Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(5):1455-1466. doi:10.7150/jca.51604 This issue Cite
Review
Changes in Protein Glycosylation in Head and Neck Squamous Cell Carcinoma
Chengcheng Liao1, Jiaxing An2, Zhangxue Tan1, Fangping Xu1, Jianguo Liu1, Qian Wang1,3 ![]()
1. Oral Disease Research Key Laboratory of Guizhou Tertiary Institution, School of Stomatology, Zunyi Medical University, Zunyi 563006, China.
2. Department of Gastroenterology, Affiliated Hospital of Zunyi Medical University, Zunyi 563000, China.
3. Microbial Resources and Drug Development Key Laboratory of Guizhou Tertiary Institution, Life Sciences Institute, Zunyi Medical University, Zunyi 563006, China.
Received 2020-8-6; Accepted 2020-12-9; Published 2021-1-1
Abstract

Glycosylation is an important posttranslational modification of proteins, and it has a profound influence on diverse life processes. An abnormal polysaccharide structure and mutation of the glycosylation pathway are closely correlated with human cancer progression. Glycoproteins such as EGFR, E-cadherin, CD44, PD-1/PD-L1, B7-H3 and Muc1 play important roles in the progression of head and neck squamous cell carcinoma (HNSCC), and their levels of glycosylation and changes in glycosyl structure are closely linked to HNSCC progression and malignant transformation. The regulation of protein glycosylation in HNSCC provides potential strategies to control cancer stem cell (CSC) subgroup expansion, epithelial-mesenchymal transition (EMT), tumor-related immunity escape and autophagy. Glycoproteins with altered glycosylation can be used as biomarkers for the early diagnosis, monitoring and prognostication of HNSCC. However, the glycobiology of cancer is still a new field that needs to be deeply studied, especially in HNSCC.
Keywords: Glycosylation, HNSCC, EGFR, E-Cadherin, CD44, PD-1/PD-L1, B7-H3, Muc1
Introduction
Head and neck squamous cell carcinoma (HNSCC) refers to squamous epithelial malignancies that originate in the oral cavity, nasopharynx, oropharynx, hypopharynx and larynx. The total incidence of HNSCC ranks sixth among neoplastic diseases, consisting of more than 90% of all head and neck tumors [1]. Globally, there are approximately 600,000 new cases of HNSCC each year, most of which are locally advanced [1,2].] The 5-year survival rate of HNSCC patients is approximately 43%, and most patients cannot obtain an early diagnosis even after the tumor has metastasized to the lymph nodes in the neck [1]. Therefore, it is important to explore effective molecular diagnostic indicators and design new treatment strategies for HNSCC patients with various tumor stages.
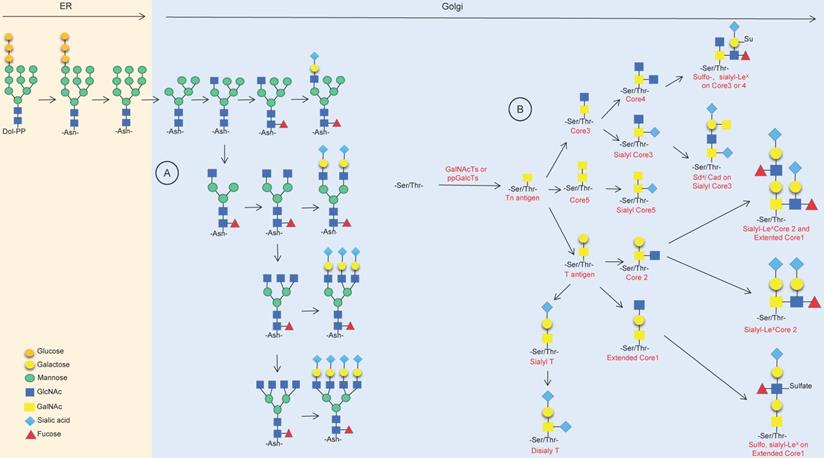
Protein glycosylation occurs during the process of peptide chain synthesis, and the sugar chain is linked to a specific glycosylation site on the peptide chain via enzyme catalysis. There are 9 kinds of glycans in mammalian cells, including glucose, N-acetylglucosamine, galactose, N-acetylgalactosamine, mannose, fucose, gluconic acid, xylose and sialic acid. The most common advanced glycation end products (AGEs) are N-glycosylation and O-glycosylation [5,6]. ]More than 2,000 proteins in human cells harbor an amino acid motif suitable for N-glycosylation, and these include membrane-bound proteins and secreted proteins but do not include cytoplasmic or nuclear proteins [7]. N-glycosylation occurs during the translation of target proteins by appending glycan structures to the amino group of asparagine (Asn); it starts in the endoplasmic reticulum (ER) and ends in the Golgi body, forming di-, tri- or tetraantennary glycans and then decorating them with various modifications, including fucosylation, salivation, galactose addition and GlcNAc, finally forming a series of highly complex and nonuniform structures of N-glycosylation (Fig. 1A) [7-10]. O-glycosylation happens in the Golgi apparatus; with the catalyst of polypeptide-GalNAc-transferases (pp-GalNAcTs, GALNTs), a single N-acetylgalactosamine (GalNAc) residue is transferred to serine (Ser) and/or threonine (Thr) residues of specific proteins, forming the Tn antigen, which finally forms five core structures named core 1 to core 5 (Fig. 1B) [7,11-13].
Synthesis of N-linked glycans and O-linked glycans. (A) Synthesis of mainly asparagine streptan (N-glycan) in the endoplasmic reticulum and Golgi apparatus. (B) Synthesis of O-linked glycans and common products in the Golgi.
Glycosylation may represent a hallmark of cancer [14] and causes the secretion of tumor-associated polysaccharides or glycoproteins, promoting their release into the bloodstream as a tumor-related marker [15]. The glycosylation process elevates the complexity of protein function regulation and has therapeutic significance for many diseases, including cancer, in which it regulates the proliferation, invasion and angiogenesis of tumors [16,17]. ]Alterations in glycosylation directly affect tumor growth and survival and facilitate tumor-induced immunomodulation and eventual metastasis [18]. Accurate diagnosis and treatment based on tumor glycosylation is a hot topic in biomedical research. Understanding HNSCC-related glycosylation alterations is critical for developing therapeutic interventions that aim to recover the normal glycosylation patterns of cancer cells. In this review, we focused on changes in protein glycosylation and its effects on HNSCC and discussed aberrant glycosylation levels and their potential use as diagnostic markers of HNSCC. According to the latest studies of protein glycosylation, this review introduces recent advances in EGFR, E-cadherin, CD44, PD-1/PD-L1, B7-H3 and Muc1.
The effect of EGFR glycosylation on the biological behavior of HNSCC
Epidermal growth factor receptor (EGFR/HER1/ErbB1) belongs to the tyrosine kinase receptor ErbB family [19]. It is overexpressed in most epithelial-derived malignancies, such as HNSCC [20]. EGFR is involved in many crucial signaling pathways, such as RAS/RAF/MEK/MAPK/ERK, PI3K/Akt, and JAK/STAT. These signaling cascades widely influence tumor growth, metastasis, poor prognosis, resistance to chemotherapy and radiation in HNSCC [21,22]. ]EGFR is widely expressed in HNSCC with few genetic mutations, making it an attractive therapeutic target for HNSCC patients who have a poor prognosis and are prone to recurrence and metastasis [23]. Understanding the effect of EGFR glycosylation on the interaction between EGFR and its related ligands will help us to elucidate the activation mechanisms of EGFR and facilitate the computational design of efficient inhibitors.
EGFR N-glycosylation correlation with malignant progression of HNSCC
N-glycosylation influences both the structure and stability of EGFR. The N-glycosylation of the glycosyl group next to the EGF binding site in EGFR could cause its noncovalent interactions with the EGFR extracellular domain, stabilizing the EGF binding site, leading to a stronger interaction between EGF and EGFR; N-glycosylation also helps maintain the binding of the EGFR dimer and plays a special role in the binding of antibodies and the extracellular domain of EGFR [24].
The N-glycosylation inhibitor 2-deoxyglucose (2DG) could change the N-glycosylation state of EGFR in HNSCC, induce the expression of the endoplasmic reticulum stress (ERS) markers CHOP and BiP, and finally affect the activity of EGFR; therefore, 2DG can enhance the antitumor effects of cisplatin and radiotherapy and overcome erlotinib resistance [25]. In addition, the disruption of glycolysis by 2DG is also a way to exert a tumor-suppressive effect [26,27]. Tunicamycin (TM), which is another N-glycosylation inhibitor, transfers N-acetylglucosaminyl-1-phosphate to phosphorylated dolichol, inhibiting N-linked glycosylation by blocking GlcNAc phosphotransferase (GPT) [28]. Wang et al. [29] found that after incubation with TM, ERS was increased and the EGFR signaling pathway was suppressed by inhibition of EGFR N-glycosylation.
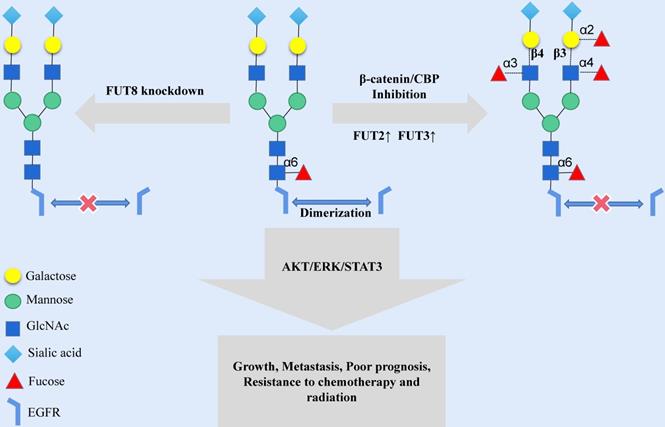
Modifying the terminus of N-glycosylation is another way to change the N-glycosylation-mediated activity of EGFR. Human tumor-specific N-glycan modifications include fucosylation, salivary acidification and lactose addition [7]. The fucosylation of N-glycans is mainly catalyzed by fucosyltransferase (FUT); the FUT family is a group of fucosylation synthetases, including FUT1 to FUT11, which catalyze the transfer of fucose from GDP-fucose to oligosaccharides on the substrate, sugars of glycoproteins chain or glycolipid [30]. In human malignant oral squamous cell carcinoma (OSCC) cells, the fucosylation of N-glycan antennae is lacking in EGFR, while indolent cells have high levels of fucosylation at the N420 and N579 sites of EGFR. ICG-001 and E7386, inhibitors of β-catenin/CBP signaling, may cause an increase in the transcription expression of FUT2 and FUT3 and then increase the fucosylation level of N-glycan antennae in EGFR [31] (Fig. 2). The structural modification of EGFR N-glycan might trigger a change in the cellular localization and signal transduction of EGFR. FUT1 is a key enzyme for LewisY (Ley) synthesis [32], and the Ley precursor is related to the motility of oral mucosa cells [33]. The increased expression of Ley was significantly associated with poor prognosis, and Ley of EGFR could stabilize the expression EGFR and downstream signals and promote the migration of OSCC cells [34]. FUT4 is involved in the synthesis of LewisX (LeX) in HNSCC, and LeX is related to the drug resistance response and the prognosis of HNSCC patients [35]. FUT8 is a typical N-glycan branching enzyme and plays a critical role in regulating the signal transduction of cell surface receptors [36]. Although FUT8 does not affect the expression of EGFR on the cell membrane, the FUT8-mediated core fucosylation of N-glycans is necessary for EGF to bind its receptor, and the lack of FUT8 could influence the activity of EGFR (Fig. 2). However, it should be noted that the inhibition of FUT8 may considerably impact normal human cells, thus limiting the application of FUT8 inhibitors in tumor therapy [37].
Increasing the expression of FUT2 and FUT3 or decreasing the expression of FUT8 in the Golgi can destroy the extracellular N-glycan structure of EGFR protein and affect the transmission of EGFR signal cascade.
Abnormal sialylation of EGFR is associated with the malignancy, metastasis and aggressiveness of tumors [38]. Sialylation could suppress EGFR dimerization, autophosphorylation, and EGF-induced lung cancer cell invasion [39]. When compared with the primary tumor of HNSCC, the sialic acid content in metastatic tumors is lower [40]. Sialidase regulates cellular sialic acid by removing the a-glycosidically linked sialic acid residues of glycoproteins and glycolipids and is involved in the development of cancer. The plasma membrane-associated sialidase NEU3 plays a unique role in transducing EGFR transmembrane signals by regulating the hydrolysis of gangliosides [41]. NEU3 could enhance the phosphorylation of EGFR and promote the migration and invasion of HNSCC cells. The expression of NEU3 is significantly upregulated in HNSCC tissues compared with in normal epithelial tissues, and its increased mRNA is positively correlated with lymph node metastasis [42]. According to the above results, NEU3 might possess the ability to increase the malignancy of HNSCC cells by inhibiting sialylation on EGFR.
To date, cetuximab is the only FDA-approved EGFR-targeted therapy drug for the treatment of HNSCC [43]. However, the treatment efficacy of cetuximab is low, with an objective response rate of 13% as a monotherapy and 36% in combination with chemotherapy [44,45].] Friederike et al. [46] reported that after chemotherapy or radiotherapy combined with cetuximab, HNSCC patients with higher EGFR-K521 (k-allele) expression levels had a significantly shorter progression-free survival period compared with those without expression of EGFR-K521. Cetuximab cannot inhibit the downstream cascade of the EGFR signaling pathway in HNSCC cells with high k-allele expression. EGFR-K521 N-glycan is sialic acid deficient compared to EGFR protein, and EGFR-K521 protein N-glycan sialic acid deficiency may be a structural basis for reducing the effectiveness of cetuximab. This study was important for exploring the mechanisms of cetuximab resistance and suggested that N-glycan sialic acid deficiency EGFR-K521 can be used as a biomarker to predict the prognosis of HNSCC patients.
Aberrant O-glycosylation interferes with EGFR vitality
GalNAc-type O-glycosylation is initiated by the transfer of N-acetylgalactosamine (GalNAc) to a serine or threonine residue and forms the Thomsen-nouvelle (Tn) antigen; this process is catalyzed by the GalNAc transferase family (GALNTs) [47,50]. GALNT2 is overexpressed in cancer cells at the invasive front of OSCC and enhances the invasiveness of OSCC cells by modifying EGFR O-glycans [48]. C1GALT1 (Thomsen-Friedenreich antigen) is the only enzyme that catalyzes the transfer of UDP-galactose to the Tn antigen and forms the core 1 structure [49]; moreover, C1GALT1 is the precursor of various GalNAcl-type O-glycans that are extensively distributed on secreted glycoproteins and the cell surface. The knockdown of C1GALT1 could inhibit the O-glycan extension of EGFR, reduce the binding affinity of EGF-EGFR, and thereby inhibit the signal transduction of EGFR. Itraconazole is a C1GALT1 inhibitor and may be therapeutic for HNSCC [50]. Although the O-glycosylation of EGFR seems to be less important than N-glycosylation, it is necessary to know that the change in EGFR activity is caused by aberrant O-glycosylation in HNSCC.
The influence of aberrant glycosylation on cancer stem cells of HNSCC
Cancer stem cells (CSCs) are a small subgroup of tumor cells with the ability to self-renew and differentiate. HNSCC CSCs are highly tumorigenic and participate in tumor differentiation, treatment resistance, relapse and metastasis [51]. In CSCs, the glycosylation of specific markers affects diverse fundamental processes of cells, such as adhesion, survival, invasion, metastasis, pluripotency, stemness, drug resistance and apoptosis [52]. Therapy targeting glycosyl groups on CSCs combined with surgical treatment, radiotherapy and chemotherapy will help improve the prognosis of patients.
The influence of glycosylated CD44 on the stemness of HNSCC CSCs
CD44 is a biomarker of CSCs and regulates cancer stemness in solid tumors [53]. CD44 members contain CD44s (standard CD44) and CD44 v1-v10, and they both have unique cell adhesion properties that could cause the interaction between two different cell types or one cell type and its surrounding matrix, sequentially accelerating the aggregation and migration of tumor cells [54]. CD44 can be glycosylated through N-glycans and O-glycans, and the molecular weight of the CD44 subtype depends on its glycosylation level [50,55].] The functional diversity of CD44 is derived from its various glycosylation patterns in the extracellular matrix and the multiple subtypes of protein produced by alternative splicing, which also contributes to the metastasis of human tumors [56,57].] The expression of CD44v4 leads to the activation of ERK1/2 and resistance to cisplatin. CD44v6 is mainly related to the activation of PI3K/Akt/GSK3β and the invasion and migration of HNSCC; CD44s and CD44v3 participate in the growth and migration of HNSCC [58,59] but the differences in CD44s and CD44 V1-V10 glycosylation structures and their corresponding functions have not been fully elucidated. TM can lessen the expression of CD44 in HNSCC by inhibiting N-glycosylation [30]. In addition, when a glycosylated, conformation-dependent CD44 epitope is targeted by monoclonal antibody RO5429083, it can increase the number of natural killer (NK) cells, reduce the EGFR signaling of CSCs and increase the targeting efficiency of RO5429083 towards CSC populations [60].
Hyaluronic acid (HA) is one of the main components of the extracellular matrix. HA accumulation is closely related to the poor prognosis of patients with advanced cancer and is also conducive to tumor angiogenesis, invasion and metastasis [61]. It has been proven that HA can stimulate the self-renewal, clonal formation and differentiation of CSCs [62]. The extracellular N-terminal hyaluronic acid binding domain (HABD) of CD44 could prompt its bond to HA. The terminal sialic acid on CD44 N-glycan can form a charged paired hydrogen with the basic amino acid of HABD, interrupting the bond of HA to glycosylated CD44 HABD [63]. In addition, the integrity of potential N-linked glycosylation sites of CD44 is critical for hyaluronic binding [64]. The c-Jun signal induced by HA/CD44 leads to the production of survival protein (cIAP-1/cIAP-2) and chemotherapy resistance in HPV16+ HNSCC cells, thus promoting the development of HNSCC [65].
Except for CD44, more than 30 CSC markers can be used to identify different cancer and tissue types of CSCs, and most of them are cell surface glycoproteins [66]. Although further investigations are still needed, some of these markers, in addition to CD44, may be affected by glycosylation changes. For example, when comparing the mass spectra and lectin binding in CD133+ and CD133- cells, there was a greater than 10% difference in the N-glycan structures [67]. When CSCs of colon cancer differentiate into tumor cells, the glycosylation of CD133 may change and lead to different folding patterns of CD133, thus covering different specific epitopes [68].
The influence of altered fucosylation on the stemness of HNSCC CSCs
It has been proven that fucosylation is closely associated with pancreatic CSCs [69], and the expression of FUT9 in colorectal cancer cell lines is positively correlated with the phenotypic characteristics of CSCs [70]. In addition, the miR-29b/Sp1/FUT4 axis can promote the malignant behaviors of leukemia stem cells by regulating fucosylated CD44 via the Wnt/β-catenin pathway [71]. These results suggest that adjusting the level of fucosylation in tumor cells may affect the stemness of CSCs.
Sialyl Lewis-X (SLeX) is closely related to CSCs in HNSCC, while FUT3, FUT5 and FUT6 participate in the synthesis of SLeX [72]. In OSCC CSCs, the expression of fucosyltransferases FUT3 and FUT6 on the spherical cell membrane is higher when compared with that in adherent cells, and the increased expression of SLeX might lead to greater aggression, tumorigenicity, tumorigenicity, chemoresistance and lower radiosensitivity of CSCs [73]. Moreover, the content of secreted SLeX is very high in OSCC and increases with the progression of disease. The SLeX positive rate in OSCC cells cultured in suspension is 95-100%, while that in adherent cells is 10-40% [74].
Aberrant N-glycosylation status affects E-cadherin-mediated EMT
Epithelial cadherin (E-cadherin or cadherin 1) belongs to the cadherin family, and the reduction in E-cadherin expression is involved in the progression and metastasis tumors [75]. E-cadherin is highly expressed in the normal squamous epithelial cells of the oral cavity and oropharynx and inhibits the disengagement-dependent growth and EMT transformation of HNSCC cells [76]. The absence of E-cadherin might result in the dissociation of invasive tumor cells at the edge of tongue carcinomas and regional metastasis of lymph nodes [77]. In normal epithelial cells, the inhibitory effect of E-cadherin against tumorigenesis is maintained by sequestering the binding of β-catenin to lymphoid enhancer factor (LEF)/T cell factor (TCF); this process could impede the transcription of genes involved in the proliferation-associated Wnt signaling pathway [78]. The E-cadherin/β-catenin complex helps maintain the integrity of epithelial cells, disrupts the Wnt signaling pathway, and participates in a variety of human malignancies and fibrosis disorders caused by EMT [79].
The posttranslational modifications of E-cadherin include phosphorylation modification, O-glycan modification and N-glycan modification. β1,6-GlcNAc branched N-glycans are of great importance to the regulation of E-cadherin-mediated adhesion and signal transduction [80]. The N-glycosylation of E-cadherin could influence the progression of tumors and transformation towards a malignant phenotype [81]. N-acetylglucosamine transferase III (GnT-III), N-acetylglucosamine transferase V (GnT-V) and FUT8 are related to the reconstruction of E-cadherin N-glycan [82]. Aberrant N-glycosylation at the Asn-554 [83,84] Asn-566 [84] and Asn-633 [85] sites of E-cadherin could strengthen its critical function in cancer.
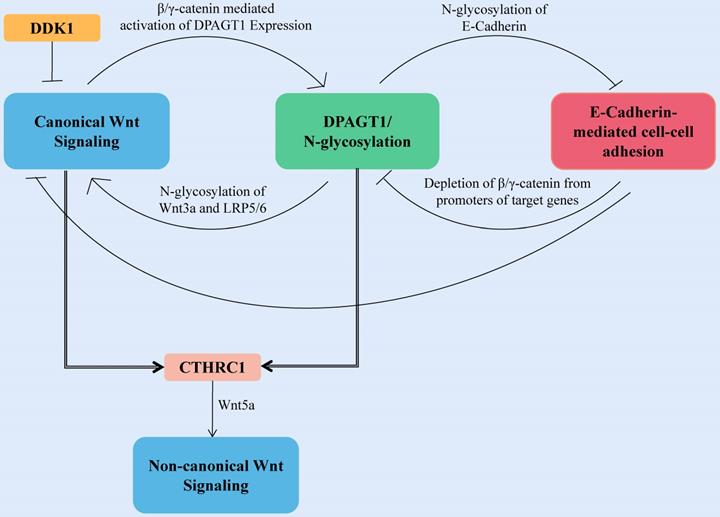
The first committed step of protein N-glycosylation is catalyzed by the dolichyl-phosphate N-acetylglucosamine-phosphotransferase DPAGT1 [86]. DPAGT1 is a key node that regulates the loss of E-cadherin and the activation of the Wnt pathway induced by aberrant N-glycosylation-related networks (Fig. 3). DPAGT1 and Wnt/β-catenin control the N-glycosylation status of E-cadherin through positive and negative feedback mechanisms, reducing the localization of E-cadherin on the cytomembrane of HNSCC (Fig. 3) [87-90]. The Wnt signal intensity is regulated by the N-glycosylation degree of Wnt3a and low-density lipoprotein-related receptors 5 and 6 (LRP5/6) because Wnt3a and LRP5/6 can be secreted and expressed effectively on the cell membrane only under proper N-glycosylation (Fig. 3) [91].
Canonical Wnt signaling activates DPAGT1 expression and protein N-glycosylation, leading to extensive N-glycosylation of E-cadherin. In HNSCC, the positive feedback loop between Wnt signaling and DPAGT1 is amplified and partially inhibited by wnt pathway inhibitor DDK1. Furthermore, extensive N-glycosylation of E-cadherin prevents it from depleting nuclear β/γ-catenins allowing the positive feedback between Wnt and DPAGT1 to operate without controls. CTHRC1 is upregulated by DPAGT1 and canonical Wnt signaling, affecting the noncanonical Wnt pathway.
Glycosylation-related immune checkpoints and HNSCC immune escape
Aberrant glycan structures and mutations of the glycosylation pathway are associated with the immune escape ability of tumor cells [92]. Specific glycan signatures on tumor cells can be considered a novel type of immune checkpoink [93]. In parallel, the glycosylation of tumor proteins produces neoantigens that masquerade as normal parts of the body to evade immune cells [93,94]. PD-1, CTLA-4, TIM-3, IDO and other inhibitory immune checkpoints have been proven to participate in the construction of the HNSCC immunosuppressive microenvironment [95]. Many immune checkpoints, such as PD-1 [96], B7-H3 [97] and TIM-3 [96] are glycoproteins with varying degrees of glycosylation.
N-glycosylated PD-1/PD-L1 and HNSCC immune escape
The programmed death 1 (PD-1)/programmed death-ligand 1 (PD-L1) axis could suppress antitumor immunity [98]. PD-1 interacts with PD-L1 to inhibit the proliferation of T cells and the production of cytokines [99]. PD-L1 combined with CD80 impedes the activation of T cells [100]. PD-L1 protein stability, translocation and protein-protein interactions can be altered by glycosylation, phosphorylation, ubiquitination, sumoylation and acetylation [101]. Current research indicates that N-glycosylation and ubiquitination are the major posttranslational modifications involved in the immunosuppressive activity of PD-L1 [102].
Let-7a/b can inhibit PD-L1 glycosylation and promote PD-L1 degradation in HNSCC, and the process is achieved via the β-catenin/STT3 pathway [103]. EMT can induce the N-glycosyltransferase STT3 through β-catenin transcription, stabilize the N-glycosylation of PD-L1 and increase its expression, finally helping CSCs escape from the immune system [104]. Deglycosylation significantly improves the binding affinity and signal intensity of anti-PD-L1 antibodies to PD-L1, making quantitative clinical outcome predictions based on PD-L1 more accurate [105].
N-glycosylation can stabilize the protein structure of PD-1, thus compromising the antitumor immune responses, while the inhibition of Fut8 can reduce the expression of PD-1 on the cell surface and enhance the activation of T cells, leading to more efficient cancer destruction [106]. A recent study showed that PD-1 is extensively N-glycosylated in T cells; glycosylation of PD-1, especially at site N58, is the key to mediating its interaction with PD-L1 [107]. Therefore, inhibiting the glycosylation of PD-1/PD-L1 would help to suppress the immune escape of HNSCC and improve the efficacy of antibodies (Fig. 4). Designing antibodies against the glycosylation sites of PD-1/PD-L1 provides a potential way to confront PD-1/PD-L1-related immune escape.
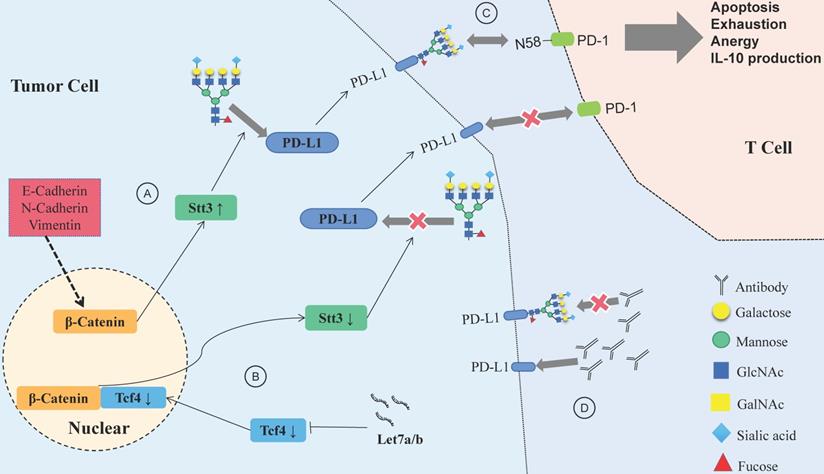
Relationship between glycosylation changes and immune escape of HNSCC. (A) E-cadherin, N-cadherin, and vimentin expression in HNSCC affects PD-L1 N-glycosylation, which is stabilized through the β-catenin/STT3 signaling axis. (B) The Let-7 family of miRNAs inhibits the expression of TCF-4, suppresses β-catenin/STT3-mediated PD-L1 glycosylation, reduces PD-L1 stability, promotes the ubiquitination and degradation of PD-L1, and improves the ability of T cells to recognize HNSCC cells. (C) Glycosylation of PD-1 mediates its interaction with glycosylated PD-L1, and the PD-1/PD-L1 axis has a central role in suppression of antitumor immunity. (D) Deglycosylation significantly improves the binding affinity and signal intensity of anti-PD-L1 antibodies to PD-L1.
N-glycosylated B7-H3 and HNSCC immune escape
B7 homolog 3 (B7-H3), an immune checkpoint protein of the B7 family, is an important regulator of the adaptive immune response; B7-H3 is mainly expressed on the surface of tumor cells, antigen-presenting cells, NK cells, and tumor endothelial cells. B7-H3 has a common inhibitory effect on T cells and helps tumor cells escape from the immune system; it also participates in cell proliferation, migration, invasion, angiogenesis, metastasis and anticancer drug resistance [108,109].]
B7-H3 is overexpressed in OSCC, promoting aerobic glycolysis in OSCC via the PI3K/Akt/mTOR pathway [110]. Compared with those of normal oral mucosal epithelial cells, the glycans of B7-H3 in OSCC contain terminal α-galactoses and more diverse N-glycan structures with higher fucosylation; with the action of B7-H3, OSCC cells could develop more effective interactions with DC-SIGN[DC-specific intercellular adhesion molecule-3 (ICAM-3)-grabbing nonintegrin] and Langerin on immune cells [111]. These results suggest that glycosylation changes of B7-H3 affect the occurrence and development of OSCC and change the immune microenvironment of OSCC tumors.
O-GlcNAc mediates HNSCC autophagy
The function of autophagy-related proteins, in particular their interaction with macroautophagic regulators, is modulated by phosphorylation, glycosylation, ubiquitination, acetylation, lipidation and proteolysis [112]. Glycan signaling in the extracellular matrix affects the location, activity and/or expression of key autophagy regulators, such as AMPK and mTORC1. Within the intracellular space, the components of the autophagosome membrane include ganglion glycosides, a subset of proteins composing the autophagic machinery are regulated by glycosylation, and exposure of oligosaccharides to the cytoplasm can also trigger autophagy [113]. The intracellular O-linked β-N-acetylglucosamine (O-GlcNAc) modification can regulate cell autophagy, as well as transcription, translation, protein degradation and signal transduction [114]. It has been reported that O-GlcNAc regulates autophagy by modifying autophagy core protein Beclin 1 [115], the forkhead family of transcription factors [116], SNARE protein SNAP-29 [117], and autophagy modulators such as Bcl-2 [115], AMPK [118]. In HNSCC, the influence of various hypoxia factors and the expression pattern of HIF-1α leads to an increasing number of CSC subgroups, driving tumor growth, invasion and therapy resistance [119]. In addition, HIF-1α inhibitors can suppress the autophagy of OSCC by decreasing the expression of O-GlcNAc and O-GlcNAc transferase (OGT) and increasing the expression of O-GlcNAcase (OGA) [120]. However, in another study, the levels of O-GlcNAcylation did not increase significantly in OSCC tissues, which was also not connected with the histological grading of OSCC [121].
Aberrant MUC1 glycosylation in HNSCC
MUC1 is a heterodimeric glycoprotein consisting of a highly glycosylated extracellular part and a small cytoplasmic tail [122]. Due to the higher expression of MUC1 in OSCC than in normal mucosal tissues, it is considered a reliable biomarker for the diagnosis of OSCC. Silencing the MUC1 gene could induce apoptosis and inhibit the proliferation, invasion, migration and EMT of OSCC cells [123,124]. T]his membrane-localized glycoprotein is overexpressed and aberrantly glycosylated in most epithelial cancers [125], and the aberrant glycosylation of MUC1 might result in the shortening of the sugar chain, causing the exposure of hidden antigens; the hidden antigens usually have peptidic and carbohydrate properties, making the MUC1 epitope tumor-specific [126]. In addition, extensive O-glycosylation of MUC1 contributes to cell resistance to anoikis, increasing cell adhesion and modulating the tumor immunological microenvironment through engagement of the lectin Siglec-9 [127,128]. Glycosylated MUC1 also has a positive sense and could stabilize an extended bioactive conformation of the peptide recognized by the antibody [129].
The biochemical functions and characteristics of the MUC1 protein have been identified. Due to the highly variable structure of MUC1, clarifying the specific effect of various MUC1 subtypes on tumor cells may have greater clinical significance [125]. N-glycosylated MUC1-C contributes to the upregulation of galectin-3 expression and interaction of galectin-3 and MUC1 could promote the dimerization and activation of EGFR in human epithelial cancer cells [130,131].] In addition, phosphorylation of the MUC1-C cytoplasmic domain could regulate the function of MUC1-C in the Wnt/β-catenin pathway [132].
Potential clinical applications of aberrant glycosylation as an HNSCC biomarker
Based on the abnormal glycosylated type of HNSCC, we can design corresponding treatment protocols and identify potential diagnostic biomarkers. Specific N-glycopeptide could be used as a serum biomarker to identify the clinical status of HNSCC patients. For the N-glycopeptides of IgG1, IgG4, HPT and TRFE, their abundances are significantly different between patients and controls, making them ideal candidates for future diagnostic modalities of OSCC [133]. In another study, the relative abundances of fucosylated, triantennary and tetraantennary glycans were significantly increased in OSCC patient serum compared with normal human serum, and OSCC patients showed significantly elevated levels of two IgM antibodies and decreased levels of nine IgG antibodies (Table 1) [134].
Aberrant glycosylation markers with clinical application value in OSCC
| Antibody class | Glycan name | P-value | Application | Compared with normal level | Reference |
|---|---|---|---|---|---|
| IgM | SSEA3 | < 0.01 | Potential candidate OSCC biomarkers | ↑ | 134 |
| CD2 | 0.005 | ↑ | |||
| IgG | GHC | 0.01 | ↓ | ||
| LeY | 0.01 | ↓ | |||
| SiaLeX | < 0.001 | ↓ | |||
| αNeuAc-OCH2C6H4-p-NHCOOCH2 | 0.04 | ↓ | |||
| NeuAcα2-8NeuAcα (NeuAcα2-8)2 | 0.01 | ↓ | |||
| NeuAcα2-8NeuAcα2-8NeuAc (NeuAcα2-8)3 | 0.001 | ↓ | |||
| Neu5Acα2-3Galβ1-4(Fucα1-3)(6-HSO3)GlcNAcβ (6GlcNAc-HSO3-SiaLeX) | < 0.01 | ↓ | |||
| (NeuAcα2-6Gal1-4GlcNAc1-2Man)2α1-3,6Manα1-4GlcNAcβ1-4GlcNAc (α2-6 sialylated diantennary N-glycans) | 0.001 | ↓ | |||
| GD2 | 0.02 | ↓ | |||
| IgG1 | N-glycan | Unknown but < 0.05 | Diagnosis of OSCC as early as stage I | ↑ | 133 |
| IgG4 | ↑ | ||||
| HPT | ↓ | ||||
| TRFE | ↓ | ||||
| EGFR-K521 | Sialyl glycan | 0.049 | Prediction of the prognosis of patients with HNSCC | ↑ | 46 |
| CEACAM6 | N-glycan | 0.0276 | Diagnosis of early recurrence in OSCC patients | ↑ | 135 |
Note: The P-value is the comparison between normal samples and OSCC patient samples. ↑, Increased compared to normal levels; ↓, decreased compared to normal levels; -, unchanged from normal levels.
Carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6), a glycophosphoinositol-anchored protein, is a heavily glycosylated tumor antigen. N-glycosylated CEACAM6 protein is a tumor marker for early recurrence in OSCC patients. In addition, the complex N-glycosylation of CEACAM6 is essential for EGFR-mediated OSCC cell invasion and transfer [135]. As mentioned above, aberrantly glycosylated proteins in serum and N-glycosylated CEACAM6 could be used as potential biomarkers for the diagnosis of OSCC. These biomarkers with abnormal glycosylation are summarized in Table 1.
Conclusion and prospects
The posttranslational modification of proteins is a major proteomics challenge in the post-gene era, and glycosylation lies in the heart of the problem. We found that the glycosylation changes of EGFR, E-cadherin, CD44 and PD-1/PD-L1 and the other glycoproteins have profound impacts on EMT, stemness, immune escape and other key metabolic steps of HNSCC. However, these studies have not fully revealed the detailed effects of protein glycosylation changes on HNSCC. For example, it is acknowledged that there are various O-glycan-decorated Notch receptors distributed in the extracellular domain epidermal growth factor-like (EGF) repeats [136], and many human congenital diseases are caused by O-glycosylation defects on Notch receptors; however, the relationship between the O-glycosylation defect of the Notch receptor and HNSCC has not been reported. Due to the complex mechanisms and structures of glycosylation and the large number of glycoproteins, the understanding of glycosylation in HNSCC still has a long way to go.
The glycobiology of HNSCC is an important but understudied field. Initially, monoclonal antibodies were used to identify tumor-related glycosyl changes; currently, more advanced technologies, such as ultrahigh-performance liquid chromatography (UPLC), mass spectrometry (MS), glycan microarrays, lectin histochemistry and agglutination cell count, are used to identify tumor polysaccharides [137,138]. These emerging research methods integrate glycomics, proteomics, genomics, lipidomics and metabolomics, making the study of glycobiological systems possible [139]. In the past, the basic methods of analysis, verification and construction of glycoproteins developed slowly. The emergence of electron cryomicroscopy provides an efficient tool to study the key structures of glycobiology [140]. However, these emerging methods have rarely appeared in existing HNSCC-related research.
The glycopeptide expression levels in the serum of HNSCC patients and changes in the protein glycosyl structure can be used as potential diagnostic indicators. IgG, HPT, and TRFE are abundant in serum, making them easier to identify. It is an expectation for us to diagnose HNSCC by assessing aberrant protein glycosylation, and the achievement of the goal should depend on the establishment and improvement of the glycosylation database. Moreover, it is feasible to design corresponding antibodies according to the target glycosyl sites of glycoproteins to treat HNSCC; in addition, regulating glycosyltransferase expression in HNSCC is also an important pathway for tumor suppression.
Acknowledgements
This study was supported by the Outstanding Young Talent Project of Zunyi Medical University (17zy-002) (F-801); the Top Science and Technology Talent Project in Higher Education Institution of Guizhou Province (grant No. Qian-Jiao-He KY Zi [2016]080); the Sixth Talent Foundation in Guizhou province (rcjd2019-9); the Youth Science and Technology Talents Growth Project of Guizhou Education Department (Qian-Jiao-He KY ZI [2018]236); and the Graduate Research Fund of Guizhou Province (Qian-Jiao-He YJSCXJH [2019]087).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Marur S, Forastiere AA. Head and neck cancer: changing epidemiology, diagnosis, and treatment. Mayo Clin Proc. 2008;83:489-501
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. Ca A Cancer Journal for Clinicians. 2018;68:7-30
3. Ferlay J, Soerjomataram I, Dikshit R. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:359-86
4. Marur S, Forastiere AA. Head and Neck Squamous Cell Carcinoma: Update on Epidemiology, Diagnosis, and Treatment. Mayo Clin Proc. 2016;91:386-96
5. Spiro RG. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology. 2002;12:43-56
6. Stepper J, Shastri S, Loo TS. et al. Cysteine -glycosylation, a new post-translational modification found in glycopeptide bacteriocins. FEBS Lett. 2011;585:645-50
7. Oliveira-Ferrer L, Legler K, Milde-Langosch K. Role of protein glycosylation in cancer metastasis. Semin Cancer Biol. 2017;44:141-52
8. Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer. 2015;15:540-55
9. Shah MH, Telang SD, Shah PM. et al. Tissue and serum alpha 2-3- and alpha 2-6-linkage specific sialylation changes in oral carcinogenesis. Glycoconj J. 2008;25:279-90
10. Shah M, Telang S, Raval G. et al. Serum fucosylation changes in oral cancer and oral precancerous conditions: alpha-L-fucosidase as a marker. Cancer. 2008;113:336-46
11. Helenius A. Intracellular Functions of N-Linked Glycans. Science. 2001;291:2364-69
12. Mathys L, François KO, Quandte M. et al. Deletion of the highly conserved N-glycan at Asn260 of HIV-1 gp120 affects folding and lysosomal degradation of gp120, and results in loss of viral infectivity. PLoS One. 2014;9:101181
13. Dunphy WG, Brands R, Rothman JE. Attachment of terminal N-acetylglucosamine to asparagine-linked oligosaccharides occurs in central cisternae of the Golgi stack. Cell. 1985;40:463-72
14. Vajaria BN, Patel PS. Glycosylation: a hallmark of cancer? Glycoconj J. 2017;34:147-56
15. Silsirivanit A. Glycosylation markers in cancer. Adv Clin Chem. 2019;89:189-213
16. Bektas M, Rubenstein DS. The role of intracellular protein O-glycosylation in cell adhesion and disease. J Biomed Res. 2011;25:227-36
17. Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer. 2005;5:526-42
18. Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annu Rev Pathol. 2015;10:473-510
19. Cohen SN. Isolation of a mouse submaxillary gland protein accelerating incisor eruption and eyelid opening in the new-born animal. J Biol Chem. 1962;237:1555-62
20. Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341-54
21. Yarden Y, Shilo B Z. SnapShot: EGFR signaling pathway. Cell. 2007;131:1018
22. Kalyankrishna S, Grandis JR. Epidermal growth factor receptor biology in head and neck cancer. J Clin Oncol. 2006;24:2666-72
23. Karamouzis MV, Grandis JR, Argiris A. Therapies directed against epidermal growth factor receptor in aerodigestive carcinomas. JAMA. 2007;298:70-82
24. Azimzadeh Irani M, Kannan S, Verma C. Role of N-glycosylation in EGFR ectodomain ligand binding. Proteins. 2017;85:1529-49
25. Sobhakumari A, Orcutt KP, Love-Homan L. et al. 2-Deoxy-d-glucose Suppresses the In vivo Antitumor Efficacy of Erlotinib in Head and Neck Squamous Cell Carcinoma Cells. Oncol Res. 2016;24:55-64
26. Simons AL, Fath MA, Mattson DM. et al. Enhanced response of human head and neck cancer xenograft tumors to cisplatin combined with 2-deoxy-D-glucose correlates with increased 18F-FDG uptake as determined by PET imaging. Int J Radiat Oncol Biol Phys. 2007;69:1222-30
27. Simons AL, Ahmad IM, Mattson DM. et al. 2-Deoxy-D-glucose combined with cisplatin enhances cytotoxicity via metabolic oxidative stress in human head and neck cancer cells. Cancer Res. 2007;67:3364-70
28. Duksin D, Mahoney WC. Relationship of the structure and biological activity of the natural homologues of tunicamycin. J Biol Chem. 1982;257:3105-09
29. Wang Y, Zhang L, He Z. et al. Tunicamycin induces ER stress and inhibits tumorigenesis of head and neck cancer cells by inhibiting N-glycosylation. Am J Transl Res. 2020;12:541-50
30. Mizuochi T, Taniguchi T, Shimizu A. et al. Structural and numerical variations of the carbohydrate moiety of immunoglobulin G. J Immunol. 1982;129:2016-20
31. Chandler KB, Alamoud KA, Stahl VL. et al. β-Catenin/CBP inhibition alters epidermal growth factor receptor fucosylation status in oral squamous cell carcinoma. Mol Omics. 2020;16:195-209
32. Li FF, Sha D, Qin XY. et al. Alpha1,2-fucosyl transferase gene, the key enzyme of Lewis y synthesis, promotes Taxol resistance of ovarian carcinoma through apoptosis-related proteins. Neoplasma. 2018;65:515-22
33. Dabelsteen E, Gao S. ABO blood-group antigens in oral cancer. J Dent Res. 2005;84:21-28
34. Lin WL, Lin YS, Shi GY. et al. Lewisy promotes migration of oral cancer cells by glycosylation of epidermal growth factor receptor. PLoS One. 2015;10:0120162
35. Rabassa ME, Pereyra A, Pereyra L. et al. Lewis x Antigen is Associated to Head and Neck Squamous Cell Carcinoma Survival. Pathol Oncol Res. 2018;24:525-31
36. Takahashi M, Kizuka Y, Ohtsubo K. et al. Disease-associated glycans on cell surface proteins. Mol Aspects Med. 2016;51:56-70
37. Wang X, Gu J, Ihara H. et al. Core fucosylation regulates epidermal growth factor receptor-mediated intracellular signaling. J Biol Chem. 2006;281:2572-77
38. Hakomori SI, Handa K. GM3 and cancer. Glycoconj J. 2015;32:1-8
39. Liu YC, Yen HY, Chen CY. et al. Sialylation and fucosylation of epidermal growth factor receptor suppress its dimerization and activation in lung cancer cells. Proc Natl Acad Sci U S A. 2011;108:11332-37
40. Bergler W, Riedel F, Schwartz-Albiez R. et al. A new histobiochemical method to analyze sialylation on cell-surface glycoproteins of head and neck squamous-cell carcinomas. Eur Arch Otorhinolaryngol. 1997;254:437-41
41. Miyagi T, Wada T, Yamaguchi K. et al. Plasma membrane-associated sialidase as a crucial regulator of transmembrane signalling. J Biochem. 2008;144:279-85
42. Shiga K, Takahashi K, Sato I. et al. Upregulation of sialidase NEU3 in head and neck squamous cell carcinoma associated with lymph node metastasis. Cancer Sci. 2015;106:1544-53
43. Taberna M, Oliva M, Mesía R. Cetuximab-Containing Combinations in Locally Advanced and Recurrent or Metastatic Head and Neck Squamous Cell Carcinoma. Front Oncol. 2019;9:383
44. Vermorken JB, Mesia R, Rivera F. et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116-27
45. Vermorken JB, Trigo J, Hitt R. et al. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol. 2007;25:2171-77
46. Braig F, Kriegs M, Voigtlaender M. et al. Cetuximab Resistance in Head and Neck Cancer Is Mediated by EGFR-K521 Polymorphism. Cancer Res. 2017;77:1188-99
47. Ten Hagen K G, Fritz T A, Tabak L A. All in the family: the UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Glycobiology. 2003;13:1-16
48. Lin M C, Huang M J, Liu C H. et al. GALNT2 enhances migration and invasion of oral squamous cell carcinoma by regulating EGFR glycosylation and activity. Oral Oncol. 2014;50:478-84
49. Tarp M A, Clausen H. Mucin-type O-glycosylation and its potential use in drug and vaccine development. Biochim Biophys Acta. 2008;1780:546-63
50. Lin M C, Chien P H, Wu H Y. et al. C1GALT1 predicts poor prognosis and is a potential therapeutic target in head and neck cancer. Oncogene. 2018;37:5780-93
51. Ailles L, Prince M. Cancer Stem Cells in Head and Neck Squamous Cell Carcinoma. Journal of Clinical Oncology Official Journal of the American Society of Clinical Oncology. 2009;26:2871
52. Underhill C. CD44: the hyaluronan receptor. J Cell Sci. 1992;103:293-8
53. Wang L, Zuo X, Xie K. et al. The Role of CD44 and Cancer Stem Cells. Methods Mol Biol. 2018;1692:31-42
54. Mishra M N, Chandavarkar V, Sharma R. et al. Structure, function and role of CD44 in neoplasia. J Oral Maxillofac Pathol. 2019;23:267-72
55. Camp R L, Kraus T A, Pure E. Variations in the cytoskeletal interaction and posttranslational modification of the CD44 homing receptor in macrophages. J Cell Biol. 1991;115:1283-92
56. Hakomori S. Tumor malignancy defined by aberrant glycosylation and sphingo(glyco)lipid metabolism. Cancer Res. 1996;56:5309-18
57. Athanassiou-Papaefthymiou M, Shkeir O, Kim D. et al. Evaluation of CD44 variant expression in oral, head and neck squamous cell carcinomas using a triple approach and its clinical significance. Int J Immunopathol Pharmacol. 2014;27:337-49
58. Kashyap T, Pramanik K K, Nath N. et al. Crosstalk between Raf-MEK-ERK and PI3K-Akt-GSK3β signaling networks promotes chemoresistance, invasion/migration and stemness via expression of CD44 variants (v4 and v6) in oral cancer. Oral Oncol. 2018;86:234-43
59. Patel S, Shah K, Mirza S. et al. Circulating tumor stem like cells in oral squamous cell carcinoma: An unresolved paradox. Oral Oncol. 2016;62:139-46
60. Perez A, Neskey D M, Wen J. et al. Abstract 2521: Targeting CD44 in head and neck squamous cell carcinoma (HNSCC) with a new humanized antibody RO5429083. Cancer Research. 2012;72(Suppl 8):S2521
61. Karousou E, Misra S, Ghatak S. et al. Roles and targeting of the HAS/hyaluronan/CD44 molecular system in cancer. Matrix Biol. 2017;59:3-22
62. Bourguignon LYW, Earle C, Shiina M. Activation of Matrix Hyaluronan-Mediated CD44 Signaling, Epigenetic Regulation and Chemoresistance in Head and Neck Cancer Stem Cells. Int J Mol Sci. 2017;18:1849
63. Chanmee T, Ontong P, Itano N. Hyaluronan: A modulator of the tumor microenvironment. Cancer Lett. 2016;375:20-30
64. Bartolazzi A, Nocks A, Aruffo A. et al. Glycosylation of CD44 is implicated in CD44-mediated cell adhesion to hyaluronan. J Cell Biol. 1996;132:1199-1208
65. Bourguignon LYW, Earle C, Shiina M. Hyaluronan-CD44 interaction promotes HPV 16 E6 oncogene-mediated oropharyngeal cell carcinoma survival and chemoresistance. Matrix Biol. 2019;78-79:180-200
66. Mallard BW, Tiralongo J. Cancer stem cell marker glycosylation: Nature, function and significance. Glycoconj J. 2017;34:441-52
67. Hemmoranta H, Satomaa T, Blomqvist M. et al. N-glycan structures and associated gene expression reflect the characteristic N-glycosylation pattern of human hematopoietic stem and progenitor cells. Exp Hematol. 2007;35:1279-92
68. Kemper K, Sprick MR, de Bree M. et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010;70:719-29
69. Sawanobori A, Moriwaki K, Takamatsu S. et al. A glycoproteomic approach to identify novel glycomarkers for cancer stem cells. Proteomics. 2016;16:3073-80
70. Blanas A, Zaal A, van der Haar Àvila I. et al. FUT9-Driven Programming of Colon Cancer Cells towards a Stem Cell-Like State. Cancers. 2020;12:2580
71. Liu B, Ma H, Liu Q. et al. MiR-29b/Sp1/FUT4 axis modulates the malignancy of leukemia stem cells by regulating fucosylation via Wnt/β-catenin pathway in acute myeloid leukemia. J Exp Clin Cancer Res. 2019;38:200
72. Nordén R, Nyström K, Olofsson S. Activation of host antiviral RNA-sensing factors necessary for herpes simplex virus type 1-activated transcription of host cell fucosyltransferase genes FUT3, FUT5, and FUT6 and subsequent expression of sLe(x) in virus-infected cells. Glycobiology. 2009;19:776-88
73. Desiderio V, Papagerakis P, Tirino V. et al. Increased fucosylation has a pivotal role in invasive and metastatic properties of head and neck cancer stem cells. Oncotarget. 2015;6:71-84
74. Czerwinski MJ, Desiderio V, Shkeir O. et al. In vitro evaluation of sialyl Lewis X relationship with head and neck cancer stem cells. Otolaryngol Head Neck Surg. 2013;149:97-104
75. Mendonsa AM, Na TY, Gumbiner BM. E-cadherin in contact inhibition and cancer. Oncogene. 2018;37:4769-80
76. Wong TS, Gao W, Chan JY. Interactions between E-cadherin and microRNA deregulation in head and neck cancers: the potential interplay. Biomed Res Int. 2014;2014:126038
77. Kudo Y, Kitajima S, Ogawa I. et al. Invasion and metastasis of oral cancer cells require methylation of E-cadherin and/or degradation of membranous beta-catenin. Clin Cancer Res. 2004;10:5455-63
78. Wong SHM, Fang CM, Chuah LH. et al. E-cadherin: Its dysregulation in carcinogenesis and clinical implications. Crit Rev Oncol Hematol. 2018;121:11-22
79. Tian X, Liu Z, Niu B. et al. E-cadherin/β-catenin complex and the epithelial barrier. J Biomed Biotechnol. 2011;2011:567305
80. Zhu W, Leber B, Andrews DW. Cytoplasmic O-glycosylation prevents cell surface transport of E-cadherin during apoptosis. EMBO J. 2001;20:5999-6007
81. Pinho SS, Osório H, Nita-Lazar M. et al. Role of E-cadherin N-glycosylation profile in a mammary tumor model. Biochem Biophys Res Commun. 2009;379:1091-6
82. Pinho SS, Seruca R, Gärtner F. et al. Modulation of E-cadherin function and dysfunction by N-glycosylation. Cell Mol Life Sci. 2011;68:1011-20
83. Carvalho S, Catarino TA, Dias AM. et al. Preventing E-cadherin aberrant N-glycosylation at Asn-554 improves its critical function in gastric cancer. Oncogene. 2016;35:1619-31
84. Zhao H, Sun L, Wang L. et al. N-glycosylation at Asn residues 554 and 566 of E-cadherin affects cell cycle progression through extracellular signal-regulated protein kinase signaling pathway. Acta Biochim Biophys Sin. 2008;40:140-8
85. Zhou F, Su J, Fu L. et al. Unglycosylation at Asn-633 made extracellular domain of E-cadherin folded incorrectly and arrested in endoplasmic reticulum, then sequentially degraded by ERAD. Glycoconj J. 2008;25:727-40
86. Dong YY, Wang H, Pike ACW. et al. Structures of DPAGT1 Explain Glycosylation Disease Mechanisms and Advance TB Antibiotic Design. Cell. 2018;175:1045-58
87. Vargas DA, Sun M, Sadykov K. et al. The Integrated Role of Wnt/β-Catenin, N-Glycosylation, and E-Cadherin-Mediated Adhesion in Network Dynamics. PLoS Comput Biol. 2016;12:1005007
88. Varelas X, Bouchie MP, Kukuruzinska MA. Protein N-glycosylation in oral cancer: dysregulated cellular networks among DPAGT1, E-cadherin adhesion and canonical Wnt signaling. Glycobiology. 2014;24:579-91
89. Liu G, Sengupta PK, Jamal B. et al. N-glycosylation induces the CTHRC1 protein and drives oral cancer cell migration. J Biol Chem. 2013;288:20217-27
90. Sengupta PK, Bouchie MP, Nita-Lazar M. et al. Coordinate regulation of N-glycosylation gene DPAGT1, canonical Wnt signaling and E-cadherin adhesion. J Cell Sci. 2013;126:484-96
91. Langer MD, Guo H, Shashikanth N. et al. N-glycosylation alters cadherin-mediated intercellular binding kinetics. J Cell Sci. 2012;125:2478-85
92. Cadena AP, Cushman TR, Welsh JW. Glycosylation and Antitumor Immunity. Int Rev Cell Mol Biol. 2019;343:111-27
93. Li B, Chan HL, Chen P. Immune Checkpoint Inhibitors: Basics and Challenges. Curr Med Chem. 2019;26:3009-25
94. Manukian G, Bar-Ad V, Lu B. et al. Combining Radiation and Immune Checkpoint Blockade in the Treatment of Head and Neck Squamous Cell Carcinoma. Front Oncol. 2019;9:122
95. Mandal R, Şenbabaoğlu Y, Desrichard A. et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight. 2016;1:89829
96. Lee MJ, Heo YM, Hong SH. et al. The Binding Properties of Glycosylated and Non-Glycosylated Tim-3 Molecules on CD4CD25 T Cells. Immune Netw. 2009;9:58-63
97. Chapoval AI, Ni J, Lau JS. et al. B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol. 2001;2:269-74
98. Baumeister SH, Freeman GJ, Dranoff G. et al. Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol. 2016;34:539-73
99. Freeman GJ, Long AJ, Iwai Y. et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027-34
100. Butte MJ, Keir ME, Phamduy TB. et al. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111-22
101. Hsu JM, Li CW, Lai YJ. et al. Posttranslational Modifications of PD-L1 and Their Applications in Cancer Therapy. Cancer Res. 2018;78:6349-53
102. Li CW, Lim SO, Xia W. et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632
103. Yu D, Liu X, Han G. et al. The let-7 family of microRNAs suppresses immune evasion in head and neck squamous cell carcinoma by promoting PD-L1 degradation. Cell Commun Signal. 2019;17:173
104. Hsu JM, Xia W, Hsu YH. et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. 2018;9:1908
105. Lee HH, Wang YN, Xia W. et al. Removal of N-Linked Glycosylation Enhances PD-L1 Detection and Predicts Anti-PD-1/PD-L1 Therapeutic Efficacy. Cancer Cell. 2019;36:168-78
106. Okada M, Chikuma S, Kondo T. et al. Blockage of Core Fucosylation Reduces Cell-Surface Expression of PD-1 and Promotes Anti-tumor Immune Responses of T Cells. Cell Rep. 2017;2:1017-28
107. Sun L, Li CW, Chung EM. et al. Targeting Glycosylated PD-1 Induces Potent Antitumor Immunity. Cancer Res. 2020;80:2298-310
108. Flem-Karlsen K, Fodstad Ø, Nunes-Xavier CE. B7-H3 Immune Checkpoint Protein in Human Cancer. Curr Med Chem. 2020;27:4062-86
109. Castellanos JR, Purvis IJ, Labak CM. et al. B7-H3 role in the immune landscape of cancer. Am J Clin Exp Immunol. 2017;6:66-75
110. Li Z, Liu J, Que L. et al. The immunoregulatory protein B7-H3 promotes aerobic glycolysis in oral squamous carcinoma via PI3K/Akt/mTOR pathway. J Cancer. 2019;10:5770-84
111. Chen JT, Chen CH, Ku KL. et al. Glycoprotein B7-H3 overexpression and aberrant glycosylation in oral cancer and immune response. Proc Natl Acad Sci U S A. 2015;112:13057-62
112. Xie Y, Kang R, Sun X. et al. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11:28-45
113. Fahie K, Zachara NE. Molecular Functions of Glycoconjugates in Autophagy. J Mol Biol. 2016;428:3305-24
114. Hart GW. Three Decades of Research on O-GlcNAcylation - A Major Nutrient Sensor That Regulates Signaling, Transcription and Cellular Metabolism. Front Endocrinol (Lausanne). 2014;5:183
115. Marsh SA, Powell PC, Dell'italia LJ. et al. Cardiac O-GlcNAcylation blunts autophagic signaling in the diabetic heart. Life Sci. 2013;92:648-56
116. Housley MP, Udeshi ND, Rodgers JT. et al. A PGC-1alpha-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J Biol Chem. 2009;284:5148-57
117. Guo B, Liang Q, Li L. et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat Cell Biol. 2014;16:1215-26
118. Bullen JW, Balsbaugh JL, Chanda D. et al. Cross-talk between two essential nutrient-sensitive enzymes: O-GlcNAc transferase (OGT) and AMP-activated protein kinase (AMPK). J Biol Chem. 2014;289:10592-606
119. Gammon L, Mackenzie IC. Roles of hypoxia, stem cells and epithelial-mesenchymal transition in the spread and treatment resistance of head and neck cancer. J Oral Pathol Med. 2016;45:77-82
120. Li YN, Hu JA, Wang HM. Inhibition of HIF-1α Affects Autophagy Mediated Glycosylation in Oral Squamous Cell Carcinoma Cells. Dis Markers. 2015;2015:239479
121. Kongkaew T, Aung WPP, Supanchart C. et al. O-GlcNAcylation in oral squamous cell carcinoma. J Oral Pathol Med. 2018;47:260267
122. Taylor-Papadimitriou J, Burchell JM, Plunkett T. et al. MUC1 and the immunobiology of cancer. J Mammary Gland Biol Neoplasia. 2002;7:209-21
123. Zhang AM, Chi XH, Bo ZQ. et al. MUC1 gene silencing inhibits proliferation, invasion, and migration while promoting apoptosis of oral squamous cell carcinoma cells. Biosci Rep. 2019;39:BSR20182193
124. Thakur A, Tupkari JV, Joy T. et al. Expression of mucin-1 in oral squamous cell carcinoma and normal oral mucosa: An immunohistochemical study. J Oral Maxillofac Pathol. 2018;22:210-5
125. Sousa AM, Grandgenett PM, David L, Almeida R, Hollingsworth MA, Santos-Silva F. Reflections on MUC1 glycoprotein: the hidden potential of isoforms in carcinogenesis. APMIS. 2016;124:913-24
126. Gendler SJ, Spicer AP, Lalani EN. et al. Structure and biology of a carcinoma-associated mucin, MUC1. Am Rev Respir Dis. 1991;144:42-7
127. Piyush T, Rhodes JM, Yu LG. MUC1 O-glycosylation contributes to anoikis resistance in epithelial cancer cells. Cell Death Discov. 2017;3:17044
128. Beatson R, Tajadura-Ortega V, Achkova D. et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec-9. Nat Immunol. 2016;17:1273-81
129. Movahedin M, Brooks TM, Supekar NT. et al. Glycosylation of MUC1 influences the binding of a therapeutic antibody by altering the conformational equilibrium of the antigen. Glycobiology. 2017;27:677-87
130. Piyush T, Chacko AR, Sindrewicz P. et al. Interaction of galectin-3 with MUC1 on cell surface promotes EGFR dimerization and activation in human epithelial cancer cells. Cell Death Differ. 2017;24:1937-47
131. Ramasamy S, Duraisamy S, Barbashov S. et al. The MUC1 and galectin-3 oncoproteins function in a microRNA-dependent regulatory loop. Mol Cell. 2007;27:992-1004
132. Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer. 2009;9:874-85
133. Saraswat M, Mäkitie A, Tohmola T. et al. Tongue Cancer Patients Can be Distinguished from Healthy Controls by Specific N-Glycopeptides Found in Serum. Proteomics Clin Appl. 2018;12:1800061
134. Guu SY, Lin TH, Chang SC. et al. Serum N-glycome characterization and anti-carbohydrate antibody profiling in oral squamous cell carcinoma patients. PLoS One. 2017;12:0178927
135. Chiang WF, Cheng TM, Chang CC. et al. Carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6) promotes EGF receptor signaling of oral squamous cell carcinoma metastasis via the complex N-glycosylation. Oncogene. 2018;37:116-27
136. Urata Y, Takeuchi H. Effects of Notch glycosylation on health and diseases. Dev Growth Differ. 2020Jan;62(1):35-48
137. Dube DH, Bertozzi CR. Glycans in cancer and inflammation-potential for therapeutics and diagnostics. Nat Rev Drug Discov. 2005;4:477-88
138. Gao C, Wei M, McKitrick TR. et al. Glycan Microarrays as Chemical Tools for Identifying Glycan Recognition by Immune Proteins. Front Chem. 2019;7:833
139. Mariño K, Bones J, Kattla JJ. et al. A systematic approach to protein glycosylation analysis: a path through the maze. Nat Chem Biol. 2010;6:713-23
140. Atanasova M, Bagdonas H, Agirre J. Structural glycobiology in the age of electron cryo-microscopy. Curr Opin Struct Biol. 2020;62:70-78
Author contact
![]() Corresponding author: Qian Wang, E-mail: qianwang07com.
Corresponding author: Qian Wang, E-mail: qianwang07com.