Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(13):3887-3899. doi:10.7150/jca.51467 This issue Cite
Research Paper
High SEC61G expression predicts poor prognosis in patients with Head and Neck Squamous Cell Carcinomas
Leifeng Liang1*, Qingwen Huang2*, Mei Gan1, Liujun Jiang1, Haolin Yan1, Zhan Lin1, Haisheng Zhu1, Rensheng Wang3 ![]() , Kai Hu3
, Kai Hu3 ![]()
1. Department of Oncology, The Sixth Affiliated Hospital of Guangxi Medical University, Yulin, Guangxi, China.
2. Department of Pathology, The Sixth Affiliated Hospital of Guangxi Medical University, Yulin, Guangxi, China.
3. Department of Radiation Oncology, The First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, China.
*These authors contributed equally to this work.
Received 2020-8-3; Accepted 2021-4-23; Published 2021-5-5
Abstract

Background: Overexpression of the membrane protein SEC61 translocon gamma subunit (SEC61G) has been observed in a variety of cancers; however, its role in head and neck squamous cell carcinomas (HNSCC) is unknown. This study aimed to elucidate the relationship between SEC61G and HNSCC based on data from The Cancer Genome Atlas (TCGA) database.
Methods: Data for HNSCC patients were collected from TCGA and the expression level of SEC61G was compared between paired HNSCC and normal tissues using the Wilcoxon rank-sum test. The relationship between clinicopathologic features and SEC61G expression was also analyzed using the Wilcoxon rank-sum test and logistic regression. Receiver operating characteristic (ROC) curves were generated to evaluate the value of SEC61G as a binary classifier using the area under the curve (AUC value). The association of clinicopathologic characteristics with prognosis in HNSCC patients was assessed using Cox regression and the Kaplan-Meier methods. A nomogram, based on Cox multivariate analysis, was used to predict the impact of SEC61G on prognosis. Functional enrichment analysis was performed to determine the hallmark pathways associated with differentially expressed genes in HNSCC patients exhibiting high and low SEC61G expression.
Results: The expression of SEC61G was significantly elevated in HNSCC tissues compared to normal tissues (P < 0.001). The high expression of SEC61G was significantly correlated with the T stage, M stage, clinical stage, TP53 mutation status, PIK3CA mutation status, primary therapy outcome, and cervical lymph node dissection (all P < 0.05). Meanwhile, ROC curves suggested the significant diagnostic ability of SEC61G for HNSCC (AUC = 0.923). Kaplan-Meier survival analysis showed that patients with HNSCC characterized by high SEC61G expression had a poorer prognosis than patients with low SEC61G expression (hazard ratio = 1.95, 95% confidence interval 1.48-2.56, P < 0.001). Univariate and multivariate analyses revealed that SEC61G was independently associated with overall survival (P = 0.027). Functional annotations indicated that SEC61G is involved in pathways related to translation and regulation of SLITs/ROBOs expression, SRP-dependent co-translational protein targeting to the membrane, nonsense-mediated decay, oxidative phosphorylation, and Parkinson's disease.
Conclusion: SEC61G plays a vital role in HNSCC progression and prognosis; it may, therefore, serve as an effective biomarker for the prediction of patient survival.
Keywords: SEC61G, prognosis, biomarker, head and neck squamous cell carcinoma
Introduction
HNSCC, including oral, oropharyngeal, and laryngeal cancers, is among the top 10 most common diseases worldwide, and with more than 65,000 new cases and approximately 15,000 deaths each year in the United States alone [1]. Despite active comprehensive treatment and recent advances in therapeutic options, the prognosis for HNSCC patients remains unsatisfactory, with a 5-year survival rate estimated at 65%, even lower in the advanced diseases [1]. The low survival rates associated with HNSCC are partly due to the failure of early diagnosis, which is primarily attributed to the lack of appropriate screening and diagnostic biomarkers [2, 3]. Although various biomarkers have been applied to HNSCC, such as human papillomavirus status and programmed cell death protein 1 [4], their reliability remains controversial. Thus, identification of new biomarkers related to tumor stage and prognosis is exceedingly important to facilitate early diagnosis, prognosis evaluation, and treatment of HNSCC.
SEC61G is one of three subunits comprising the SEC61 membrane protein complex [5], which represents the central module of the protein translocation apparatus in the endoplasmic reticulum membrane [6]. SEC61G participates in protein folding, modification, translocation, and the unfolded protein response, particularly under the conditions of hypoxia and nutrient deprivation in tumor microenvironment [7-9]. SEC61G was also found to be overexpressed in gastric cancer [10], breast cancer [11], and glioblastoma [12]. However, the association of SEC61G with HNSCC has not yet been characterized.
In this study, we sought to demonstrate the correlation between SEC61G and HNSCC and to analyze the prognostic role of SEC61G in HNSCC based on RNA-sequencing (RNA-seq) data from TCGA. Moreover, we analyzed the expression levels of SEC61G in HNSCC and normal tissues and determined the correlation between SEC61G expression and patient prognosis in terms of overall survival (OS). Further, we performed prognosis and clinical correlations to explore the potential diagnostic and prognostic value of SEC61G; while its biological significance was defined using enrichment analysis, molecular interaction network analysis and immune infiltration correlation analysis. Taken together, our study suggests that SEC61G represents a significant independent predictor for HNSCC.
Materials and methods
RNA-seq data source
A total of 501 HNSCC cases with gene expression data (HTSeq-FPKM) were collected from TCGA. Samples with RNA-seq data that lacked corresponding clinical data were excluded from the analysis. Level-3 HTSeq-FPKM data were transformed into transcripts per million reads (TPM) for subsequent analyses. After filtering, the TPM data of 500 HNSCC patients were further analyzed. Gene expression data were divided into a high expression group and a low expression group according to the median SEC61G expression level. This study met the publication guidelines stated by TCGA (https://cancer genome.nih.gov/publications/publication guidelines). All data used were acquired from TCGA, and hence, ethical approval and informed consent of the patients were not required.
Pathological sample collection
Samples were collected for paired tumor and normal tissues from 60 patients with HNSCC diagnosed between January 2015 and December 2015. All patient-derived information and specimens were collected and archived under the protocols approved by the Institutional Review Boards of The Sixth Affiliated Hospital of Guangxi Medical University (Approve No. YLSY-IRB-CR-2020087). This study was performed in accordance with the principles of the Declaration of Helsinki.
Immunohistochemistry study
Immunohistochemistry (IHC) was performed on formalin-fixed, paraffin-embedded tissue sections. SEC61G was detected with the rabbit anti-SEC61G polyclonal antibody (11147-2-AP, Proteintech Group Inc., Wuhan Sanying Biotechnology Development Co. Ltd.). Paraffin slices were dewaxed in xylene. A gradient alcohol dehydration process was then performed consisting of 100%, 95%, 80%, and 70% ethanol (2 min each). The tissues were subsequently rinsed with distilled water twice for 5 min each, and with phosphate-buffered saline (PBS) thrice, for 5 min each. Next, pressure cooker-mediated antigen retrieval was performed in EDTA buffer, pH 9.0 (Fuzhou Maixin Biotechnology Development Co. Ltd.) for 30 min. Sections were incubated with a 1:100 dilution of anti-SEC61G antibody for 30 min at 37 °C and subsequently incubated with enzyme-labeled anti-mouse/rabbit lgG polymer (Fuzhou Maixin Biotechnology Development Co. Ltd.) for 30 min at 25 °C. After rinsing with PBS three times for 5 min each, the sections were incubated with DAB chromogenic reagent (Fuzhou Maixin Biotechnology Development Co., Ltd.) for 5 min, followed by counterstaining with Mayer's hematoxylin, dehydrating, and mounting.
Two independent single-blinded pathologists assessed the sections and applied a semiquantitative scoring system [13] to evaluate staining intensity (0, no staining; 1+, weak staining; 2+, moderate staining; 3+, strong staining) and the percentage of stained cells (0, < 5%; 1, 5%-25%; 2, 26%-50%; 3, 51%-75%; and 4, > 75%). The staining intensity scores were multiplied by the percentage of positive cells to generate the immunoreactivity score of each case [14]. All cases were sorted into two groups according to the immunoreactivity score. Positive of SEC61G was defined as detectable immunoreactions in the cytoplasmic or membrane with an immunoreactivity score of ≥ 1.
Enrichment analysis
The Search Tool for the Retrieval of Interacting Genes (STRING; http://strin g-db.org; version 10.0) online database was used to predict the protein-protein interaction network of SEC61G co-expressed genes and to analyze the functional interactions among proteins [15]. An interaction with a combined score > 0.4 was considered statistically significant. The expression profiles (HTSeq-Counts) between the high SEC61G expression group and the low SEC61G expression group were compared to identify differentially expressed genes (DEG) using Wilcoxon rank-sum test in the R language-related software, DESeq2 (3.8). [16]. Differences with a |log fold change (FC)|>1.5 and adjusted P-value < 0.05 were considered as threshold values for identifying DEGs [17-20]. Clusterprofiler [21] software 3.6.0 was applied to perform Gene Ontology (GO) function enrichment analyses on the DEGs identified between the high and low SEC61G expression groups.
Gene set enrichment analysis (GSEA)
GSEA [22] is a computational method that determines whether a defined set of genes exhibits statistically significant concordant differences between two biological states. In this study, GSEA was performed with the R package ClusterProfiler [21] to elucidate the significant function and pathway differences between the high and low SEC61G expression groups. Each analysis procedure was repeated 1000 times. A function or pathway term withadjusted P-value < 0.05, and false discovery rate (FDR) < 0.25 was considered to statistically significant enrichment [23-25].
Immune infiltration analysis by single-sample GSEA (ssGSEA)
Immune infiltration analysis of HNSCC samples was performed by the ssGSEA method using the GSVA package in R (http://www.biocondutor.org/package/release/bioc/html/GSVA.html) for 24 types of immune cells, including neutrophils, mast cells, eosinophils, macrophages, natural killer (NK) cells, CD56dim NK cells, CD56bright NK cells, dendritic cells (DCs), immature DCs (iDCs), activated DCs (aDCs), plasmacytoid DCs (pDCs), T cells, CD8+ T cells, T helper (Th) cells, Th1 cells, Th2 cells, Th17 cells, T follicular helper cells, regulatory T cells (Treg), central memory T cells (Tcm), effector memory T cells (Tem), gamma delta T cells (Tgd), cytotoxic cells, and B cells. Based on the reported signature genes for the 24 types of immunocytes [26], the relative enrichment score of each was quantified from the gene expression profile for each tumor sample. Spearman's correlation coefficient analysis was performed to identify relationships HNSCC and each immune cell subset. Moreover, immune cell infiltration was investigated between the high and low SEC61G expression groups using the Wilcoxon rank-sum test.
Construction and evaluation of the nomogram
To individualize the predicted survival probability for l, 3, and 5 years, a nomogram was constructed based on the results of the multivariate analysis. The RMS R package was used to generate a nomogram including clinical characteristics significantly associated with SEC61G and calibration plots. Calibration and discrimination are the most used methods for evaluating the performance of models. In this study, the calibration curves were graphically assessed by mapping the nomogram-predicted probabilities against the observed rates, and the 45-degree line represented the best predictive values. The concordance index (C-index) was used to determine the discrimination of the nomogram, which was calculated by a bootstrap approach with 1000 resamples. In addition, the predictive accuracies of the nomogram and individual prognostic factors were compared using the C-index and ROC analyses. All statistical tests were two-tailed with the statistical significance level set at 0.05.
Statistical analysis
All statistical analyses and plots were conducted using R (Version 3.6.3). Wilcoxon rank-sum test and Wilcoxon rank signed test were used to analyze the expression of SEC61G in non-paired samples and paired samples, respectively. Kruskal-Wallis test, Wilcoxon rank-sum test, and logistic regression evaluated relationships between clinical-pathologic features and SEC61G expression. Furthermore, ROC analysis and the frequently-used method for binary assessment were performed using the pROC package [27] to assess the effectiveness of SEC61G expression to discriminate HNSCC from normal samples. The computed AUC value ranging from 0.5 to 0.1 indicates the discriminative potential from 50% to 100%. The prognostic data was obtained from Cell [28], while Cox regression analyses and the Kaplan-Meier method were used to evaluate prognostic factors. In all tests, P-value < 0.05 was considered statistically significant.
Results
Elevated expression of SEC61G in HNSCC
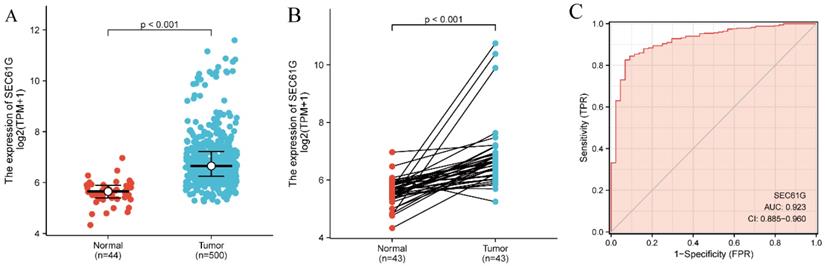
The SEC61G expression levels in 500 tumor tissues were substantially and significantly higher than those in 44 normal tissues (P < 0.001; Figure 1A) and were also higher in the 43 tumor tissues compared with paired normal tissues (P < 0.001; Figure 1B). In addition, SEC61G expression showed promising discriminative power with an AUC value of 0.923 to identify tumors from normal tissue (Figure 1C).
SEC61G expression between cancer and normal tissues in HNSCC patients (A) SEC61G expression levels in HNSCC and matched normal tissues. (B) SEC61G expression levels in HNSCC and matched normal tissues. (C) ROC analysis of SEC61G shows promising discrimination power between tumor and normal tissues.
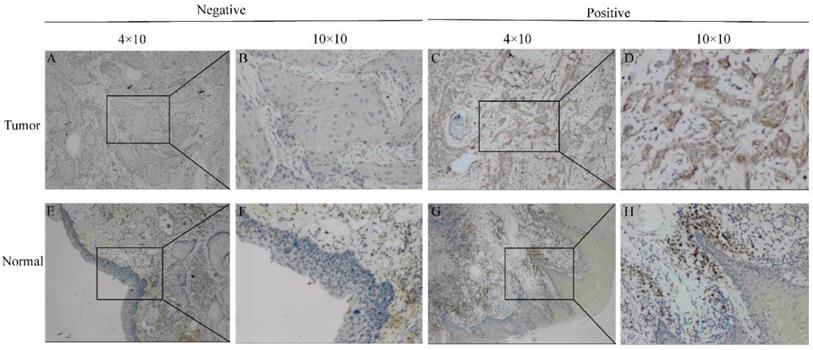
To further determine the significance of SEC61G expression, IHC staining was performed for a cohort comprising 60 cases of primary HNSCC paired with noncancerous tissue. There were 42 males and 18 females with a mean age of 59 years (range 29-80 years) involved in our HNSCC cohort. According to the SEC61G IHC staining, 78.3% (47/60) of normal tissues were negative while 63.3% (38/60) of HNSCC tissues were positive. Representative images are presented in Figure 2.
Representative images of SEC61G expression in tongue cancer tissues and their normal controls. (A, B) Negative or (C, D) positive staining for SEC61G in tongue cancer tissues. (E, F) Negative or (G, H) positive staining for SEC61G in normal tissues. Original magnifications 40× and 100× (inset panels).
Associations between SEC61G expression and clinicopathologic variables
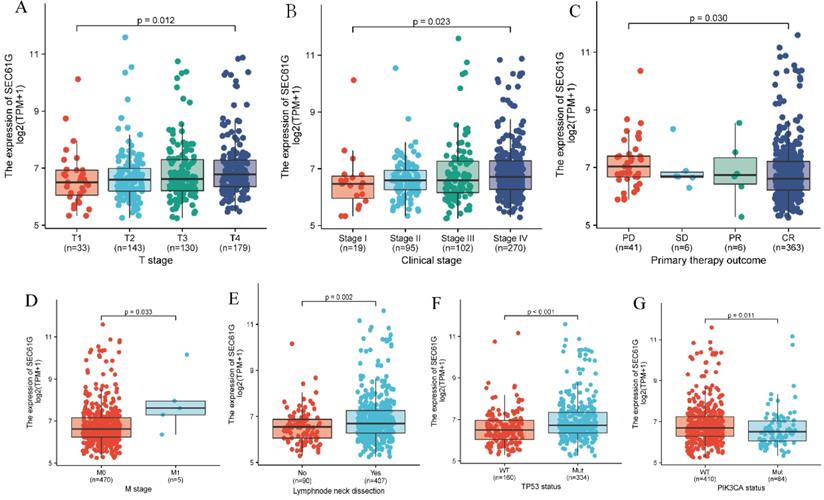
The Kruskal-Wallis rank-sum test showed that the level of SEC61G was significantly correlated with T stage (P = 0.012), clinical stage (P = 0.023), and primary therapy outcome (P = 0.030; Figure 3A-C). Meanwhile, results of the Wilcoxon rank-sum test found that SEC61G expression level was significantly correlated with M stage (P = 0.033), cervical lymph node dissection (P = 0.002), TP53 mutation status (P < 0.001), and PIK3CA mutation status (P = 0.011; Figure 3D-G).
Association of SEC61G expression with clinicopathologic characteristics. (A) T stage; (B) clinical stage; (C) primary therapy outcome; (D) M stage; (E) lymph node neck dissection; (F) TP53 mutation status; (G) PIK3CA mutation status.
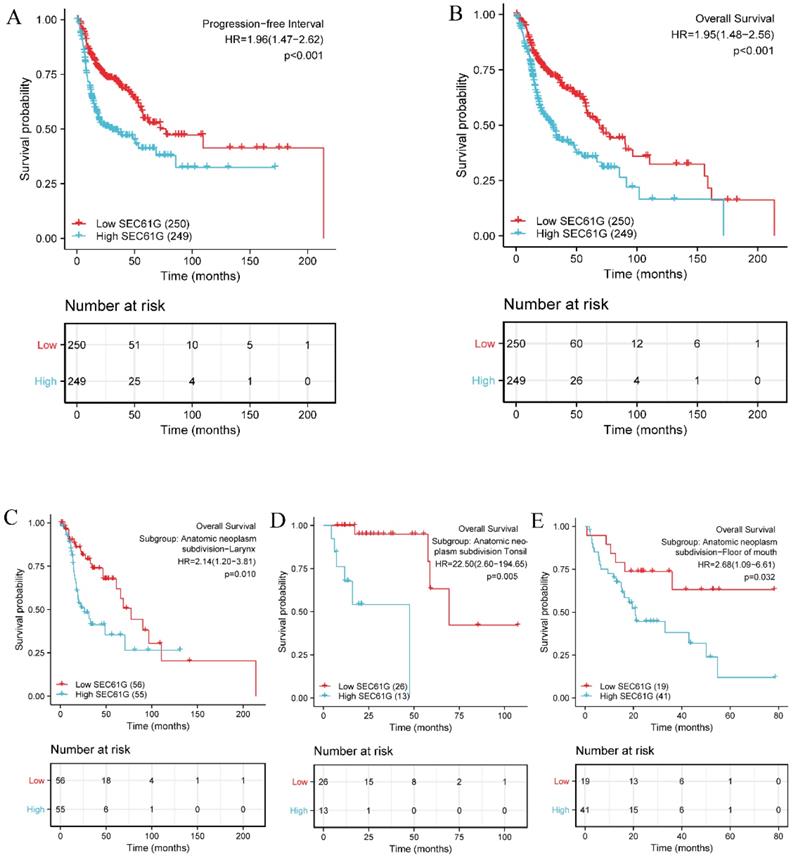
Moreover, Kaplan-Meier survival analysis showed that high SEC61G expression was more strongly associated with a poor prognosis than low SEC61G expression (Figure 4A and Figure 4B, P < 0.001). Subgroup analyses further demonstrated that SEC61G expression correlated with OS in different HNSCC anatomic sites, including the larynx (P = 0.010, Figure 4C), tonsil (P = 0.005, Figure 4D) and floor of mouth (P = 0.032, Figure 4E) cancers.
Kaplan-Meier survival curves comparing the high and low expression of SEC61G in HNSCC patients. (A) Progression-free interval. (B) Overall survival. (C-E) Overall survival for subgroup analyses in different HNSCC anatomical sites: larynx (C), tonsil (D), and floor of mouth cancer (E).
Impact of high SEC61G expression on the prognosis of HNSCC patients with different clinicopathological status
To better understand the relevance and mechanisms of SEC61G expression in HNSCC, we investigated the relationship between SEC61G expression and clinical characteristics of HNSCC patients by univariate Cox analysis. Other clinicopathologic variables that correlated with poor survival included the M stage, radiation therapy, primary therapy outcome, lymphovascular invasion, and TP53 mutation status. To further explore factors associated with survival, multivariate Cox regression analysis was performed and found that high SEC61G expression remained an independent factor associated with poor OS, along with radiation therapy, primary therapy outcome, and lymphovascular invasion (Table 1).
Association of clinicopathological characteristics with overall survival using univariate or multivariate Cox regression analysis
| Characteristic | Sample (N) | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|---|
| HR (95% CI) | P-value | HR (95% CI) | P- value** | ||
| T stage (T3 & T4 vs. T1 & T2) | 484 | 1.230 (0.921-1.642) | 0.160 | ||
| N stage (N1 & N2 & N3 vs. N0) | 477 | 1.257 (0.960-1.647) | 0.097 | 1.410 (0.864-2.303) | 0.169 |
| M stage (M1 vs. M0) | 474 | 4.794 (1.765-13.016) | 0.002 | 2.346 (0.286-19.237) | 0.427 |
| Clinical stage (Stage III & Stage IV vs. Stage I & Stage II) | 485 | 1.214 (0.875-1.683) | 0.245 | ||
| Age (> 60 vs. ≤ 60 years) | 499 | 1.238 (0.945-1.621) | 0.122 | ||
| Gender (Male vs. Female) | 499 | 0.754 (0.566-1.004) | 0.054 | 0.911 (0.565-1.469) | 0.701 |
| Histologic grade (G3 & G4 vs. G1 & G2) | 480 | 0.942 (0.690-1.287) | 0.709 | ||
| Radiation therapy (Yes vs. No) | 438 | 0.623 (0.459-0.846) | 0.002 | 0.479 (0.293-0.784) | 0.003 |
| Primary therapy outcome (CR vs. PD, SD, PR) | 415 | 0.180 (0.122-0.264) | < 0.001 | 0.270 (0.156-0.465) | < 0.001 |
| Smoker (Yes vs. No) | 489 | 1.085 (0.775-1.520) | 0.633 | ||
| Alcohol history (Yes vs. No) | 488 | 0.967 (0.727-1.288) | 0.821 | ||
| Lymphovascular invasion (Yes vs. No) | 338 | 1.688 (1.201-2.371) | 0.003 | 1.681 (1.044-2.707) | 0.032 |
| Lymph node neck dissection (Yes vs. No) | 496 | 0.728 (0.524-1.012) | 0.059 | 0.792 (0.341-1.839) | 0.587 |
| Race (White vs. Asian & Black or African American) | 483 | 0.677 (0.448-1.023) | 0.064 | 0.823 (0.426-1.589) | 0.561 |
| TP53 status (Mut vs. WT) | 494 | 1.531 (1.119-2.094) | 0.008 | 1.223 (0.742-2.014) | 0.430 |
| PIK3CA status (Mut vs. WT) | 494 | 0.988 (0.702-1.392) | 0.946 | ||
| SEC61G (High vs. Low) | 499 | 1.947 (1.482-2.559) | < 0.001 | 1.690 (1.063-2.687) | 0.027 |
HR: hazard ratio; CI: confidence interval; CR: complete response; PD: progressive disease; SD: stable disease; PR: partial response; Mut: mutation; WT: wild type.
Elevated expressions of SEC61G predict poor prognosis in different cancer stages
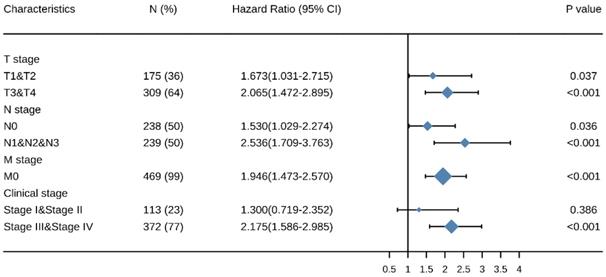
Kaplan-Meier survival analysis showed that HNSCC patients with high SEC61G expression had a worse prognosis than patients with low SEC61G expression in the different cancer stage categories: T (T1 & T2, P = 0.037, T3 & T4, P < 0.001), N (N0, P = 0.036, N1 & N2 & N3, P < 0.001), M (M0, P < 0.001), and clinical stage (III & IV, P < 0.001) (Figure 5). These results suggest that the SEC61G expression level can impact prognosis in HNSCC patients at different pathological stages.
Multivariate survival analysis of overall survival probabilities concerning SEC61G expression in patients of different subgroups according to cancer stage.
Construction and validation of a nomogram based on SEC61G expression
To provide clinicians with a quantitative approach for predicting the prognosis of HNSCC patients, a nomogram was constructed that integrated the clinical characteristics determined to be independently associated with survival via multivariate analysis (radiation therapy, primary therapy outcome, lymphovascular invasion, and SEC61G; Figure 6A). A point scale was used to assign these variables to the nomogram based on multivariate Cox analysis: a straight line was used to determine the points for the variables, and the sum of the points awarded to each variable was rescaled on a range from 0 to 100. The positions of the variables were accumulated and recorded as the total points. The probability of HNSCC patients' survival at 1, 3, and 5 years was determined by drawing vertical lines from the total point axis downward to the outcome axis. Using survival ROC package, the time-dependent ROC for SEC61G to 1-year, 3-year, and 5-year OS was analyzed (Supplementary Figure S1A), as well as that for the established nomogram prognostic model (Supplementary Figure S1B). According to the median value (-0.292), the score of the nomogram prognostic model was divided into high and low groups. Kaplan-Meier survival analysis revealed that high nomogram scores significantly correlated with a worse prognosis than low nomogram score expression (P < 0.001; Supplementary Figure S2).
Relationship between SEC61G and other clinical factors with overall survival (OS). (A) Nomogram for predicting the probability of 1-, 3-, and 5-year OS for HNSCC patients. (B) Calibration plot of the nomogram for predicting the OS likelihood.
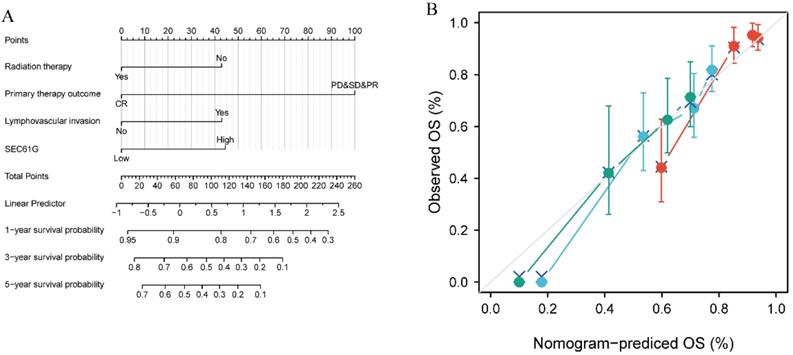
Moreover, within the nomogram, SEC61G expression was found to contribute the most extreme data points (ranging from 0 to 100) compared with the other clinical variables, which was consistent with the results of multivariate Cox regression. The C-index of the nomogram was 0.681 with 1000 bootstrap replicates (95% confidence interval: 0.658-0.703). The bias-corrected line in the calibration plot was close to the ideal curve (i.e., the 45-degree line), indicating good agreement between the predicted and observed values (Figure 6B). Overall, the nomogram was found to be a superior model for predicting long-term survival in HNSCC patients than individual prognostic factors.
Identification of DEGs between the high and low SEC61G expression groups
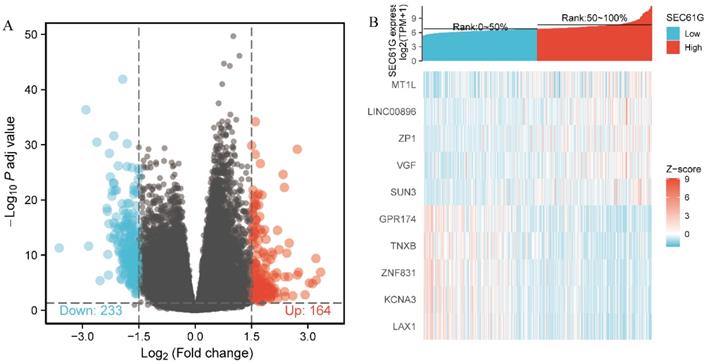
The data from TCGA were analyzed using the DSEeq2 package in R (|logFC|>1.5, adjusted P-value < 0.05) and 397 DEGs were identified between the groups with high and low SEC61G expression, including 164 upregulated and 233 downregulated genes in the high expression group (Figure 7).
Differentially expressed genes between patients with high and low SEC61G expression. (A) Volcano plot of differentially expressed genes between the high and low SEC61G expression groups. Normalized expression levels are shown in descending order from green to red. (B) Heatmap of the top ten significant differentially expressed genes between the high and low SEC61G expression groups. Green and red dots represent downregulated and upregulated genes, respectively.
Functional annotation and predicted signaling pathways
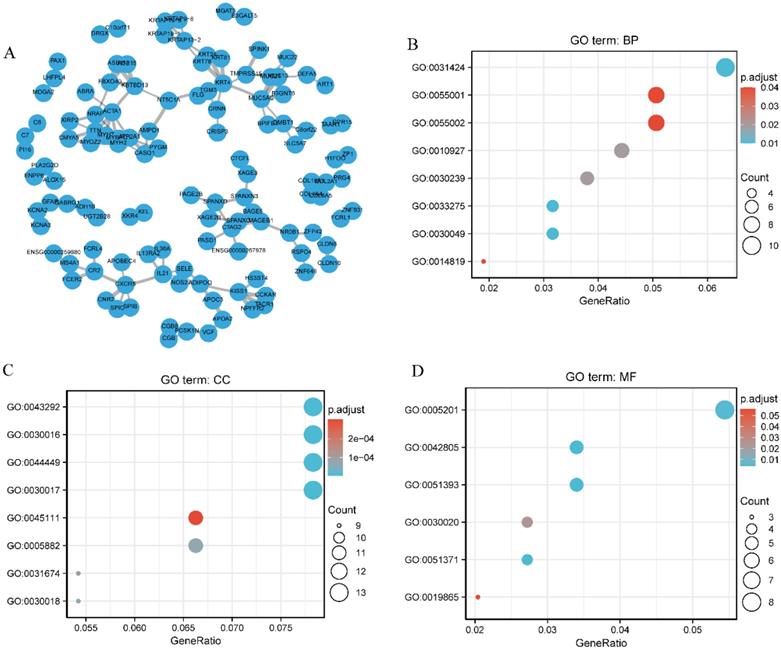
A network of SEC61G and its potential co-expressed genes in SEC61G are shown in Figure 8A. To better understand the functional implication of SEC61G in HNSCC from the 397 DEGs identified between the high and low expression groups, GO enrichment analysis was performed using the ClusterProfile package. Sixteen enriched terms were identified in the GO “biological process” category, including keratinization, muscle cell development, and cellular component assembly involved in morphogenesis (Figure 8B). These results suggest a link between the aberrant expression of SEC61G and keratinization. Meanwhile, 29 GO terms within the “cellular component” category were associated with contractile fibers (Figure 8C). Furthermore, the “molecular function” category revealed significant enrichment in GO terms related to the extracellular matrix structural constituents (Figure 8D).
Protein-protein interaction network and functional enrichment analysis. (A) Protein-protein interaction network of SEC61G and its co-expressed genes. (B) Enriched GO terms in the “biological process” category. (C) Enriched GO terms in the “cellular component” category. (D) Enriched GO terms in the “molecular function” category. The x-axis represents the proportion of differentially expressed genes (DEGs) and the y-axis represents different categories. Blue and red tones represent adjusted P values from 0.0 to 0.05, respectively, and different circle sizes represent the number of DEGs.
SEC61G-related signaling pathways based on GSEA
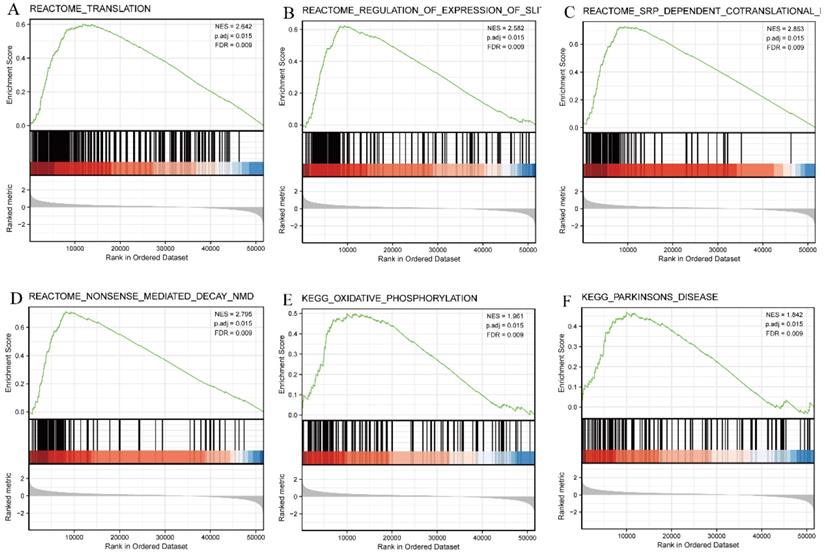
GSEA was then used to identify signaling pathways associated with HNSCC between the high and low SEC61G expression groups, based on significant differences (adjusted P-value < 0.05, FDR < 0.25) in the enrichment of MSigDB Collection (c2.all.v7.0.symbols.gmt [Curated]). Six pathways, including translation, regulation of expression of the SLITs/ROBOs pathway, SRP-dependent co-translational protein targeting the membrane, nonsense-mediated decay pathway, oxidative phosphorylation, and Parkinson's disease, were identified as significantly different between the two groups (Figure 9).
Enrichment plots from GSEA. Several pathways were differentially enriched in HNSCC patients according to high and low SEC61G expression. (A) Translation. (B) Regulation of the SLITs/ROBOs pathway expression. (C) SRP-dependent co-translational protein targeting the membrane. (D) Nonsense-mediated decay pathway. (E) Oxidative phosphorylation. (F) Parkinson disease. ES, enrichment score; NES, normalized enrichment score; ADJ p-Val, adjusted P-value; FDR, false discovery rate.
Correlation between SEC61G expression and immune infiltration
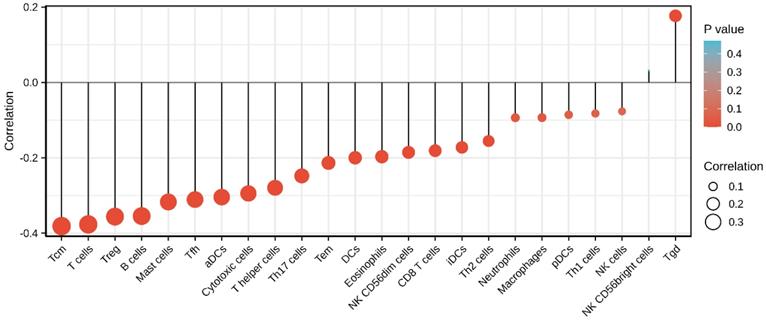
Finally, we analyzed the correlation between the expression level (TPM) of SEC61G and immune cell enrichment (generated by ssGSEA) based on the Spearman correlation coefficient. SEC61G expression was negatively correlated with the abundance of Tcm, T cells, Tregs, B cells, mast cells, Tfh, cytotoxic cells, Th cells, Th17 cells, Tem, DCs, eosinophils, NK CD56dim, CD8 T cells, and iDCs, and was positively correlated with the abundance of NK CD56bright cells and Tgd. (Figure 10).
Correlations between the relative abundance of 24 immune cells and SEC61G expression levels. The size of the dots represents the absolute Spearman's correlation coefficient values.
Discussion
An effective prognostic biomarker provides important information regarding cancer aggressiveness and/or the clinical outcome of a specific patient in the absence of treatment. However, they are also crucial components in personalized medicine and precision medicine, as they can prevent undertreatment or overtreatment. Previous studies have also revealed that SEC61G participates in the regulation of multiple disease states including diabetes, neurodegeneration, and cancer [7]. More recently, SEC61G was found to be overexpressed in gastric cancer, breast cancer, and glioblastoma [10-12]. However, little is known regarding its prognostic value in HNSCC. Our results consistently demonstrate that SEC61G expression serves as a robust predictor of HNSCC clinical outcome.
In the current study, we performed a bioinformatics analysis to assess the prognostic value of SEC61G in HNSCC using high-throughput RNA-seq data obtained from TCGA. We observed that SEC61G was more highly expressed in tumor tissues compared to normal samples. Moreover, overexpression of SEC61G in cancer tissues correlated with poor clinicopathologic factors, suggesting that SEC61G functioned as an oncogene. Previous in vitro functional assays revealed that SEC61G knockdown inhibited non-small-cell lung cancer cell proliferation and invasion while promoted apoptosis [29]. In addition, compared with low SEC61G expression, high expression was significantly correlated with a poor OS in HNSCC patients. Similarly, Liu et al. reported that SEC61G expression may represent a potential prognostic marker for poor survival in glioblastoma patients [9]. Hence, we conjectured that SEC61G may also serve as a biomarker for HNSCC.
To further investigate the biological function of SEC61G in HNSCC, we used TCGA data for GSEA and found that genes associated with regulation of the SLITs/ROBOs pathway were differentially enriched in the SEC61G high expression HNSCC phenotype. SLITs interact with ROBOs, the complexes of which subsequently play a considerable role in muscle cell formation, angiogenesis, cell migration, stem cell growth, organ development, and tumor formation by recruiting different adaptor molecules or proteins to activate a cascade of signaling pathways [30, 31]. However, little is known how accurately SLIT binding to ROBO is communicated across the cell membrane [32]. Meanwhile, increasing evidence suggests that SLITs can also bind to extracellular matrix molecules [33, 34]. The mobile combinations of SLITs and ROBOs depend on different environments. Notably, P-cadherin was shown to be involved in regulating cell-cell adhesion by combining with ROBO3 in oral squamous cell carcinoma [35]. In addition to weakening cell-cell adhesion, the assembly of cytoskeletal actin and dissolution of the extracellular matrix can be affected by SLIT/ROBO signaling to regulate cancer cell metastasis [36, 37]. Overexpression of SLITs/ROBOs has also been observed in melanoma [38], gastric cancer [39], pancreatic cancer tissues and cell lines [40], as well as hepatocellular carcinoma [41], thereby demonstrating that SLITs/ROBOs signaling has a facilitating effect in certain cancers. It is, therefore, reasonable to conclude that blockade of SLITs/ROBOs signaling may potently inhibit tumor angiogenesis and effectively prevent malignant transformation for suppression of tumor growth and metastasis, as observed for colorectal carcinoma [42]. However, studies have also reported that the expression of SLIT is downregulated, or not detected, in most tumors, including breast cancer [43], gastric cancer [44], lung cancer [45], liver cancer [46], esophageal cancer [47], among others, and is largely related to promoter hypermethylation, demonstrating the inhibitory effect of SLITs/ROBOs in these cancers [43-47]. Meanwhile, in the current study, we found that SEC61G may exhibit crosstalk with pathways regulating the expression of SLITs/ROBOs. However, it is unclear whether SEC61G and regulation of the SLITs/ROBOs pathway have a synergistic effect or a complementary effect, and the detailed regulation network has not been reported. Moreover, our protein-protein interaction network revealed that SEC61G participates in sophisticated crosstalk with numerous other genes. Thus, further investigations are warranted to elucidate the detailed molecular mechanisms underlying these interactions. In addition, SEC61G expression is regulated by the long non-coding RNA LINC02418 [29], suggesting that SEC61G may be involved in a more complex regulatory network.
The tumor microenvironment is regarded as a crucial interface mediating physiological reactions in cancer cells. Tumor infiltrating immune cells account for an indispensable component of the tumor microenvironment with their composition and distribution considered to be related to cancer prognosis. Given the crucial role of the tumor microenvironment in mediating cancer progression and tumor-infiltrating immune cells account for an indispensable component of the tumor microenvironment [48, 49], we sought to investigate the relationship between SEC61G and immune infiltration in HNSCC. As shown in figure 10, it shows that increased SEC61G expression was negatively correlated with the abundances of B cells, CD8+ T cells, and Tregs in HNSCC. Previous studies have revealed that low levels of infiltrating B cells and cytotoxic CD8+ T in tumor tissues were associated with poor prognosis of cancer patients including HNSCC [50-54]. On the contrary, several other studies have reported that high levels of tumor-infiltrating Tregs were significantly associated with worse outcome in breast cancer [55], hepatocellular carcinoma [56], pancreatic ductal adenocarcinoma [57], lung cancer [58], gastric cancer [59], and ovarian cancer [60]. But interestingly, in the context of HNSCC, patients with elevated Treg levels reportedly have a significantly better OS [61]. We, therefore, inferred that SEC61G might affect the prognosis of patients by modulating immune infiltration in HNSCC.
Although our approaches can provide new insights into the correlation between SEC61G and HNSCC, certain limitations were noted in this study. First, only one dataset was assessed, which may cause sample bias. Second, to increase the credibility of the results, the sample size should be further expanded. Third, to improve the clinical application, additional clinical factors should be included. Fourth, further experimental verification is required to elucidate the biological functions of SEC61G in vitro and in vivo.
In summary, our study revealed the prognostic value of SEC61G in HNSCC for the first time. Our findings strongly suggest that SEC61G offers potential as a biomarker to predict the treatment outcome and prognosis in HNSCC patients. Further experimental validation is warranted, however, to elucidate the biological impact and underlying mechanism of SEC61G.
Abbreviations
aDCs: activated dendritic cells; AUC: area under the curve; C-index: concordance index; DCs: dendritic cells; DEG: differentially expressed gene; iDCs: immature dendritic cells; FC: fold change; FDR: false discovery rate; GO: gene ontology; GSEA: gene set enrichment analysis; HNSCC: head and neck squamous cell carcinomas; NK cell: natural killer cell; OS: overall survival; pDCs: plasmacytoid dendritic cells; RNA-seq: RNA-sequencing; ROC: receiver operating characteristic; ssGSEA: single-sample gene set enrichment analysis; TCGA: The Cancer Genome Atlas; Tcm: central memory T cells; Tems: effector memory T cells; Tgd: gamma delta T cells; Th1: type-1 T helper cells; Th2: type-2 T helper cells; Th17: type-17 T helper cells; TPM: transcripts per million; Tregs: regulatory T cells.
Supplementary Material
Supplementary figures.
Acknowledgements
We acknowledge the support received from Mr. Lao, who is the founder of helixlife. cn.
Funding
This work was supported by the Cultivation Project of “139” Program for Medicine High-level Key Talents of Guangxi and the National Natural Science Foundation of China (No. 82060019).
Ethics approval
This research had been approved by the Institutional Review Boards of the Sixth Affiliated Hospital of Guangxi Medical University. This project was in accordance with the principles of the Helsinki Declaration.
Author contributions
Leifeng Liang and Qingwen Huang organized and wrote the manuscript. Leifeng Liang designed and produced the figures. Mei Gan and Liujun Jiang contributed to the literature search for the manuscript. Haolin Yan, Zhan Lin, Haisheng Zhu, Rensheng Wang, and Kai Hu revised the manuscript. All authors reviewed the manuscript and approved the manuscript for publication.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA: a cancer journal for clinicians. 2019;69:7-34
2. Zhang W, Han Y, Li W, Cao L, Yan L, Qin C. et al. Clinical data analysis reveals the role of OGR1 (GPR68) in head and neck squamous cancer. Animal models and experimental medicine. 2020;3:55-61
3. Yu B, Zhang Y, Wu K, Wang L, Jiang Y, Chen W. et al. CD147 promotes progression of head and neck squamous cell carcinoma via NF-kappa B signaling. Journal of cellular and molecular medicine. 2019;23:954-66
4. Economopoulou P, de Bree R, Kotsantis I, Psyrri A. Diagnostic Tumor Markers in Head and Neck Squamous Cell Carcinoma (HNSCC) in the Clinical Setting. Front Oncol. 2019;9:827
5. Hartmann E, Sommer T, Prehn S, Görlich D, Jentsch S, Rapoport TA. Evolutionary conservation of components of the protein translocation complex. Nature. 1994;367:654-7
6. Greenfield JJ, High S. The Sec61 complex is located in both the ER and the ER-Golgi intermediate compartment. J Cell Sci. 1999;112( Pt 10):1477-86
7. Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annual review of pathology. 2015;10:173-94
8. Liu Y, Ji W, Shergalis A, Xu J, Delaney AM, Calcaterra A. et al. Activation of the Unfolded Protein Response via Inhibition of Protein Disulfide Isomerase Decreases the Capacity for DNA Repair to Sensitize Glioblastoma to Radiotherapy. Cancer Res. 2019;79:2923-32
9. Liu B, Liu J, Liao Y, Jin C, Zhang Z, Zhao J. et al. Identification of SEC61G as a Novel Prognostic Marker for Predicting Survival and Response to Therapies in Patients with Glioblastoma. Medical science monitor: international medical journal of experimental and clinical research. 2019;25:3624-35
10. Tsukamoto Y, Uchida T, Karnan S, Noguchi T, Nguyen LT, Tanigawa M. et al. Genome-wide analysis of DNA copy number alterations and gene expression in gastric cancer. The Journal of pathology. 2008;216:471-82
11. Reis-Filho JS, Pinheiro C, Lambros MBK, Milanezi F, Carvalho S, Savage K. et al. EGFR amplification and lack of activating mutations in metaplastic breast carcinomas. The Journal of pathology. 2006;209:445-53
12. Lu Z, Zhou L, Killela P, Rasheed AB, Di C, Poe WE. et al. Glioblastoma proto-oncogene SEC61gamma is required for tumor cell survival and response to endoplasmic reticulum stress. Cancer Res. 2009;69:9105-11
13. Li Q, Wu J, Wei P, Xu Y, Zhuo C, Wang Y. et al. Overexpression of forkhead Box C2 promotes tumor metastasis and indicates poor prognosis in colon cancer via regulating epithelial-mesenchymal transition. Am J Cancer Res. 2015;5:2022-34
14. Xu Q, Chang H, Tian X, Lou C, Ma H, Yang X. Hypoxia-induced MFAP5 Promotes Tumor Migration and Invasion via AKT Pathway in Head and Neck Squamous Cell Carcinoma. Journal of Cancer. 2020;11:1596-605
15. Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607-D13
16. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550
17. Dalman MR, Deeter A, Nimishakavi G, Duan Z-H. Fold change and p-value cutoffs significantly alter microarray interpretations. BMC Bioinformatics. 2012;13(Suppl 2):S11
18. Harding T, Swanson J, Van Ness B. EZH2 inhibitors sensitize myeloma cell lines to panobinostat resulting in unique combinatorial transcriptomic changes. Oncotarget. 2018;9:21930-42
19. Martin B, Gabris-Weber BA, Reddy R, Romero G, Chattopadhyay A, Salama G. Relaxin reverses inflammatory and immune signals in aged hearts. PLoS One. 2018;13:e0190935
20. Mathers TC, Mugford ST, Percival-Alwyn L, Chen Y, Kaithakottil G, Swarbreck D. et al. Sex-specific changes in the aphid DNA methylation landscape. Mol Ecol. 2019;28:4228-41
21. Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284-7
22. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545-50
23. Min JL, Barrett A, Watts T, Pettersson FH, Lockstone HE, Lindgren CM. et al. Variability of gene expression profiles in human blood and lymphoblastoid cell lines. BMC genomics. 2010;11:96
24. van der Pouw Kraan TCTM, Wijbrandts CA, van Baarsen LGM, Voskuyl AE, Rustenburg F, Baggen JM. et al. Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: assignment of a type I interferon signature in a subpopulation of patients. Ann Rheum Dis. 2007;66:1008-14
25. Dinu I, Potter JD, Mueller T, Liu Q, Adewale AJ, Jhangri GS. et al. Improving gene set analysis of microarray data by SAM-GS. BMC Bioinformatics. 2007;8:242
26. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC. et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782-95
27. Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C. et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77
28. Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD. et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell. 2018;173:400-16.e11
29. Han B. LncRNA LINC02418 regulates proliferation and apoptosis of non-small cell lung cancer cells by regulating miR-4677-3p/SEC61G. European review for medical and pharmacological sciences. 2019;23:10354-62
30. Zhang B, Dietrich UM, Geng J-G, Bicknell R, Esko JD, Wang L. Repulsive axon guidance molecule Slit3 is a novel angiogenic factor. Blood. 2009;114:4300-9
31. Yang X-M, Han H-X, Sui F, Dai Y-M, Chen M, Geng J-G. Slit-Robo signaling mediates lymphangiogenesis and promotes tumor lymphatic metastasis. Biochem Biophys Res Commun. 2010;396:571-7
32. Tong M, Jun T, Nie Y, Hao J, Fan D. The Role of the Slit/Robo Signaling Pathway. Journal of Cancer. 2019;10:2694-705
33. Xiao T, Staub W, Robles E, Gosse NJ, Cole GJ, Baier H. Assembly of lamina-specific neuronal connections by slit bound to type IV collagen. Cell. 2011;146:164-76
34. Wright KM, Lyon KA, Leung H, Leahy DJ, Ma L, Ginty DD. Dystroglycan organizes axon guidance cue localization and axonal pathfinding. Neuron. 2012;76:931-44
35. Bauer K, Dowejko A, Bosserhoff AK, Reichert TE, Bauer R. Slit-2 facilitates interaction of P-cadherin with Robo-3 and inhibits cell migration in an oral squamous cell carcinoma cell line. Carcinogenesis. 2011;32:935-43
36. Parray A, Siddique HR, Kuriger JK, Mishra SK, Rhim JS, Nelson HH. et al. ROBO1, a tumor suppressor and critical molecular barrier for localized tumor cells to acquire invasive phenotype: study in African-American and Caucasian prostate cancer models. Int J Cancer. 2014;135:2493-506
37. Tseng R-C, Chang J-M, Chen J-H, Huang W-R, Tang Y-A, Kuo IY. et al. Deregulation of SLIT2-mediated Cdc42 activity is associated with esophageal cancer metastasis and poor prognosis. J Thorac Oncol. 2015;10:189-98
38. Wang B, Xiao Y, Ding BB, Zhang N, Yuan Xb, Gui L. et al. Induction of tumor angiogenesis by Slit-Robo signaling and inhibition of cancer growth by blocking Robo activity. Cancer Cell. 2003;4:19-29
39. Shi R, Liu W, Liu B, Xu Z, Chen L, Zhang Z. Slit2 expression and its correlation with subcellular localization of β-catenin in gastric cancer. Oncol Rep. 2013;30:1883-9
40. Han S, Cao C, Tang T, Lu C, Xu J, Wang S. et al. ROBO3 promotes growth and metastasis of pancreatic carcinoma. Cancer Lett. 2015;366:61-70
41. Ao J-Y, Chai Z-T, Zhang Y-Y, Zhu X-D, Kong L-Q, Zhang N. et al. Robo1 promotes angiogenesis in hepatocellular carcinoma through the Rho family of guanosine triphosphatases' signaling pathway. Tumour Biol. 2015;36:8413-24
42. Zhou W-J, Geng ZH, Chi S, Zhang W, Niu X-F, Lan S-J. et al. Slit-Robo signaling induces malignant transformation through Hakai-mediated E-cadherin degradation during colorectal epithelial cell carcinogenesis. Cell Res. 2011;21:609-26
43. Sharma G, Mirza S, Prasad CP, Srivastava A, Gupta SD, Ralhan R. Promoter hypermethylation of p16INK4A, p14ARF, CyclinD2 and Slit2 in serum and tumor DNA from breast cancer patients. Life Sci. 2007;80:1873-81
44. Kim M, Kim J-H, Baek S-J, Kim S-Y, Kim YS. Specific expression and methylation of SLIT1, SLIT2, SLIT3, and miR-218 in gastric cancer subtypes. Int J Oncol. 2016;48:2497-507
45. Tseng R-C, Lee S-H, Hsu H-S, Chen B-H, Tsai W-C, Tzao C. et al. SLIT2 attenuation during lung cancer progression deregulates beta-catenin and E-cadherin and associates with poor prognosis. Cancer Res. 2010;70:543-51
46. Jin J, You H, Yu B, Deng Y, Tang N, Yao G. et al. Epigenetic inactivation of SLIT2 in human hepatocellular carcinomas. Biochem Biophys Res Commun. 2009;379:86-91
47. Dickinson RE, Dallol A, Bieche I, Krex D, Morton D, Maher ER. et al. Epigenetic inactivation of SLIT3 and SLIT1 genes in human cancers. Br J Cancer. 2004;91:2071-8
48. Nguyen N, Bellile E, Thomas D, McHugh J, Rozek L, Virani S. et al. Tumor infiltrating lymphocytes and survival in patients with head and neck squamous cell carcinoma. Head & neck. 2016;38:1074-84
49. Wolf GT, Chepeha DB, Bellile E, Nguyen A, Thomas D, McHugh J. Tumor infiltrating lymphocytes (TIL) and prognosis in oral cavity squamous carcinoma: a preliminary study. Oral oncology. 2015;51:90-5
50. Sandoval F, Terme M, Nizard M, Badoual C, Bureau M-F, Freyburger L. et al. Mucosal imprinting of vaccine-induced CD8⁺ T cells is crucial to inhibit the growth of mucosal tumors. Sci Transl Med. 2013;5:172ra20
51. Chen X, Yan B, Lou H, Shen Z, Tong F, Zhai A. et al. Immunological network analysis in HPV associated head and neck squamous cancer and implications for disease prognosis. Molecular immunology. 2018;96:28-36
52. Cao B, Wang Q, Zhang H, Zhu G, Lang J. Two immune-enhanced molecular subtypes differ in inflammation, checkpoint signaling and outcome of advanced head and neck squamous cell carcinoma. Oncoimmunology. 2018;7:e1392427
53. Fousek K, Ahmed N. The Evolution of T-cell Therapies for Solid Malignancies. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:3384-92
54. Li L, Wang X-L, Lei Q, Sun C-Z, Xi Y, Chen R. et al. Comprehensive immunogenomic landscape analysis of prognosis-related genes in head and neck cancer. Scientific reports. 2020;10:6395
55. Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL. et al. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24:5373-80
56. Wang Y, Liu T, Tang W, Deng B, Chen Y, Zhu J. et al. Hepatocellular Carcinoma Cells Induce Regulatory T Cells and Lead to Poor Prognosis via Production of Transforming Growth Factor-β1. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2016;38:306-18
57. Tang Y, Xu X, Guo S, Zhang C, Tang Y, Tian Y. et al. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PloS one. 2014;9:e91551
58. Petersen RP, Campa MJ, Sperlazza J, Conlon D, Joshi M-B, Harpole DH. et al. Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer. 2006;107:2866-72
59. Hu M, Li K, Maskey N, Xu Z, Peng C, Wang B. et al. Decreased intratumoral Foxp3 Tregs and increased dendritic cell density by neoadjuvant chemotherapy associated with favorable prognosis in advanced gastric cancer. International journal of clinical and experimental pathology. 2014;7:4685-94
60. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P. et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942-9
61. Lukesova E, Boucek J, Rotnaglova E, Salakova M, Koslabova E, Grega M. et al. High level of Tregs is a positive prognostic marker in patients with HPV-positive oral and oropharyngeal squamous cell carcinomas. BioMed research international. 2014;2014:303929
Author contact
![]() Corresponding authors: Kai Hu, M.D and Prof. Rensheng Wang. Department of Radiation Oncology, The First Affiliated Hospital of Guangxi Medical University, 6 Shuangyong Road, Nanning 530021, Guangxi, China; E-mail: hukaigxmu.edu.cn (KH); 13807806008com (RW).
Corresponding authors: Kai Hu, M.D and Prof. Rensheng Wang. Department of Radiation Oncology, The First Affiliated Hospital of Guangxi Medical University, 6 Shuangyong Road, Nanning 530021, Guangxi, China; E-mail: hukaigxmu.edu.cn (KH); 13807806008com (RW).