Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(6):1804-1814. doi:10.7150/jca.50509 This issue Cite
Review
CAR-T cells for Colorectal Cancer: Target-selection and strategies for improved activity and safety
Huali Li, Chao Yang, Huangrong Cheng, Shuoyang Huang, Yongbin Zheng ![]()
Department of Gastrointestinal Surgery, Renmin Hospital of Wuhan University, Wuhan, Hubei 430060, P.R. China.
Received 2020-7-9; Accepted 2020-12-29; Published 2021-1-21
Abstract

Chimeric antigen receptor-T (CAR-T) cell immunotherapy is a novel method that is genetically engineered to recruit T cells against malignant disease. Administration of CAR-T cells has led to progress in hematological malignancies, and it has been proposed for solid tumors like colorectal cancer (CRC) for years. However, this method was not living up to expectations for the intrinsic challenges posed to CAR-T cells by solid tumors, which mainly due to the lacking of tumor-restricted antigens and adverse effects following treatment. New approaches are proposed to overcome the multiple challenges to alleviate the difficult situation of CAR-T cells in CRC, including engineering T cells with immune-activating molecules, regional administration of T cell, bispecific T cell engager, and combinatorial target-antigen recognition. In this review, we sum up the current stage of knowledge about target-selection, adverse events like on/off-tumor toxicity, the preclinical and clinical studies of CAR-T therapy, and the characteristics of strategies applied in CRC.
Keywords: colorectal cancer, chimeric antigen receptor-T (CAR-T) cell, tumor associated antigens, toxicity, strategies
Introduction
Colorectal cancer (CRC) is a major public health problem globally, according to statistics, approximately 147,950 individuals will be diagnosed and 53,200 will die with this disease in 2020 [1]. According to the data, CRC is the third commonly diagnosed malignant disease in males with 9% and females with 8%, the third for mortality (10.7%), and metastatic CRC (mCRC) is usually associated with poor prognosis, with 5-year relative survival rate ranges from 14% to 90%, corresponding to patients diagnosed with advanced disease and localized disease, respectively [1, 2]. Multiple pieces of research have demonstrated the critical role of the immune system in controlling cancer growth, more importantly, has led the way to new strategies that promote active or passive immunotherapy against cancer [3, 4]. The application of targeted immunotherapies can promote a better clinical benefit for patients with CRC. But the tumor type of most CRC patients with microsatellite instability-high (MSI-h) which has a markedly higher mutational load than microsatellite stable (MSS) is a barrier to the benefit of immunotherapy in those patients [5]. Despite the immunogenicity of the subtype with a higher mutational load, CRC is prone to escape immunosurveillance for several mechanisms [6]. Hence a series of strategies are recommended to rescue the therapeutic activity of the immune system, including active methods like immune checkpoint blockade, co-stimulatory signal domain, cancer vaccines [7-9], and passive approaches like adoptive cell therapy (ACT) and monoclonal antibodies [10, 11]. And chimeric antigen receptor-T (CAR-T) cell therapy has demonstrated remarkable progress in cancer in the last decade.
CAR-T cell therapy is a form of cancer treatment in which the recipient's own T cells are extracted and genetically modified before reinfusion. It is an extraordinary concept proposed by Z Eshhar in 1989 [12], which attempts to express functional chimeric receptors that recognize tumor antigen in a non-histocompatibility complex molecules (MHC)-restricted manner, paving the way for the design at the will of T-cell receptors of any desired specificity. As compared to the traditional cell-mediated therapies, CAR-T cells possess improved specificity and cytotoxicity from major MHC through single-chain variable fragment (scFv) in addition to the T cell receptor [13, 14]. The CAR construct is composed of an extracellular scFv linked via a spacer, transmembrane domain, and intracellular signaling molecules that are capable to trigger the effector functions of T cells [15]. T cells are transfected with CAR structures through mRNA, viral vector transduction, or plasmid transfection to redirect them toward pre-defined target cells [16]. The CAR structure initially contained only the CD3ζ domain which is equipped to recapitulate “signal 1” of T cell activation, known as “first-generation” CAR [17]; it later evolved into a more complex form in which intracellular costimulatory domains were added (CD3ζ plus 4-1BB or CD28 signaling domains), giving rise to second-generation [18]; and the third-generation added 4-1BB and CD28 domains simultaneously, which have augmented T cell functions including cell proliferation and persistence [19]; the latest fourth-generation CAR is with a transgenic 'payload' to shape the tumor environment (TME) by the inducible release of transgenic immune modifiers [20]. Such transferred genetically-modified T cells are of unique pharmacokinetic properties, due to their ability to replicate and persist long-term, following a single administration. Furthermore, the use of synthetic immune receptors may impact signaling pathways involved in T cell function and survival in unexpected ways.
The method of adoptive transfer of CAR-T cells that takes advantage of the cellular immune system against cancer has demonstrated impressive success in hematologic malignancies in particular of CD19 positive B cell malignancies, and it has been proposed for solid tumors like CRC, targeting surface proteins such as natural-killer group 2, member D (NKG2D), the ligands of which partially upregulate in tumor cells, and carcinoembryonic antigen (CEA) [21, 22]. Whereas increased studies dedicated to improving the treatment efficacy of CAR-T cells have shown encouraging progress, results concerning solid tumors are not sufficient to advance their clinical application. The development of CAR-T cell therapy in solid tumors has stagnated as a result of the lack of tumor-restricted antigens which are also called tumor specific antigens (TSAs), the hostile TME, and inadequate T cell persistence [23, 24]. Continuous efforts are driving the development of basic research in this field, and there are several studies entered clinical trials (shown in Table 1) and a variety of combinational strategies are proposed for further improving efficacy and safety. Each strategy has its mechanism of action and has various advantages or limitations as summarized in Table 2.
The antigens of CAR-T cells entered clinical trials in CRC
| Antigen | Expression profile (normal) | Identifier | Phase stage | Type of cancer | Study start | Sponsor | Status |
|---|---|---|---|---|---|---|---|
| CEA | Epithelial of colon, tongue, esophagus and cervix, prostate; mucous cells of neck; secretory epithelia and duct cells of sweat glands; bronchial mucus [41, 42] | NCT02349724 | I | Colorectal, lung, gastric, breast and pancreas cancer | December 2014 | Southwest Hospital | Unknown |
| NCT03682744 | I | Colorectal, peritoneal, gastric, breast and pancreas cancer, peritoneal metastases | September 2018 | Sorrento Therapeutics | Active, not recruiting | ||
| EGFR | The majority of normal epithelial tissues (epidermal cells, fibroblasts, liver, kidney, ovary, testis, prostate, and uterus) [43, 44] | NCT03542799 | I | Metastatic CRC | May 2018 | ShenZhen Second People's Hospital | Not yet recruiting |
| NCT03152435 | I/II | CRC | June 2017 | Shenzhen Second People's Hospital | Unknown | ||
| MUC1 | The apical surface of most epithelial cells (glandular epithelia, lactating mammary gland, many simple epithelial cells lining glands or ducts) [45, 46] | NCT02617134 | I/II | CRC; malignant glioma of brain; gastric carcinoma | November 2015 | Hefei Binhu Hospital | Unknown |
| NKG2DL | Presenting at low or undetectable levels in normal human tissues [30, 31] | NCT03310008 | I | Colon Cancer Liver Metastasis | August 2017 | Celyad (formerly named Cardio3 Bio-Sciences) | Active, not recruiting |
| NCT03692429 | I | CRC | November 2018 | Celyad (formerly named Cardio3 Bio-Sciences) | Recruiting | ||
| HER2 | Developing epithelial structures of the respiratory and digestive systems, the ependymal cells lining the ventricles of the central nervous system [33] | NCT02713984 | I/II | CRC, breast, ovarian, lung, gastric, glioma and pancreatic cancer | March 2016 | Southwest Hospital of Third Millitary Medical University | Withdraw |
| NCT03740256 | I | CRC, bladder, lung, breast, gastric, pancretic and esophageal cancer, HNSC, cancer of the salivary gland | June 2020 | Baylor College of Medicine | Not yet recruiting | ||
| CD133 | Hematopoietic stem cells, CD34+ progenitor cells; kidney, nervous system, and epithelial tissues of the prostate [47] | NCT02541370 | I/II | CRC, liver, pancreatic, brain, breast, ovarian and acute myeloid and lymphoid leukemias | June 2015 | Chinese PLA General Hospital | Completed |
The advantages and limitations of various strategies
| Strategy | Advantages | Limitations | |
|---|---|---|---|
| Combination with immune-activating molecules | Cytokine | Improve the activation, proliferation, and cytotoxicity of T cell | Risk of toxicity caused by high level of serum cytokines |
| Chemokine receptor | Improve the capability of migration, effective homing of T cell and achieving access to tumor cells | Chronic stimulation of T cell may lead to high levels of intracellular calcium resulting T cell hypofunction | |
| Infusion method | Regional administration | Avoid the traffic load of T cells, reduced engineered T cell depletion | Without a focus on toxicity caused by the high level of systemic cytokines like IFN-γ following infusion |
| Switch-mediated CAR | Bispecific T cell engager | Redirect and regulate T cell activity toward target cells in a dose-dependent and specific manner | Few available TSAs |
| dCAR system | CCR-CAR-modified T cell | Improved safety and enhanced specificity for combining with dual antigens | The system must be precisely controlled, including the signaling intensity, the level and ratio relative to each synergic receptor |
Targets and Toxicities
The possible targets of CARs
Targeting truly tumor-restricted antigen is the key to successful CAR-T therapy. As expected, CAR-T cell therapy can not only eliminate tumor cells but also reach the specificity which ensures clinical safety if the targets belong to TSAs. But little is known about viable TSA targets, and the targets recognized by the CAR are always tumor associated antigens (TAAs). Here, we sum up the possible targets applied in CAR-T therapy for CRC, with corresponding expression profile, preclinical and (or) clinical studies (shown in Table 1).
Studies have described guanylyl cyclase2C (GUCY2C) as a possible target for CARs in mice models [25]. GUCY2C is a membrane-bound cyclase and its cell surface expression is confined to the apical surfaces of intestinal epithelial cells and a subset of hypothalamic neurons [25, 26]. The anti-tumor activity of GUCY2C-specific CAR-T cells has been confirmed in both human and syngeneic CRC xenograft models which used murine T cells, but CARs produced from 5F9 scFv (GUCY2C-scFv) didn't show cross-reactivity with murine GUCY2C, limiting the quantification of intestinal toxicity in mice models [25]. And there has not been seen the clinical application of GUCY2C CAR-T cells in patients with CRC yet.
Epithelial cell adhesion molecule (EpCAM) is proposed to be an emerging biomarker for circulating tumor cells (CTCs) and is recognized as a novel target for adoptive T cell therapy [27]. As reported, third-generation EpCAM CAR-T cells were generated to identify the specificity of EpCAM to CRC cells and models, demonstrated lytic cytotoxicity to target cells and secreted cytotoxic cytokines including tumor necrosis factor α (TNF-α) and interferon γ (IFN-γ) in an EpCAM-dependent manner. Infusion with these CAR-T cells significantly restrained tumor growth and development in xenograft mice models [28]. However, EpCAM is expressed in most healthy adult tissues, mainly in the basolateral cell membrane of simple, transitional and pseudo-stratified epithelial [29].
Another NKG2D-based CAR-T cell therapy also showed cytotoxicity against CRC cells in a dose-dependent manner, significantly suppressed tumor growth and extended overall survival of mice. Furthermore, NKG2D-positive lymphocyte infiltration was found in the tumor site of NKG2D CAR-T cells-treated mice, but it was also accompanied by gradual weight loss [30]. A clinical trial of NKG2D CAR-T cells targeting NKG2D ligands (NKG2DLs) has also been reported recently in which two autologous and allogeneic NKG2D CAR-T cells were investigated in mCRC patients through a 3+3 design evaluating 3 DL (1×108, 3×108 and 1×109 cells per infusion), showed that DL-1 and DL-2 were completed without any dose-limiting toxicity occurrence [10]. In humans, NKG2DLs consist of MHC class I-related chain A (MICA) and B (MICB), and six unique long 16 binding protein (ULBP1-6). These molecules are present at low or undetectable levels on normal tissues but significantly upregulated in multiple malignant tumors including CRC [30, 31].
Human epidermal growth factor receptor 2 (HER2) is an oncogene that encodes for a transmembrane glycoprotein receptor that functions as an intracellular tyrosine kinase and is a member of the HER2 (HER2/ERBB) family [32]. In humans, HER2 is present in developing epithelial structures of the respiratory and digestive systems, the ependymal cells lining the ventricles of the central nervous system [33]. In a recent report, HER2-targeted CAR-T cell confirmed its target potential, showed a similar line of efficacy against HER2+ tumors, including tumor regression or even elimination of CRC xenograft and protection of relapse, achieving improved survival benefit compared with the corresponding control group [34].
As is known, CEA is an acid glycoprotein which is the most common tumor marker in CRC currently. In a phase I trial, CAR-T cells were applied in 10 CEA positive CRC patients with escalating dose level(DL) that ranged from 1×105 to 1×108 (CAR+/kg cells). Seven patients who had experienced progressive disease during previous treatment were stable following treatment with CAR-T cells, moreover, two of them remained stable for more than 30 weeks, another two even showed tumor regression [21]. But there were several adverse events observed after cell therapy, including fever, lymphocyte count decrease and duodenum perforation. It is unclear whether the general expression of CEA in normal human tissues (shown in Table 1) is responsible for these adverse events, but the potential safety risk does exist.
According to Hege K.M [35], tumor associated glycoprotein 72 (TAG-72) was applied as tumor targets in CAR therapy, and cells were administered using two methods, intravenous infusion and hepatic artery infusion. Detectable persistence of CAR-T cells was observed in blood, and the trafficking to tumor sites was recognized through tumor biopsy [35]. Their findings demonstrated the anti-tumor efficacy of TAG-72 CAR-T cells but accompanied with the limitation that large metastatic deposits were resistant to T cells and evaded the immune attack. CAR-T cell therapy applied in patients with solid tumors has encountered many challenges due to the own nature of solid tumors and the presence of immunosuppressive immune cells, which prevent the successful application of this method in the clinic. And there is a series of ongoing trials targeting antigens like MUC1 (NCT02617134) or CD133 (NCT02541370) that are established to further assess the safety and feasibility of CAR-T cells for CRC (shown in Table 1).
In addition to targeting proteins exposed on tumor cell-surface, researchers also considered tumor stem cells (TSCs) and the process of epithelial-mesenchymal transition (EMT) as possible intervention targets, as both of which are involved in tumor progression [36, 37]. Given the invasive nature of CRC, doublecortin-like kinase 1(DCLK1) which plays a critical role in tumor growth and regulating the process of EMT may be a novel target for immunotherapy in CRC [38]. As reported, T cells engineered with DCLK1-scFv(CBT-511) induced cytotoxicity and increased IFN-γ release when co-incubated with CRC cells in two-dimensional, furthermore, higher levels of IFN-γ were observed when cells were cultured in three-dimensional [39]. In vivo, CBT-511 CAR-T cells blocked the growth of subcutaneous xenograft tumors derived from LoVo CRC cells, but there was no further detection for histotoxicity when a dramatic release of IFN-γ was observed [39]. And it is reported that DCLK1 not only upregulates in tumor cells but also scattered in the intestinal epithelium where the lower crypts in normal murine and human intestines are most frequently observed, heightening concern about toxicity [40]. Although there are few TSA available or even reported in CRC, increasing designs are focused on dual TAA targeting, and even by optimizing the CAR structure and combining the density difference of target antigen to indirectly regulate the immune-related toxicity caused by lack of tumor-specific antigen.
The off-tumor toxicity
Apart from the considerable advantage of CAR-T therapy showed in solid tumor models, the inherent divergence between mice and humans, the sharing-expression pattern of most antigens in the human body that is comparable to distribution in mice are two gaps difficult to bridge [26, 30, 48, 49]. Adoptive T cell therapy has enormous potential against cancer, it is also a double-edged sword in solid tumors where non-malignant tissues suffered 'indiscriminate attacks' in some cases during treatment. The most typical resulting “on-target off-tumor” toxicity is a case report, in which a mCRC patient received 1×1010 third-generation CAR-T cells intravenously targeted ERBB2 (also referred to as HER2) overexpressing tumors [50]. The patient suffered respiratory distress, displayed a dramatic pulmonary infiltrating within 15 minutes after cell infusion, and died 5 days after infusion despite intensive medical intervention [50]. The most important reason for this case is that a large number of ERBB2 CAR-T cells localized to the lung immediately following infusion, and the synchronous recognition of ERBB2 that exposed on normal epithelial cells triggered the release of cytokines. The analogous toxicity has also been shown in a recent report, in which two advanced CRC patients experienced lymphopenia after administrated with CD133 CAR-T cells, one of them even experienced hyperbilirubinemia (direct bilirubin) with grade 3 and lasted 3 weeks [51]. The simultaneous expression of CD133 in a variety of hematocytes such as hematopoietic stem cells and progenitor cells may be the main reason for hematopoietic system toxicity, and bilirubinemia toxicity may be partially put down to CD133 that was recognized as a marker of endothelial cells. It raises concern that such “on target off-target” toxicity may also occur on antigens with a similarly sharing-expression profile. For instance, TAG-72 described above is expressed in a variety of normal tissues including the normal mucosa, the endometrium during the secretory phase, and the fetal tissues [26, 52-54].
The on-tumor toxicity
Besides the “on target off-target” toxicity summarized above which damaged both malignant and normal tissues, cytokine release syndrome(CRS) is one of the most frequently observed toxicity of CAR-T cell therapy as a result of overactivation of T cells [55, 56]. CRS initially manifests with symptoms including fatigue, fever, and myalgias, which may be accompanied with hypoxia and hypotension, even can develop into life-threatening outcomes with capillary leak [57, 58]. As previously reported, the levels of several cytokines like interleukin 2 (IL-2), IFN-γ and TNF-α are markedly elevated in patient's serum after treatment with CAR-T cells [59-61]. The most commonly studied CAR therapy in hematological malignancies is usually with various adverse symptoms caused by such high levels of cytokine in serum. In terms of CRC, symptoms in accord with low-grade CRS were observed in both trial C-9701 (intravenous infusion) and trial C-9702 (hepatic artery infusion) when administered TAG-72 CAR-T cells [35]. The most frequently observed grade 1 to 2 adverse events in trail C-9701 included chills accounted for about 70%, and dizziness, fever, paresthesia, each term was about 30%, all suggesting low-grade toxicity with IFN or CRS. Other symptoms attributed to IFN-α were nausea, malaise, flu syndrome and abdominal pain, accounting for 50%, 40%, and 30%, respectively. In trial C-9702, one patient experienced a recurrent transient syndrome of fever, increased bilirubin and anemia 2 months following the last infusion[35]. Infusion-related toxicities were common, including fever, nausea, vomiting, increased bilirubin, headache and anemia. It is recognized that grading CRS is of great benefit to guide the management of adverse events. Moreover, attempts to alleviate and even to prevent toxicities are proposed, and various safety strategies that focus on optimizing CAR structure are proposed, including combinatorial target-antigen recognition, switch-mediated CAR. Synthetic Notch receptor as a novel method has demonstrated efficacy in hematological malignancies [62, 63]. All these approaches to alleviating toxicity have further promoted the treatment of CAR-T cells in malignant tumors.
Strategies to improve safety
Bispecific T cell engager
As many unfavorable factors exist between CAR-T cells and solid tumors, a series of bispecific antibodies emerged that bind to signal domain CD3ζ directly, to recruit T cells. Bispecific single-chain antibodies (bscAbs) that are referred to as “BiTE” molecules are one kind of such antibodies [64]. In a preliminary study, a kind of bscAbs combining various scFv recognizing human CEA with a de-immunized scFv recognizing human CD3 were constructed for treatment against CEA positive tumors [65]. Results showed that CEA/CD3-bscAbs redirected human T cells to lyse CEA+ tumor cells in vitro and in vivo, and the bscAbs could mediate efficient regression in different tumor cell models, though the available CEA-binding site is under sufficient. Furthermore, the cytotoxic activity of a subset of CEA/CD3-bscAbs was not competitively inhibited by soluble CEA at concentrations that exceeded levels found in the serum of most patients [65]. As shown in previous reports, these bispecific molecules could transiently connect resting T cells to tumor cells, leading to activation of T cells and lysis of tumor cells [65-67]. The bscAbs showed cross-reactivity with normal human tissues, however, and there is still the possibility of damaging normal tissues. Interestingly, progress has been made in the recombinant bispecific molecules. Engineering T cells with CARs that are activated by bifunctional small molecule “switch” consisting of a scFv recognizing antigen expressed on tumor cells conjugated to a protein combined with CAR-scFv, can redirect and regulate the activity of CAR-T cells toward target cells both in a dose-dependent and specific manner, and induced immune activity against cancer in vitro and in vivo [68, 69] (Fig. 1A). The system which used fluorescein isothiocyanate (FITC)-specific CAR-T cell and CD19- or CD22-directed bifunctional molecule showed high cytotoxicity to CD19- or CD22-positive cells [69]. The activation and proliferation of T cells not only depend on the presence of tumor cells that express the target antigen, but also are strictly regulated and dose titratable by bifunctional molecules. The safety of CAR-T therapy in solid tumors may be significantly improved through such a BiTE-based method.
Summarized strategies for CAR-T cells to overcome toxicity in CRC. A: CAR-T cells were engineered to express CARs that do not directly recognize antigens on target cells, but are recruited to effector cells through bifunctional small molecule “switch”. B: CAR-T cell are transduced with optimized constructions include the CAR structures that provide inefficient activation signals of T cells when bind to one antigen and chimeric costimulatory receptors(CCR) recognize another antigen.
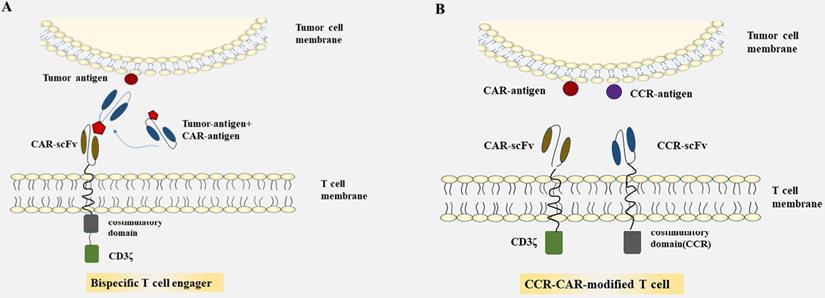
Dual-CAR-modified T cell
Most of the CARs are designed with single target, which may lead to immune escape and selective survival of antigen-negative tumor cells on account of tumor heterogeneity [70-72]. The therapeutic effect of single CAR treatment correlates with the own properties of the target antigen, and with its role in tumorigenesis and tumor growth. Researchers have proposed strategies to render T cells specifically in the lack of TSAs, where T cells are engineered with optimized constructions including the CAR structures that provide inefficient activation signals of T cells when binding to one antigen and chimeric costimulatory receptors (CCR) recognize another antigen [73, 74] (Fig. 1B). T cells expressing PSMA-CCR and suboptimal activation receptor (low-affinity PSCA-specific scFv) were effectively activated when co-cultured with PSCA+PSMA+ instead of PSCA+PSMA- cells, and showed enhanced protection against tumor cells in vitro and in vivo [74]. In a dual-receptor CAR (dCAR) study, they constructed a tandem structure consisted of CEA-CD3ζ and MSLN-4/1BB signaling domains that are physically separated, and the activation of CAR-T cells was efficiently regulated by CEA and MSLN, respectively. And dCAR-T cells exerted reinforced efficacy compared to single-receptor CAR with only one signaling pathway, under the existence of CEA+MSLN+ tumor cells [75]. Nevertheless, multiple parameters of such dCAR system have to be precisely controlled for practical application, including the signaling intensity, the level and ratio relative to each synergic receptor. Moreover, the ability to truly exhibiting switch controllability of the dCAR system can also be complicated, due to the different levels of target antigen expressed on various tissues and organs in the human body.
The design of “switch” mediated CARs through small molecules that control the therapeutic effect of T cells while still retaining antigen specificity, may make it possible for these parameters controlling of CAR-T immunotherapy in solid tumors [76-78]. In these physically separated receptors, antigen-binding site and intracellular signaling domain integrate into a complete signal chain only with the help of heterodimerizing small molecules, including rapamycin analog AP21967 (rapalog) that induces heterodimerization of FK506 binding protein (FKBP) domain and T2089L mutant of FKBP-rapamycin binding component (FRB*) [76], gibberellin analog containing an acetoxymethyl group (GA3-AM) induced dimerization system which consists of gibberellin insensitive dwarf1 (GID1) and gibberellin insensitive (GAI) [78], and rimiducid (AP1903) induced inducible MyD88/CD40 (iMC) and antigen-CD3ζ signaling domain [77]. This titratable pharmacologic regulation can not only combine with dual antigens, or even multiple antigens, but make it possible for precisely controlling the intensity and timing of T cell activity, and even the location of T cells, thereby improving antigen-specificity and dose-dependent safety, if this dimerization system cooperates with the dCAR system described above.
Strategies to enhance efficacy
Although studies have demonstrated promising prospect of CAR-T therapy in some mice models, the full and persistent function of CAR-T cells has been limited for some unique challenges posed by solid tumors: (a) the chemokine/chemokine receptor mismatches, antigen loss or heterogeneity; (b) the barriers characterized by physical and chemical nature of the TME: high tissue pressure, hypoxia, abnormal vasculature, and unfavorable immune cells; (c) the inherent inhibitory mechanisms of T cells [79, 80]. Since then, attempts to strengthen the functions of CAR-T cells in preclinical models and to further prolong the overall survival of CRC patients emerged.
Engineered T cell with immune-activating molecules
Cytokines that play a crucial part in T cell activation, proliferation, and cytotoxicity are one of the best-characterized molecules and delivered by CAR-T cells [81, 82]. Preclinical studies have demonstrated that the involvement of cytokines like IL-2, IL-7, IL-12, and IL-21 enhanced the immune activity of the immune system, and may reverse the inhibitory TME to an immune stimulatory environment [83-86]. Of these immune-activating molecules, IL‐12 is a central one to assist in cancer treatment [87]. In Chi's group, they confirmed that combination use of CEA CAR‐T cells and recombinant human IL-12 (the dose of IL-12 were 1, 10, 50, 100, 200, 500, and 1000 U/mL, respectively) significantly enhanced anti-tumor efficacy in vivo, showed improved T cell efficacy and increased level of serum cytokines than CEA CAR‐T cells alone. The cytotoxicity of CAR-T cells improved with the increase of IL-12 dose within 50 U/mL and is close to saturation when IL-12 dose is more than 50 U/mL [87]. Another kind of “two in one” approach in which researchers engineered mesenchymal stem cells (MSCs) to release both IL-7 and IL-12 for the tremendous plasticity of MSCs and their capability to secrete a mass of immune-activating molecules, also showed a mutual interaction between interleukins and CAR-T cells [86, 88-90]. The improved CAR-T cell activation through modified MSCs was judged by cytokine production and cytolytic activities, and the enhanced elimination of tumor cells in a xenograft mouse model of CRC was confirmed. The safety and feasibility of EGFR and EGFR IL-12 CAR-T cells in the treatment of mCRC are evaluated currently in phase I and II trials (NCT03542799, NCT02959151). Other interleukins combined with CAR-T cell include IL-18, IL-23, IL-15, which have yet been seen in CRC [91-93]. Such a combinatory strategy has great potential to boost CAR-T cells for stronger and safer efficacy, but the significant increase of serum cytokines levels resulted from enhanced T cell activation was also observed [87, 94].
As is known, IL-12 contributes to cell differentiation of Th1 and proliferation of natural killer (NK) cell, while IL-2 and IL-21 work co-operatively to enhance the cytolytic activity of CD8+T cells and NK cells, induce a sustainable CD8+ response [95]. Engineering T cells to secrete these molecules which are driven by the CAR in tumor site to avoid the systematical increase of cytokines would be desirable [96, 97]. The interleukins delivered by CAR-T cells deposit in tumor site could attract innate immune cells like macrophages and NK cells attacking tumor cells that escape from CAR-T cells due to the lack of targets recognized by CAR [98] (Fig. 2A). Some bench studies also showed that CAR-T cells genetically engineered to constitutively secrete cytokines can reverse the unfavorable TME not only by enhancing the proliferation and expansion of T cells but also by activating the immune effector cells [82, 98, 99].
Summarized strategies for CAR-T cells to enhance efficacy in CRC. A: Engineering T cells to secrete inducible IL-12(iIL-12) which are driven by the CAR in tumor site. The interleukins delivered by CAR-T cells deposit in the tumor lesion could attract innate immune cells, like NK cells ,endogenous T cells, dendritic cells and macrophages modulating the tumor microinvironment. B: Furnishing CAR-T cells with genes of chemokine-corresponding receptors to chemokines secreted by solid tumors improves the homing of T cells and the capability of migration and achieving access to tumor site.
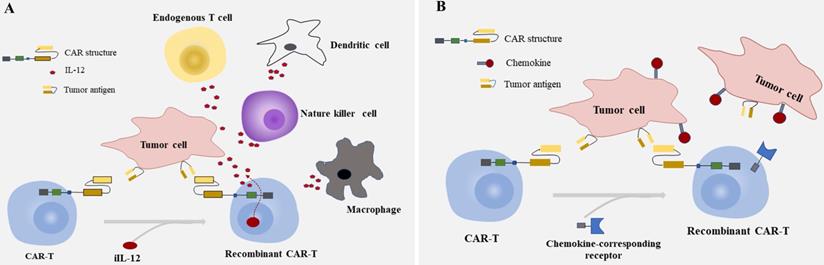
Chemokine and chemokine receptor mismatch is considered to be one of the key factors that limit T cell location in tumor deposits, as active trafficking of T cells to tumor sites in part be attributable to the correct match between chemokine receptors on T cells and chemokines exposed on tumor cells [80, 100, 101]. To overcome the above factor impeding the migration of CAR-T cells to tumor mass, furnishing CAR-T cells with chemokine (C-C-motif) receptor (CCR), C-X-C motif chemokine receptor (CXCR), or other receptors to chemokines secreted by tumor cells is another option to achieve effective homing of CAR-T cells and improve the capability of migration [102-104] (Fig. 2B). As shown in recent research, CXCR1/CXCR2 modified CARs significantly improved T cell persistence and migration, induced durable response, and long-lasting immunologic memory in preclinical models of aggressive tumors [105]. The identical cytotoxicity was also observed in a case of hepatocellular carcinoma (HCC), in which the migration ability of T cells engineered with CXCR2 significantly enhanced in vitro, showed accelerated cell trafficking and tumor-specific accumulation in vivo [102]. Given the characteristics between T cells and tumors, it would be of great prospects for engineering T cells with CXCR modified CARs in solid tumors like CRC. But caution should be raised as some preliminary data indicate that the continuous secretion of high levels of intracellular calcium resulted from chronic stimulation of T cells through chemokine receptors may induce T cell hypofunction [106, 107].
Regional administration of T cell
An opportunity to avoid the traffic load in solid tumors might be regional administration of T cell, which has made modest success in some cancer [108-110]. In CRC with liver metastases, Burga has demonstrated that cell delivery to liver metastases was optimized through regional intrahepatic infusion [111]. Furthermore, the efficacy of CAR-T cells could be enhanced when mice received cells in tandem with blockage of inhibitory immune molecules, including myeloid-derived suppressor cells (MDSC) depletion, GM-CSF neutralization that are to prevent MDSC expansion, and PD-L1 inhibition. They later carried out a preclinical study about intraperitoneal (IP) delivery of CEA CAR-T cells against peritoneal carcinomatosis on the basis above, showed superior anti-tumor efficacy against CEA+ peritoneal tumors, prolonged protection against tumor re-challenges, and effector memory T (Tcm) cells increased over time when compared with systemic infusion [112]. Moreover, in combination with depleting antibodies for MDSC and regulatory T cells, IP delivery of CAR-T cells further protected mice from peritoneal deposits. Data also showed that IP delivery of T cell did lead to high levels of systemic IFN-γ following treatment. Regional administration of T cells can indeed improve the situation of inadequate trafficking and persistence of T cell, and the blockage of inhibitory molecules can further reverse the unfavorable TME, rescue the efficacy of T cell [113, 114]. Whether patients with CRC can generally benefit from strategies combined with immune checkpoint blockage remains to be confirmed in further studies.
Conclusions and Further perspectives
In light of the recent progress of CAR therapy, especially in hematological malignancies, a series of optimization strategies for more specific and safer benefits in solid tumors are in need even with lots of limitations to this method existed. As summarized in this review, there are various targets to choose from for the CAR treatment in CRC and many promising therapeutic strategies have been proposed and shown success in preclinical models. The important role of CAR therapy in solid tumors is confirmed in a variety of studies. As discussed above, an ideal target candidate for the CAR structure should meet the conditions including significantly up-regulated in tumor cells and at low or even undetectable levels on normal tissues. But it is usually hard to find out. We hold the opinion that it is a promising way to combine the dCAR system with heterodimerizing small molecules, to binding multiple antigens, thus the activation of T cells can be controlled by adjusting the density of antigens to achieve accurate targeting of tumor cells, under the control of small molecules. When clinicians hold the controllable “switch”, these impressive results confirmed by in vivo and in vitro studies may be “replicated” to patients with solid tumors more safely.
Besides engineered with T cells, NK cells are proposed to be another vector of the CAR structure and considered to be less prone to cause graft-versus-host disease (GVHD), in addition, the CAR-NK cells may expand the horizon of treatment in solid tumors and extend to allogenic CAR therapies [115, 116]. Personalized modification of CAR-T cells, such as integrating with anti-tumor cytokines, changing cell infusion path, and a series of novel CAR structure-design are some assumed changes that can make a better prospect. Moreover, engineering T cells with a more suitable subset, manipulating immunosuppressive checkpoints such as PD1 or CTLA4, and incorporating soluble immune-unfavorable cytokines like TGF-β may also achieve durable response in solid tumors [117-119]. These strategies have opened the field for further development of adoptive cell immunotherapy, and extensive works are needed for implementing these methods in CRC. As for CRC, there are varieties of researches combined PD-1 antibodies that have been identified as a possible target for immunotherapy in MSI-h CRC patients, or PD-1 disruption like CRISPR/Cas9 ribonucleoprotein-mediated editing to break up the PD-1 gene which is located in human primary T cells [120-122]. Furthermore, the introduction of PD-1-specific scFvs has generated CAR-T cells with improved safety and efficacy against poorly responding tumor cells for its unique ability to reactivate the host's anti-tumor immunity and protected T cells from immunosuppression via disrupting the binding to its ligand [118]. Engineering CAR-T cells to co-express a PD-1 decoy receptor that replaces the PD-1 transmembrane and intracellular signaling domains with the costimulatory domain of CD28 or IL-7 receptor is proved to show superior and persistent antitumor activity against various solid tumors, converting or competing possible inhibitory signal to improve T cell function [123, 124]. And the superior CAR-T cells showed a significantly greater number of CD8+CAR-TE/EM and CD8+CAR-TCM cells. Given these favorable mechanisms, the poor efficacy of method aiming at immune checkpoint blocking alone in MSS or mismatch repair proficient (pMMR) patients with CRC may be rescued by combination with CAR-T cell therapy.
Adoptive T cell immunotherapy is a method that reactivates the immune system against cancer and has been in development for decades. The progress and experience obtained from hematological malignancies with this method have strengthened the impression of immunotherapy in cancer and make it possible for treatment in solid tumors. In this review, we have summarized CAR-T cells as monotherapy or in combination with other methods currently applied in CRC, and it is believed to bring substantial clinical benefit to CRC patients with prolonged endeavor.
Abbreviations
CEA: carcinoembryonic; EGFR: epidermal growth factor receptor; MUC1: transmembrane mucins; EpCAM: epithelial cell and adhesion molecule; NKG2DL: natural killer group 2, member D ligand; HER2: human epidermal growth factor receptor 2; IFN-γ: interferon γ; dCAR: dual chimeric antigen receptor; TSAs: tumor specific antigens; CCR: chimeric costimulatory receptor.
Authors' Contributions
Huali Li conceived and drafted the initial manuscript. Chao Yang, Huangrong Cheng, Shuoyang Huang taken charge of Literature searching and classification. Yongbin Zheng made substantial contributions to manuscript modifications. All authors read and approved the final manuscript for publication.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7-30
2. Siegel RL, Miller KD, Goding Sauer A. et al. Colorectal cancer statistics, 2020. CA Cancer J Clin. 2020;70(3):145-164
3. Janssen LME, Ramsay EE, Logsdon CD. et al. The immune system in cancer metastasis: friend or foe? J Immunother Cancer. 2017;5(1):79
4. Silver DJ, Sinyuk M, Vogelbaum MA. et al. The intersection of cancer, cancer stem cells, and the immune system: therapeutic opportunities. Neuro-oncology. 2016;18(2):153-9
5. Emambux S, Tachon G, Junca A. et al. Results and challenges of immune checkpoint inhibitors in colorectal cancer. Expert Opin Biol Ther. 2018;18(5):561-73
6. Yokoyama Y, Lew ED, Seelige R. et al. Immuno-oncological Efficacy of RXDX-106, a Novel TAM (TYRO3, AXL, MER) Family Small-Molecule Kinase Inhibitor. Cancer Res. 2019;79(8):1996-2008
7. Passardi A, Canale M, Valgiusti M. et al. Immune Checkpoints as a Target for Colorectal Cancer Treatment. Int J Mol Sci. 2017;18(6):1324
8. Wang S, Mou J, Cui L. et al. Astragaloside IV inhibits cell proliferation of colorectal cancer cell lines through down-regulation of B7-H3. Biomed Pharmacother. 2018;102:1037-44
9. Wang X, Wang N, Yang Y. et al. Polydopamine nanoparticles carrying tumor cell lysate as a potential vaccine for colorectal cancer immunotherapy. Biomater Sci. 2019;7(7):3062-75
10. Van Cutsem E, Machiels J, Van den Eynde M. et al. Phase 1 studies assessing the safety and clinical activity of autologous and allogeneic NKG2D-based CAR-T therapy in metastatic colorectal cancer. Ann Oncol. 2019;30(Suppl 4):iv124-iv5
11. Cremolini C, Morano F, Moretto R. et al. Negative hyper-selection of metastatic colorectal cancer patients for anti-EGFR monoclonal antibodies: the PRESSING case-control study. Ann Oncol. 2017;28(12):3009-14
12. GROSS G, WAKS T, ESHHAR Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A. 1989;86(24):10024-8
13. Liu B, Yan L, Zhou M. Target selection of CAR T cell therapy in accordance with the TME for solid tumors. Am J Cancer Res. 2019;9(2):228-41
14. Li J, Li W, Huang K. et al. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: lessons learned and strategies for moving forward. J Hematol Oncol. 2018;11(1):22
15. Stoiber S, Cadilha BL, Benmebarek M-R. et al. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells. 2019;8(5):472
16. Newick K, O'Brien S, Moon E. et al. CAR T Cell Therapy for Solid Tumors. Annu Rev Med. 2017;68:139-52
17. Thistlethwaite FC, Gilham DE, Guest RD. et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother. 2017;66(11):1425-36
18. Li S, Zhang J, Wang M. et al. Treatment of acute lymphoblastic leukaemia with the second generation of CD19 CAR-T containing either CD28 or 4-1BB. Br J Haematol. 2018;181(3):360-71
19. Enblad G, Karlsson H, Gammelgård G. et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin Cancer Res. 2018;24(24):6185-94
20. Chmielewski M, Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. 2015;15(8):1145-54
21. Zhang C, Wang Z, Yang Z. et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA Metastatic Colorectal Cancers. Mol Ther. 2017;25(5):1248-58
22. Han Y, Xie W, Song D-G. et al. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells. J Hematol Oncol. 2018;11(1):92
23. Martinez M, Moon EK. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front Immunol. 2019;10:128
24. Zhao Z, Xiao X, Saw PE. et al. Chimeric antigen receptor T cells in solid tumors: a war against the tumor microenvironment. Sci China Life Sci. 2020;63(2):180-205
25. Magee MS, Abraham TS, Baybutt TR. et al. Human GUCY2C-Targeted Chimeric Antigen Receptor (CAR)-Expressing T Cells Eliminate Colorectal Cancer Metastases. Cancer Immunol Res. 2018;6(5):509-16
26. Magee MS, Kraft CL, Abraham TS. et al. GUCY2C-directed CAR-T cells oppose colorectal cancer metastases without autoimmunity. Oncoimmunology. 2016;5(10):e1227897
27. Joosse SA, Gorges TM, Pantel K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol Med. 2015;7(1):1-11
28. Zhang B-L, Li D, Gong Y-L. et al. Preclinical Evaluation of Chimeric Antigen Receptor-Modified T Cells Specific to Epithelial Cell Adhesion Molecule for Treating Colorectal Cancer. Hum Gene Ther. 2019;30(4):402-12
29. Schnell U, Cirulli V, Giepmans BNG. EpCAM: structure and function in health and disease. Biochim Biophys Acta. 2013;1828(8):1989-2001
30. Deng X, Gao F, Li N. et al. Antitumor activity of NKG2D CAR-T cells against human colorectal cancer cells in vitro and in vivo. Am J Cancer Res. 2019;9(5):945-58
31. Antonangeli F, Soriani A, Cerboni C. et al. How Mucosal Epithelia Deal with Stress: Role of NKG2D/NKG2D Ligands during Inflammation. Front Immunol. 2017;8:1583
32. Greally M, Kelly CM, Cercek A. HER2: An emerging target in colorectal cancer. Curr Probl Cancer. 2018;42(6):560-71
33. KOKAI Y, COHEN JA, DREBIN JA. et al. Stage- and tissue-specific expression of the neu oncogene in rat development. Proc Natl Acad Sci U S A. 1987;84(23):8498-501
34. Teng R, Zhao J, Zhao Y. et al. Chimeric Antigen Receptor-modified T Cells Repressed Solid Tumors and Their Relapse in an Established Patient-derived Colon Carcinoma Xenograft Model. J Immunother. 2019;42(2):33-42
35. Hege KM, Bergsland EK, Fisher GA. et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunother Cancer. 2017;5:22
36. Clarke MF, Dick JE, Dirks PB. et al. Cancer stem cells-perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66(19):9339-44
37. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178-96
38. Sureban SM, May R, Mondalek FG. et al. Nanoparticle-based delivery of siDCAMKL-1 increases microRNA-144 and inhibits colorectal cancer tumor growth via a Notch-1 dependent mechanism. J Nanobiotechnology. 2011;9:40
39. Sureban SM, Berahovich R, Zhou H. et al. DCLK1 Monoclonal Antibody-Based CAR-T Cells as a Novel Treatment Strategy against Human Colorectal Cancers. Cancers (Basel). 2019;12(1):54
40. Nakanishi Y, Seno H, Fukuoka A. et al. Dclk1 distinguishes between tumor and normal stem cells in the intestine. Nat Genet. 2013;45(1):98-103
41. Hammarström S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999;9(2):67-81
42. Matsuoka Y, Endo K, Kawamura Y. et al. Normal bronchial mucus contains high levels of cancer-associated antigens, CA125, CA19-9, and carcinoembryonic antigen. Cancer. 1990;65(3):506-10
43. Habban Akhter M, Sateesh Madhav N, Ahmad J. Epidermal growth factor receptor based active targeting: a paradigm shift towards advance tumor therapy. Artif Cells Nanomed Biotechnol. 2018;46(sup2):1188-98
44. St-Arnaud R, Chabot JG, Pelletier G. et al. Epidermal growth factor: structure, location, phosphorylation and regulation of its receptor. Biochimie. 1984;66(7-8):515-30
45. Betge J, Schneider NI, Harbaum L. et al. MUC1, MUC2, MUC5AC, and MUC6 in colorectal cancer: expression profiles and clinical significance. Virchows Arch. 2016;469(3):255-65
46. Peat N, Gendler SJ, Lalani N. et al. Tissue-specific expression of a human polymorphic epithelial mucin (MUC1) in transgenic mice. Cancer Res. 1992;52(7):1954-60
47. Charruyer A, Strachan LR, Yue L. et al. CD133 is a marker for long-term repopulating murine epidermal stem cells. J Invest Dermatol. 2012;132(11):2522-33
48. Frick GS, Pitari GM, Weinberg DS. et al. Guanylyl cyclase C: a molecular marker for staging and postoperative surveillance of patients with colorectal cancer. Expert Rev Mol Diagn. 2005;5(5):701-13
49. Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol. 2003;3(10):781-90
50. Morgan RA, Yang JC, Kitano M. et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843-51
51. Wang Y, Chen M, Wu Z. et al. CD133-directed CAR T cells for advanced metastasis malignancies: A phase I trial. Oncoimmunology. 2018;7(7):e1440169
52. Magee MS, Abraham TS, Baybutt TR. et al. Human GUCY2C-Targeted Chimeric Antigen Receptor (CAR)-Expressing T Cells Eliminate Colorectal Cancer Metastases. Cancer Immunol Res. 2018;6(5):509-16
53. Xu M, Real FX, Welt S. et al. Expression of TAG-72 in normal colon, transitional mucosa, and colon cancer. Int J Cancer. 1989;44(6):985-9
54. Cho J, Kim K-M, Kim HC. et al. The prognostic role of tumor associated glycoprotein 72 (TAG-72) in stage II and III colorectal adenocarcinoma. Pathol Res Pract. 2019;215(1):171-6
55. Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019;34:45-55
56. Neelapu SS, Tummala S, Kebriaei P. et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol. 2018;15(1):47-62
57. Frey N, Porter D. Cytokine Release Syndrome with Chimeric Antigen Receptor T Cell Therapy. Biol Blood Marrow Transplant. 2019;25(4):e123-e7
58. Wei J, Han X, Bo J. et al. Target selection for CAR-T therapy. J Hematol Oncol. 2019;12(1):62
59. Fitzgerald JC, Weiss SL, Maude SL. et al. Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy for Acute Lymphoblastic Leukemia. Crit Care Med. 2017;45(2):e124-e31
60. Liu D, Zhao J. Cytokine release syndrome: grading, modeling, and new therapy. J Hematol Oncol. 2018;11(1):121
61. Chou CK, Turtle CJ. Insight into mechanisms associated with cytokine release syndrome and neurotoxicity after CD19 CAR-T cell immunotherapy. Bone Marrow Transplant. 2019;54(Suppl 2):780-4
62. Roybal KT, Rupp LJ, Morsut L. et al. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 2016;164(4):770-9
63. Roybal KT, Williams JZ, Morsut L. et al. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell. 2016;167(2):419-432.e16
64. Wolf E, Hofmeister R, Kufer P. et al. BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov Today. 2005;10(18):1237-44
65. Lutterbuese R, Raum T, Kischel R. et al. Potent control of tumor growth by CEA/CD3-bispecific single-chain antibody constructs that are not competitively inhibited by soluble CEA. J Immunother. 2009;32(4):341-52
66. Osada T, Hsu D, Hammond S. et al. Metastatic colorectal cancer cells from patients previously treated with chemotherapy are sensitive to T-cell killing mediated by CEA/CD3-bispecific T-cell-engaging BiTE antibody. Br J Cancer. 2010;102(1):124-33
67. Oberst MD, Fuhrmann S, Mulgrew K. et al. CEA/CD3 bispecific antibody MEDI-565/AMG 211 activation of T cells and subsequent killing of human tumors is independent of mutations commonly found in colorectal adenocarcinomas. mAbs. 2014;6(6):1571-84
68. Kim MS, Ma JSY, Yun H. et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J Am Chem Soc. 2015;137(8):2832-5
69. Ma JSY, Kim JY, Kazane SA. et al. Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci U S A. 2016;113(4):E450-E8
70. Bagley SJ, O'Rourke DM. Clinical investigation of CAR T cells for solid tumors: Lessons learned and future directions. Pharmacol Ther. 2019;205:107419
71. Liu E, Marin D, Banerjee P. et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med. 2020;382(6):545-53
72. Kloss CC, Lee J, Zhang A. et al. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol Ther. 2018;26(7):1855-66
73. Wilkie S, van Schalkwyk MCI, Hobbs S. et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32(5):1059-70
74. Kloss CC, Condomines M, Cartellieri M. et al. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71-5
75. Zhang E, Yang P, Gu J. et al. Recombination of a dual-CAR-modified T lymphocyte to accurately eliminate pancreatic malignancy. J Hematol Oncol. 2018;11(1):102
76. Wu C-Y, Roybal KT, Puchner EM. et al. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science (New York, NY). 2015;350(6258):aab4077
77. Foster AE, Mahendravada A, Shinners NP. et al. Regulated Expansion and Survival of Chimeric Antigen Receptor-Modified T Cells Using Small Molecule-Dependent Inducible MyD88/CD40. Mol Ther. 2017;25(9):2176-88
78. Miyamoto T, DeRose R, Suarez A. et al. Rapid and orthogonal logic gating with a gibberellin-induced dimerization system. Nat Chem Biol. 2012;8(5):465-70
79. Miliotou AN, Papadopoulou LC. CAR T-cell Therapy: A New Era in Cancer Immunotherapy. Curr Pharm Biotechnol. 2018;19(1):5-18
80. Wang Z, Chen W, Zhang X. et al. A long way to the battlefront: CAR T cell therapy against solid cancers. J Cancer. 2019;10(14):3112-23
81. Liu Y, Di S, Shi B. et al. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3-Targeted Chimeric Antigen Receptor-Engineered T Cells in Hepatocellular Carcinoma. J Immunol. 2019;203(1):198-207
82. Yeku OO, Brentjens RJ. Armored CAR T-cells: utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem Soc Trans. 2016;44(2):412-8
83. Sim GC, Radvanyi L. The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev. 2014;25(4):377-90
84. Kueberuwa G, Kalaitsidou M, Cheadle E. et al. CD19 CAR T Cells Expressing IL-12 Eradicate Lymphoma in Fully Lymphoreplete Mice through Induction of Host Immunity. Mol Ther Oncolytics. 2018;8:41-51
85. Adachi K, Kano Y, Nagai T. et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36(4):346-51
86. Hombach AA, Geumann U, Günther C. et al. IL7-IL12 Engineered Mesenchymal Stem Cells (MSCs) Improve A CAR T Cell Attack Against Colorectal Cancer Cells. Cells. 2020;9(4):873
87. Chi X, Yang P, Zhang E. et al. Significantly increased anti-tumor activity of carcinoembryonic antigen-specific chimeric antigen receptor T cells in combination with recombinant human IL-12. Cancer Med. 2019;8(10):4753-65
88. Almalki SG, Agrawal DK. Key transcription factors in the differentiation of mesenchymal stem cells. Differentiation. 2016;92(1-2):41-51
89. Zimmerlin L, Park TS, Zambidis ET. et al. Mesenchymal stem cell secretome and regenerative therapy after cancer. Biochimie. 2013;95(12):2235-45
90. Poggi A, Varesano S, Zocchi MR. How to Hit Mesenchymal Stromal Cells and Make the Tumor Microenvironment Immunostimulant Rather Than Immunosuppressive. Front Immunol. 2018;9:262
91. Hu B, Ren J, Luo Y. et al. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017;20(13):3025-33
92. Wang D, Shao Y, Zhang X. et al. IL-23 and PSMA-targeted duo-CAR T cells in Prostate Cancer Eradication in a preclinical model. J Transl Med. 2020;18(1):23
93. Chen Y, Sun C, Landoni E. et al. Eradication of Neuroblastoma by T Cells Redirected with an Optimized GD2-Specific Chimeric Antigen Receptor and Interleukin-15. Clinical Cancer Res. 2019;25(9):2915-24
94. Zhang L, Morgan RA, Beane JD. et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clinical Cancer Res. 2015;21(10):2278-88
95. Gutting T, Burgermeister E, Härtel N. et al. Checkpoints and beyond - Immunotherapy in colorectal cancer. Semin Cancer Biol. 2019;55:78-89
96. Zimmermann K, Kuehle J, Dragon AC. et al. Design and Characterization of an “All-in-One” Lentiviral Vector System Combining Constitutive Anti-G CAR Expression and Inducible Cytokines. Cancers (Basel). 2020;12(2):375
97. Chmielewski M, Abken H. CAR T Cells Releasing IL-18 Convert to T-Bet FoxO1 Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017;21(11):3205-19
98. Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. 2014;257(1):83-90
99. Avanzi MP, Yeku O, Li X. et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018;23(7):2130-41
100. Moon EK, Wang L-CS, Bekdache K. et al. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology. 2018;7(3):e1395997
101. Ager A, Watson HA, Wehenkel SC. et al. Homing to solid cancers: a vascular checkpoint in adoptive cell therapy using CAR T-cells. Biochem Soc Trans. 2016;44(2):377-85
102. Liu G, Rui W, Zheng H. et al. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur J Immunol. 2020;50(5):712-724
103. Knochelmann HM, Smith AS, Dwyer CJ. et al. CAR T Cells in Solid Tumors: Blueprints for Building Effective Therapies. Front Immunol. 2018;9:1740
104. Mollica Poeta V, Massara M, Capucetti A. et al. Chemokines and Chemokine Receptors: New Targets for Cancer Immunotherapy. Front Immunol. 2019;10:379
105. Jin L, Tao H, Karachi A. et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019;10(1):4016
106. Heissmeyer V, Macián F, Im S-H. et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. 2004;5(3):255-65
107. Adachi Y, Hattori M, Yoshida T. Regulation of T cell activation and anergy by the intensity of the Ca²+ signal in cooperation with other signals. Biosci Biotechnol Biochem. 2010;74(9):1788-93
108. Priceman SJ, Tilakawardane D, Jeang B. et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2 Breast Cancer Metastasis to the Brain. Clin Cancer Res. 2018;24(1):95-105
109. Murad JP, Kozlowska AK, Lee HJ. et al. Effective Targeting of TAG72 Peritoneal Ovarian Tumors via Regional Delivery of CAR-Engineered T Cells. Front Immunol. 2018;9:2268
110. Nellan A, Rota C, Majzner R. et al. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J Immunother Cancer. 2018;6(1):30
111. Burga RA, Thorn M, Point GR. et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. 2015;64(7):817-29
112. Katz SC, Point GR, Cunetta M. et al. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther. 2016;23(5):142-8
113. Fujiwara K, Shigematsu K, Tachibana M. et al. Development and functional analysis of an anticancer T-cell medicine with immune checkpoint inhibitory ability. IUBMB life. 2020;72(8):1649-58
114. Jiang Y, Zhao X, Fu J. et al. Progress and Challenges in Precise Treatment of Tumors With PD-1/PD-L1 Blockade. Front Immunol. 2020;11:339
115. Zhang J, Zheng H, Diao Y. Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy. Int J Mol Sci. 2019;20(2):317
116. Zhang Q, Zhang H, Ding J. et al. Combination Therapy with EpCAM-CAR-NK-92 Cells and Regorafenib against Human Colorectal Cancer Models. J Immunol Res. 2018;2018:4263520
117. Gargett T, Truong N, Ebert LM. et al. Optimization of manufacturing conditions for chimeric antigen receptor T cells to favor cells with a central memory phenotype. Cytotherapy. 2019;21(6):593-602
118. Rafiq S, Yeku OO, Jackson HJ. et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. 2018;36(9):847-56
119. Stüber T, Monjezi R, Wallstabe L. et al. Inhibition of TGF-β-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J Immunother Cancer. 2020;8(1):e000676
120. Hu W, Zi Z, Jin Y. et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. 2019;68(3):365-77
121. Yaghoubi N, Soltani A, Ghazvini K. et al. PD-1/PD-L1 blockade as a novel treatment for colorectal cancer. Biomed Pharmacother. 2019;110:312-8
122. Xiao Y, Freeman GJ. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015;5(1):16-8
123. Huang B, Luo L, Wang J. et al. B7-H3 specific T cells with chimeric antigen receptor and decoy PD-1 receptors eradicate established solid human tumors in mouse models. Oncoimmunology. 2020;9(1):1684127
124. Liu X, Ranganathan R, Jiang S. et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016;76(6):1578-90
Author contact
![]() Corresponding author: Professor Yongbin Zheng, Department of Gastrointestinal Surgery, Renmin Hospital of Wuhan University, 99 Zhangzhidong Road, Wuhan, Hubei 430060, P.R. China.
Corresponding author: Professor Yongbin Zheng, Department of Gastrointestinal Surgery, Renmin Hospital of Wuhan University, 99 Zhangzhidong Road, Wuhan, Hubei 430060, P.R. China.