP, Tan BZ, Yu HH, Huang XS. Triptolide avoids cisplatin resistance and induces apoptosis via the reactive oxygen species/nuclear factor-kappaB pathway in SKOV3(PT) platinum-resistant human ovarian cancer cells. Oncol Lett. 2013;6:1084-92
30. Hu H, Luo L, Liu F, Zou D, Zhu S, Tan B. et al. Anti-cancer and Sensibilisation Effect of Triptolide on Human Epithelial Ovarian Cancer. J Cancer. 2016;7:2093-9
31. Huang G, Hu H, Zhang Y, Zhu Y, Liu J, Tan B. et al. Triptolide sensitizes cisplatin-resistant human epithelial ovarian cancer by inhibiting the phosphorylation of AKT. J Cancer. 2019;10:3012-20
32. Yap TA, Carden CP, Kaye SB. Beyond chemotherapy: targeted therapies in ovarian cancer. Nat Rev Cancer. 2009;9:167-81
33. Argenta PA, Um I, Kay C, Harrison D, Faratian D, Sueblinvong T. et al. Predicting response to the anti-estrogen fulvestrant in recurrent ovarian cancer. Gynecol Oncol. 2013;131:368-73
34. Lee TY, Liu CL, Chang YC, Nieh S, Lin YS, Jao SW. et al. Increased chemoresistance via snail-raf kinase inhibitor protein signaling in colorectal cancer in response to a nicotine derivative. Oncotarget. 2016;7:23512-20
35. Kurimoto R, Iwasawa S, Ebata T, Ishiwata T, Sekine I, Tada Y. et al. Drug resistance originating from a TGF-beta/FGF-2-driven epithelial-to-mesenchymal transition and its reversion in human lung adenocarcinoma cell lines harboring an EGFR mutation. Int J Oncol. 2016;48:1825-36
36. Yuan Y, Li D, Li H, Wang L, Tian G, Dong Y. YAP overexpression promotes the epithelial-mesenchymal transition and chemoresistance in pancreatic cancer cells. Mol Med Rep. 2016;13:237-42
37. Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741-51
38. Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D. et al. Pancreatic cancer: understanding and overcoming chemoresistance. Nat Rev Gastroenterol Hepatol. 2011;8:27-33
39. Faux MC, Coates JL, Kershaw NJ, Layton MJ, Burgess AW. Independent interactions of phosphorylated beta-catenin with E-cadherin at cell-cell contacts and APC at cell protrusions. Plos One. 2010;5:e14127
40. Gu A, Jie Y, Yao Q, Zhang Y, Mingyan E. Slug Is Associated With Tumor Metastasis and Angiogenesis in Ovarian Cancer. Reprod Sci. 2017;24:291-9
41. Persad S, Dedhar S. The role of integrin-linked kinase (ILK) in cancer progression. Cancer Metast Rev. 2003;22:375-84
42. Hannigan G, Troussard AA, Dedhar S. Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer. 2005;5:51-63
43. Troussard AA, Costello P, Yoganathan TN, Kumagai S, Roskelley CD, Dedhar S. The integrin linked kinase (ILK) induces an invasive phenotype via AP-1 transcription factor-dependent upregulation of matrix metalloproteinase 9 (MMP-9). Oncogene. 2000;19:5444-52
44. Liu L, Zhang S, Hu L, Liu L, Guo W, Zhang J. HMGA1 participates in MHCC97H cell proliferation and invasion through the ILK/Akt/GSK3beta signaling pathway. Mol Med Rep. 2017;16:9287-94
45. Reyes-González JM, Quiñones-Díaz BI, Santana Y, Báez-Vega PM, Soto D, Valiyeva F. et al. Downstream Effectors of ILK in Cisplatin-Resistant Ovarian Cancer. Cancers. 2020;12:880
46. Liu Q, Tan WH, Che JH, Yuan DD, Zhang LY, Sun YH. et al. 12-HETE facilitates cell survival by activating the integrin-linked kinase/NF-κB pathway in ovarian cancer. Cancer Manag Res. 2018;10:5825-38
47. Ma JW, Hung CM, Lin YC, Ho CT, Kao JY, Der T. WayAloe-emodin inhibits HER-2 expression through the downregulation of Y-box binding protein-1 in HER-2-overexpressing human breast cancer cells. Oncotarget. 2016;7:58915-30
48. Hu C, Dong T, Li R, Lu J, Wei X, Liu P. Emodin inhibits epithelial to mesenchymal transition in epithelial ovarian cancer cells by regulation of GSK-3beta/beta-catenin/ZEB1 signaling pathway. Oncol Rep. 2016;35:2027-34
49. Pooja T, Karunagaran D. Emodin suppresses Wnt signaling in human colorectal cancer cells SW480 and SW620. Eur J Pharmacol. 2014;742:55-64
50. Que L, Zhao D, Tang XF, Liu JY, Zhang XY, Zhan YH. et al. Effects of lentivirus-mediated shRNA targeting integrin-linked kinase on oral squamous cell carcinoma in vitro and in vivo. Oncol Rep. 2016;35:89-98
51. Ghasemi N, Ghobadzadeh S, Zahraei M, Mohammadpour H, Bahrami S, Ganje MB. et al. HE4 combined with CA125: favorable screening tool for ovarian cancer. Med Oncol. 2014;31:808
52. Al-Ogaidi I, Gou H, Aguilar ZP, Guo S, Melconian AK, Al-Kazaz AK. et al. Detection of the ovarian cancer biomarker CA-125 using chemiluminescence resonance energy transfer to graphene quantum dots. Chem Commun (Camb). 2014;50:1344-6
53. Zheng LE, Qu JY, He F. The diagnosis and pathological value of combined detection of HE4 and CA125 for patients with ovarian cancer. Open Med (Wars). 2016;11:125-32
54. Hamed EO, Ahmed H, Sedeek OB, Mohammed AM, Alla AAA, Abdel Ghaffar HM. Significance of HE4 estimation in comparison with CA125 in diagnosis of ovarian cancer and assessment of treatment response. Diagn Pathol. 2013;8:11
55. Partheen K, Kristjansdottir B, Sundfeldt K. Evaluation of ovarian cancer biomarkers HE4 and CA-125 in women presenting with a suspicious cystic ovarian mass. J Gynecol Oncol. 2011;22:244-252
Author contact
![]() Corresponding authors: Dr. Buzhen Tan, Department of Obstetrics & Gynecology, The Second Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, PR China. E-mail: tanbuzhencom; Tel: +86-791-86266912; Dr. Tingtao Chen, Institute of Translational Medicine, Nanchang University, Nanchang, Jiangxi 330031, PR China. E-mail: chentingtao1984com; Tel: +86-791-83827170.
Corresponding authors: Dr. Buzhen Tan, Department of Obstetrics & Gynecology, The Second Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, PR China. E-mail: tanbuzhencom; Tel: +86-791-86266912; Dr. Tingtao Chen, Institute of Translational Medicine, Nanchang University, Nanchang, Jiangxi 330031, PR China. E-mail: chentingtao1984com; Tel: +86-791-83827170.