Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(14):4307-4321. doi:10.7150/jca.54637 This issue Cite
Review
Impact of cholesterol-pathways on breast cancer development, a metabolic landscape
Alina González-Ortiz#, Octavio Galindo-Hernández, Gerson N. Hernández-Acevedo, Gustavo Hurtado-Ureta, Victor García-González ![]()
Departamento de Bioquímica, Facultad de Medicina Mexicali, Universidad Autónoma de Baja California, 21000 Mexicali, México.
#Medical degree student.
Received 2020-10-17; Accepted 2021-2-4; Published 2021-5-19
Abstract

ApoB-lipoproteins and their components modulate intracellular metabolism and have been associated with the development of neoplastic phenomena, such as proliferation, anchorage-independent growth, epithelial-mesenchymal transition, and cancer invasion. In cancer cells, the modulation of targets that regulate cholesterol metabolism, such as synthesis de novo, endocytosis, and oxidation, are contributing factors to cancer development. While mechanisms associated with sterol regulatory element-binding protein 2 (SREBP-2)/mevalonate, the low-density lipoprotein receptor (LDL-R) and liver X receptor (LXR) have been linked with tumor growth; metabolites derived from cholesterol-oxidation, such as oxysterols and epoxy-cholesterols, also have been described as tumor processes-inducers. From this notion, we perform an analysis of the role of lipoproteins, their association with intracellular cholesterol metabolism, and the impact of these conditions on breast cancer development, mechanisms that can be shared during atherogenesis promoted mainly by LDL. Pathways connecting plasma dyslipidemias in conjunction with the effect of cholesterol-derived metabolites on intracellular mechanisms and cellular plasticity phenomena could provide new approaches to elucidate the triggering factors of carcinogenesis, conditions that could be considered in the development of new therapeutic approaches.
Keywords: breast cancer, plasmatic lipoproteins, SREBP-2/mevalonate pathway, oxysterols, epoxy-cholesterols
Introduction
Metabolic syndrome (MetS) is a pathological condition characterized by the presence of least three of five of the following medical conditions: abdominal obesity, elevated blood pressure, elevated fasting plasma glucose, high serum triglycerides, and low High-density lipoprotein (HDL) concentrations, therefore a dyshomeostasis of plasmatic lipids is present (Figure 1) [1-6]. Specifically, abdominal obesity could be associated with insulin resistance and the development of dyslipidemias such as high LDL (low-density lipoprotein), low HDL concentrations, hyperglycemia and, consequently type 2 diabetes (T2D). Although its role in breast cancer (BC) is not fully elucidated, evidence supports that MetS could increase the risk of BC, and once diagnosed, impacts the prognosis of the disease. In a pioneer study, the Apolipoprotein Mortality Risk (AMORIS) comprised 812,073 Swedish individuals, compared lipid profiles from BC patients, measured by 8 years previous to diagnosis, results showed weak evidence associated with impaired triglycerides concentrations [7]. Likewise, in a report of 208 BC patients versus a control group of 176 healthy women, patients with lymph node metastasis showed plasmatic altered metabolites such as glucose, triglycerides, elevated LDL levels, as well as low HDL [8]. However, the relationship between plasmatic dyslipidemias and intracellular lipid metabolism, and their implications on BC progression is not fully clear. Nowadays, there is increasing evidence of the role of lipid metabolism alterations as biomarkers of cancer prognosis and survival [9]. Changes of expression of lipid-metabolism genes are found in several tumors, considering lipid metabolism modulates cellular processes range from plasmatic and organelle membrane organization and plasticity [10, 11], ATP production [12], to intracellular signaling alterations [13, 14].
Conditions of metabolic syndrome (MetS) could generate a favorable microenvironment for breast cancer (BC) development. A) Schematic representations of MetS conditions. B) BC cells undergo rearrangements in the pathways of cholesterol metabolism, and several cholesterol derived-metabolites have been described as promoters of cellular growth and proliferation. C) Targets that control cholesterol metabolism could promote BC cell migration and invasion through epithelial-mesenchymal transitions (EMT). Adapted from: [21, 24, 25, 32, 48, 49].
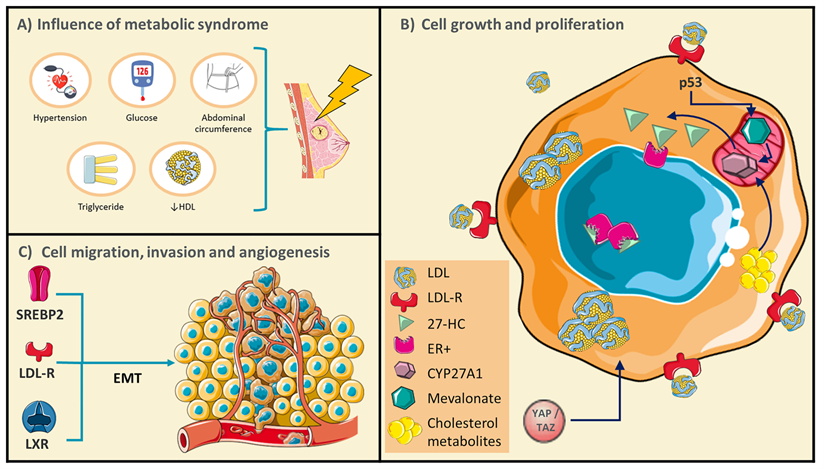
Cancer growth and proliferation have been associated with oncogene activation or loss of tumor suppressor function; nevertheless, alteration of metabolic pathways would play critical roles, enabling cancer cells to acquire and synthesize nutrients, then counteract nutrient deficit. Moreover, obesity and dyslipidemias have been considered triggering conditions of neoplastic pathology, contributing to the increase of mortality rate [15]. Interestingly, BC-cells express high levels of enzymes involved in De novo cholesterol synthesis and the treatment with statins could inhibit their proliferation in secondary tumors [16]. These studies are not conclusive since evidence indicates that the underlying mechanisms are more complex. For instance, in the Nurses' Health Study (NHS) cohort (N=79,518), postmenopausal participants without a cancer history were followed for 12 years, statin treatment was not associated with risk of invasive BC, independent of histologic subtype [17]. Indeed, in a case-control study of long-term, statin treatment was associated with increased risk of both 916 invasive ductal carcinomas (IDC) and 1,068 invasive lobular carcinomas (ILC); nonetheless of those tumor characteristics, only 6% of patients were treated with statins for more 10 years showing odds ratio (OR) of 1.83 and 1.97, respectively. The authors did not consider the association among the use of long-term statin, and BC risk diverges by estrogen receptor (ER) status, given the relatively small number of cases [18]. Furthermore, an association has been proposed among the increase in the time of BC recurrence and the early administration of lipophilic statins as a cholesterol-LDL lowering therapy [19]. Therefore, evaluation of this condition in other populations for a longer period is required. Even more, the metabolic reprogramming of tumors could be controlled by multiple mechanisms such as epigenetic factors that regulate gene expression and, critically the role of cholesterol-derived metabolites, phenomena analyzed in the following chapters.
Breast cancer and MetS association
High levels of circulating and local estrogens, altered adipokine-concentrations, disrupted insulin signaling, modifications within the microbiome, and local and systemic effects of inflammation could contribute to the obesity and cancer association [20]. Several reports suggested an increased risk of recurrence in obese versus normal weight women with BC diagnosed [21]. Obesity could be the critical pathology which allows to explain the relationship among chronic low-grade inflammatory condition and the metabolism alterations during MetS [6, 9]. For instance, in early stages, adipocyte hyperplasia has been shown to generate a low-oxygen environment, leading to the expression of inducible Hypoxia factor 1 alpha (HIF1α), resulting in processes such as infiltration of both tumor-associated neutrophils (TAN) and tumor associated macrophages (TAMs), development of epithelial-mesenchymal transitions (EMTs), as well as critically modifications in lipid metabolism and inflammation [9, 15, 20]. Hypoxia and the increased nutrient-demand promote the death of adipocytes surrounded by macrophages generating Crown-like structures, these conditions lead to the release of free fatty acids (FFAs), activating pro-inflammatory pathways through Toll-like receptor 4.
Obesity is closely linked with the presence of metabolic disorders. Excess fat triggers extensive remodeling of the adipose tissue microenvironment in terms of its size, vascularity, and cellular and matrix composition; which is accompanied by deregulated secretion of adipose cytokines or so-called adipokines. In this sense, the increase in pro-inflammatory adipokines such as TNF-α, interleukin-6 (IL-6), IL-1, IL-8 and resistin has been associated with insulin resistance and potentially T2D [22]. In turn, this condition denominated meta-inflammation could be associated with the development of BC [23]. The relationship between obesity and insulin concentrations has been documented, as a feature of the pathophysiology of T2D.
Although, a direct connection with cancer development is not conclusive; the presence of obesity before diagnosis, insulin resistance during diagnosis, and hyperinsulinemia after treatment have been associated with an increased risk of mortality and recurrence of BC [24, 25]. Data of 94,555 women free of cancer at baseline in the NIH-American Association of Retired Persons Diet and Health Study cohort (NIH-AARP), suggested individual MetS components, such as waist circumference, high cholesterol, and hypertension, were independently associated with an increased risk of BC mortality [5]. In another report, components of MetS mainly obesity were associated with an increased risk of BC-recurrence [4]. At this respect, in a retrospective cohort study, involving 4,216 women between the period of 1990 and 2008, 26 % of women had three components of MetS, and compared with MetS components-free women, the presence of ≥3 MetS components was associated with a modest risk of BC-deaths (Figure 1) [4]. Likewise, in the analysis of 116 datasets of 43 articles included 38,940 cases of several cancers, 9,643 BC cases were identified; in 5,161 women the presence of MetS was associated with breast postmenopausal cancer [26]. Even more, evidence suggests that the risk associated with various components of MetS may be synergistic, considering that the biological mechanisms that connect MetS components effects and breast cancer risk could share signaling pathways [27].
Nevertheless, among 8,641 women with local or regional invasive BC, waist circumference was associated with a higher risk of cardiovascular disease, although a direct association between BC and the risk of cardiovascular disease was not found [28]. In another report, obesity was associated with an increased risk of BC-recurrence [4]. For instance, MetS is 1.5 times more prevalent in Hispanic individuals compared with non-Hispanic whites or blacks [29]. Hispanic BC-survivors possibly show less favorable metabolic profiles compared with non-Hispanic, and activities such as clinical practice interventions may attenuate these disappointments [29]. Even more, in an observational study involving 86 women (included 46 premenopausal and 40 postmenopausal) with early-stage BC and diagnosed MetS-free, after completing (neo) adjuvant chemotherapy (12-week to 18-week), all MetS components and the overall MetS score were significantly increased after treatment [30]. Therefore, the correlation between the presence of MetS components and the risk of BC is not conclusive, consequently, evaluation of conditions such as sample size, population and the status of patients should be considered.
Role of serum lipoproteins
Alterations in cholesterol pathways such as de novo synthesis, cell internalization, and cholesterol transport mediated by plasmatic lipoproteins could regulate several mechanisms in the development of BC [31]. Very low density lipoproteins (VLDL) and LDL could be critical in several mechanisms of tumor progression [32]. For instance, the treatment of cancer cells with VLDL and LDL induces pro-tumoral processes such as proliferation, anchorage independent growth, development of EMTs, angiogenesis, and metastasis. Conversely, HDL inhibits mammosphere-formation and increases the sensitivity to radiation in mammary tumoral cells [33]. Therefore, the lipid and protein content of plasmatic lipoprotein, and alterations on intracellular cholesterol pathways in breast tissues could perform important features on phenomena associated with BC-development.
Protective mechanisms of HDL
In epidemiological studies, broad experimental evidence has shown low-HDL concentrations are connected with an increased risk of coronary artery disease [34]. The cardioprotective effect of HDL and its main protein component, apo-A1 are related with the reverse transport of cholesterol from the artery wall to the liver [35, 36], a condition which has situated HDLs for a long time as a promissory target for cardiovascular pathologies treatment [37]. In an important way, evidence suggests high levels of HDL are associated with a low risk of BC. In African-American women, an elevation of BC risk was associated with low HDL levels [38]. Likewise, in Taiwanese women low HDL concentrations were related with a three-fold increase in the risk of BC [39]. A further study compared lipid profiles of 208 BC-patients newly diagnosed without treatment, showing lower HDL levels in all stages with a diminution associated to advanced stages, whereas LDL levels were maintained elevated in every stage of BC [8].
Revision of 7,575 females during 13 years in Atherosclerosis Risk in Communities Cohort Study, suggested a positive association among low HDL in premenopausal women and an increased risk of BC [40]. In a meta-analysis of 26 studies, involucrated 24,655 individuals with cancer, patients with higher HDL had a 37% reduced risk of death compared to patients with low HDL levels [41]. In another report, in women newly diagnosed with invasive BC stage I/II, an inverse association was reported between cholesterol-HDL content, phospholipids, apolipoprotein-A1, and Ki67, a canonical proliferative marker [42]. Likewise, in a multicenter case-control study (N=690/1380), higher HDL levels-associated with reduced risk of premenopausal BC were reported, specifically HDL concentrations > 60 mg/dL decreased BC risk by 0.49-fold compared to HDL < 50 mg/dL [43]. In the characterization of the lipoprotein subfractions in BC patients (N= 56), Flote et al [42] reported a positive association between ApoA-1, HDL, and larger HDL subfractions, with expression of progesterone receptors, a marker of a good prognosis [42]. Notwithstanding, systematic review and meta-analysis of prospective studies suggested a slight inverse association between HDL and the BC risk [44]. In another report, the characterization of 1044 patients as a preoperative study in all BC stages according to TNM classification, demonstrated normal HDL-values in most stages [45]. Therefore, several reports are contradictory, and possible is critical to consider the sample size, the cancer stage and HDL concentrations.
Although the protective role of HDL in BC have been described, in conditions such as MetS, diabetes and obesity, quality of HDL could be modified, for instance, in the characterization of healthy (N = 65) and T2D patients (N = 71), diabetic HDL showed higher levels of glycation and oxidation, with 250 and 127% increments, respectively [46]. These HDL have been proposed as a risk factor related with proliferation, migration, and metastasis in BC models [46]. Other authors have found in patients with breast, colorectal and lung cancer, a decreased activity of paraoxonase, an enzyme that binds HDL and is associated with anti-inflammatory and anti-oxidant properties [47].
LDL associated mechanisms
LDL cholesterol is one of the main risk factors for cardiovascular disease [50], evidence suggests elevated serum LDL concentrations could be a contributing factor to increase recurrence and mortality in obese women with BC [50, 51]. Indeed, through univariate association studies is reported that patients with higher levels of LDL at diagnosis showed large tumors, with higher differentiation grade, frequently express HER-2 and are diagnosed in more advanced stages [52]. Plasmatic LDL concentrations greater than 144 mg/dL in BC patients are associated with high histological grade, high proliferative tumors, accompanied by a risk of lympho-vascular invasion and lymph node metastasis [52].
In the analysis of 1054 BC patients compared with 2483 healthy women stratified by age, Li et al characterized the lipid profile status at the beginning of diagnosis and during chemotherapy, lipid profiles become worsened after chemotherapy, increasing concentrations of total cholesterol, triglycerides, LDL, and apolipoprotein B; while levels of HDL and Apo-A1 decreased compared with pre-chemotherapeutic status [53]. In another report, during chemotherapy of 394 BC-patients, triglycerides, LDL and apolipoprotein-B increased significantly [53]. Although in a small study of cases (N=30) and controls (N=100), LDL concentrations were not modified [54]. In The Malmo Diet and Cancer Study, which enrolled 17,035 women (1991-1996), BC risk was inversely associated with Apo-B concentrations [55], which is the main protein of VLDL and LDL. In an important way, in a Mendelian randomization of the effects of serum lipids and BC risk from genetic datasets of the Global Lipid Genetics Consortium and the Breast Cancer Association Consortium [56] involucrated >400,000 participants, Nowak and Ärnlöv (2018) reported that genetically raised LDL-cholesterol is associated with higher BC risk [56]. The aforementioned data indicate the fundamental role of lipoprotein metabolism in response to chemotherapy, therefore describing the mechanisms involved will allow the identification of drug-targets and the design of novel therapies in BC patients. For instance, in cohort studies, under hormonal treatment or chemotherapy, patients who presented an overexpression of LDL-R showed a reduced recurrence-free survival [48]. In this case, a novel possibility is the design of strategies that could modulate the expression of LDL-R on tumoral tissues, it is recommendable taking into account the know-how of the novel treatments directed to PCSK-9, based on monoclonal antibodies [57] and RNAi [58] used in cardiovascular-risk patients.
In a complementary way, in hypercholesterolemic murine models, tumors were consistently larger, high proliferative, and showed an increased capacity to metastasize to the lung [59, 60]. Specifically, cancer cells treatment with elevated LDL concentrations overexpressed LDL-receptor (LDL-R), and induced larger size tumor compared to cell lines with basal expression. Indeed, LDL-R silencing in tumor cells induced a reduction in tumor size. In line with this notion, high expression of LDL-R in BC has been associated with decreased recurrence-free survival, predominantly in patients treated with systemic therapies [48]. The relevance of LDL-R on triple-negative growth and HER2 tumors has been highlighted [48]. Additional to the increase in the expression of LDL-R in several models, and then, in the promotion of LDL-endocytosis, the inhibition of acetyl-coenzyme A acetyltransferase (ACAT), responsible of cholesterol esterification in the cytoplasm, represents a biomarker in the regulation of intracellular cholesterol storage, and there is an implication on migration process, activating the Ras/Raf-1/MEK/ERK pathway [61]. This mechanism promotes over-expression of the Slug transcription factor, which induces an increase in the expression of phospholipase D2 (PLD2) and the formation of β-catenin-T-cell factor (TCF)-4 transcription complexes. Importantly, PLD2 has been associated with metastasis, an increase in tumor size, and a poor prognosis [62].
Currently, molecular mechanisms associated with the pro-tumoral function of LDL have been described. Characterization on in vitro and in vivo models of BC revealed a decrease in cell adhesion and an increase in cell migration induced by LDL, corroborated by a down-regulation of the expression of adhesion molecules such as member 3 of the cadherin-related family, CD22, claudin and occludin. Besides, the LDL-induced proliferative effect dependent on the activation of the Akt and ERK pathways; have been reported, and these mechanisms are characteristic of the EMT process [63]. Likewise, the treatment of L1 and L5 subfractions of LDL and VLDL increased the levels of mesenchymal markers Slug, vimentin and β-catenin, promoting migration and invasion of BC-cells, distinctive events of cells under the EMT process [32] (Figure 1). EMTs are originated in epithelial cells, which acquire mesenchymal characteristics through cytoskeleton reorganization, loss of basal-apical polarity, loss of cell-cell interactions, an increase in the secretion of metalloproteinases, promoting migratory and invasive capacity, accompanied by a fibroblastoid phenotype [64]. During this cellular phenomenon, cancer cells temporarily acquire the ability to degrade the basement membrane, invade the surrounding tissue, and perform intravasation [64]. Then, L1 and L5 subfractions could enhance the secretion of angiogenic factors in BC-cells and promote the formation of new blood vessels, resulting in a high tumor burden and a poor prognosis [32]. Notwithstanding, extensive evidence is needed to confirm the role of LDL-R overexpression in tumors, even more, the characterization of pathways that regulate the intracellular accumulation of cholesterol. For this reason, the management of plasmatic LDL-cholesterol levels as a regulator of BC-progression is highly promising, especially due to the availability of low-cost drugs such as statins. However, in clinical studies there are still disparities in the results obtained with the use of statins, with variations dependent of the type of statin [19, 65]. Therefore, this therapeutic vision must be subjected to prolonged analysis and determine the response of the different tumor phenotypes to the treatment of these drugs.
In addition, a positive correlation of higher VLDL concentrations and lymph node metastasis has been reported in BC patients, these lipoproteins are associated with the transport of cholesterol, triglycerides and metabolites such as oxysterols from liver to the tumor cells [42]. Elevated levels of VLDL have been associated with enhanced VEGF-signaling, promoting lymphatic angiogenesis, and coupled with TAMs through inflammatory cascades, could contribute to nodal metastasis disease [42]. Therefore, the exacerbated concentrations of cholesterol should promote alterations in the mechanisms that regulate its homeostasis, including alterations in de novo synthesis, endocytosis, modifications in the hydroxylation of side-chain or sterol-nucleus, and storage. Importantly, these mechanisms could impact neoplastic processes, resulting in the modulation of the microenvironment favoring cellular transformation, then tumor development.
The role of sterol regulatory element-binding protein 2 (SREBP-2)
The activity of master regulator SREBP-2 which is key in the synthesis de novo of cholesterol, has been broad described in the management of low concentrations of this steroid, activating gene expression that promotes cholesterol synthesis and endocytosis, such as HMG-CoA reductase and LDL-R, respectively [66]. SREBP-2 and SCAP (SREBP cleavage-activating protein) integrate a sensor of cholesterol in the endoplasmic reticulum, subtly regulating intracellular cholesterol concentrations. Moreover, LDL-R concentration in plasmatic membranes is modulated by three factors: the activity of SREBP-2, the pro-protein convertase subtilisin/kexin type 9 (PCSK9) expression and, the inducible degrader of LDL-R (IDOL) [67]. Considering the critical role of LDL-R in neoplastic phenomena, specifically the regulation of PCSK9 and IDOL has not been characterized in detail under a tumoral environment.
Taking into account the intracellular cholesterol concentration could be a determinant factor for the development of neoplastic processes, elevated expression of SREBP-2 has been correlated with invasive breast carcinomas and a poor prognosis condition [68]. In this sense, deletion of SREBP-2 inhibits migration and invasion phenomena in cellular models, as well as this phenomenon protects against osteolysis associated to BC [68]. BC cells secrete the receptor activator of nuclear factor-κB ligand (RANKL) which promotes osteoclast formation and this activation, resulting in excessive bone resorption [69]. Indeed, RANKL-CREB signaling induces the expression of SREBP-2, playing a key role in osteoclast formation; thus, this alteration promotes cytokines-release from the bone matrix, increasing BC proliferation [70, 71]. These data indicate the complex relationships between these cellular processes, in which positive loops are generated among osteoclast formation and intracellular cholesterol regulation.
Mevalonate synthesized by HMG-CoA reductase is a hub metabolite regulated by the SREBP-2 pathway, and then, further synthesis of geranyl-geranyl pyrophosphate is performed. YAP/TAZ are proto-oncogenes that promote tissue growth by induction of cell proliferation, stem cell amplification, as well as inhibition of apoptosis by expression of factor TEAD (Figure 2) [72]. Critically, YAP/TAZ activity is regulated by geranyl-geranyl pyrophosphate through Rho GTPase activation. Mevalonate produced through SREBP-transcriptional activity in tumor cells is stimulated by its oncogenic cofactor mutant p53 (mutations R175H and R273H) [73]. Indeed, mutant p53 in mice models is associated with metastatic tumors compared to p53 null mice [74, 75], and mutant p53 expression is correlated in preclinical cancer models with invasion, migration and metastasis [76, 77]. Importantly, depletion of mutant p53 is enough to phenotypically return BC-cells to acinar-like morphology. Genome-wide analysis has identified the mevalonate pathway as an upregulated target of mutant p53 [78], which could bind to sterol gene promoters via SREBP-transcription factors, inducing a high expression of sterol biosynthesis genes [78], then metabolites such as geranyl-geranyl pyrophosphate. Under these conditions, the role of dyslipidemias could exacerbate the metabolic dyshomeostasis in cancer cells and synthesis of precursor metabolites such as geranyl-geranyl pyrophosphate interrelates with non-canonical growth and proliferation signals dependent on cholesterol synthesis.
Role of cholesterol targets and metabolites on the regulation of breast cancer (BC) progression. During BC development, cellular concentrations of mevalonate, cholesterol, and the metabolite 27-hydroxycholesterol (27-HC) are increased; phenomenon associated with the induction of cellular proliferation, epithelial-mesenchymal transition (EMTs), invasion, and metastasis. Deregulation in 27-HC concentrations could occur through modifications in the expression of CYP27A1 and CYP7B1. Likewise, cellular targets such YAP/TAZ signaling, ERα, SREBP2, and LXR could regulate neoplastic events. Although cholesterol-derived molecules such as 7-ketocholesterol (7-KC) could show anti-tumoral functions. Adapted from: [73, 92, 100, 110-112].
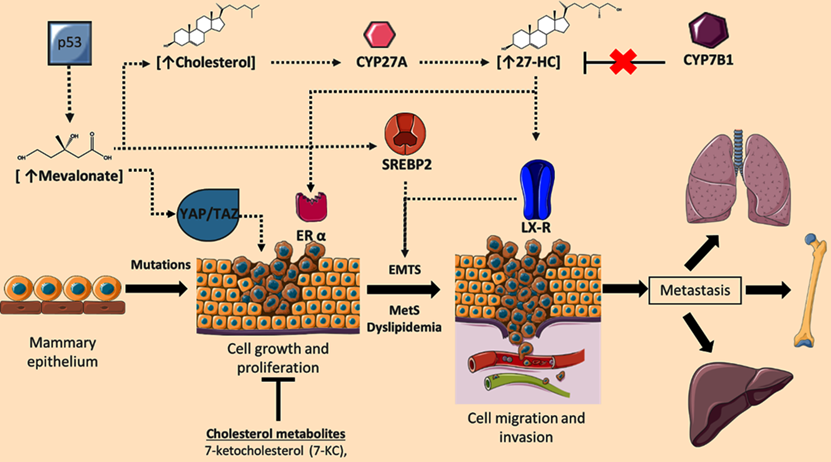
Concomitantly with SREBP-2 signaling, a contrarregulatory mechanism that responds to excessive concentrations of cholesterol has been described. Nuclear factor erythroid-derived 2-related factor 1 (Nrf1) is a direct sensor of high cholesterol concentrations wherein Nrf1 accumulates in ER, and cleavaged domain performs a function as a regulator of the Liver X receptor (LXR) and blockage of scavenger receptor (CD36) activity [79]. Therefore, the modulation of these counter-regulatory mechanisms (SREBP-2 and Nrf1), and the accumulation of metabolites such as mevalonate, geranyl-geranyl pyrophosphate or cholesterol could trigger the development of neoplastic processes. In line with this notion, there is evidence suggesting the loss of Nrf1 is associated with genomic instability, resulting in the generation of micro-nuclei, abnormal mitosis, and a decrease in protein expression of targets involved in cell cycle [80]. These observations suggest a critical function of Nrf1, both in the modulation of cholesterol metabolism, and a possible role on tumor development. Therefore, the subtle regulation of mechanisms mediated by SREBP2 and Nrf1 could negatively regulate metabolic and cellular processes present in tumor cells.
Intracellular metabolism of cholesterol
Although, several metabolites accumulate by activation of the SREBP-2 pathway, a broad family of metabolites derived from cholesterol oxidation is generated by reactions performed by cytochromes, oxidation through reactive oxygen species, or by the combination of both phenomena [81, 82]. For instance, oxysterols are oxidized molecules derivatives of cholesterol, associated with pro-atherogenic properties. Oxysterol formation is related to LDL-oxidation (oxLDL) due to the inflammatory cellular responses in tunica intima [83], also factors such as diet could favor the increase in plasmatic concentrations, which in turn are incorporated in plasmatic triglyceride-rich lipoproteins [84]. Notwithstanding, HDL could remove oxidized lipids from the surface of oxLDL, including oxysterols such as 7-ketocholesterol and 7β-hydroxycholesterol [85].
Oxysterols are 27 carbon molecules derived from cholesterol. In this case, hydroxylation occurs in the side-chain or the sterol-nucleus. Side-chain hydroxylation generates metabolites such as 24-hydroxycholesterol (24-HC), 25-hydroxycholesterol (25-HC) and 27-hydroxycholesterol (27-HC); while hydroxylation of the sterol nucleus generates 6-hydroxycholesterol (6-HC), 7α/β-hydroxycholesterol (7α/β-HC) and 7-ketocholesterol (7-KC) [86]. Moreover, oxysterols can modulate phenomena such as membrane fluidity and intracellular signaling pathways; therefore, their role has been implicated in atherosclerosis, T2D, and neurodegenerative disorders [86].
However, oxysterols and other cholesterol-metabolites such as epoxy-cholesterols have been described as the most important metabolites in the regulation of tumor development, opening new perspectives in the explanation of neoplastic phenomena. For instance, oxysterols produced by osteoblast-like MG63CM cells promote migration on MCF7 (ER+) and MDA-MB-231 (TNBC) breast cancer cells [87-89]. In addition, evidence suggests oxysterols participate in the metastatic cascade modifying the pulmonary metastatic niche and contributing to the recruitment of tumor-promoting neutrophils [90]. Although, oxysterols could modulate carcinogenesis and cancer progression, the precise effect of oxysterols on carcinogenesis, or cancer progression, remains to be evaluated considering the multiple effects of these metabolites on various cell lines.
The broad role of 27-hydroxycholesterol (27-HC)
Cholesterol 27-hydroxylase (CYP27A1) is a mitochondrial resident cytochrome-P450 enzyme which performs cholesterol hydroxylation, synthesizing 27-HC [7]. Conditions such as the disposal of cholesterol in mitochondria, and the expression and activity of CYP27A1 regulates the synthesis of 27-HC [91]. 27-HC could bind to the estrogen receptor α (ERα), promoting the oncogenic estrogen-dependent signaling, and contributing to BC-cells proliferation [92]. Although 27-HC causes a conformational change in both the ERα and ERβ [2], and ERβ is described to be expressed in both ERα+ and ERα- tumors, however a significant heterogeneity in the association of circulating 27-HC and BC risk by ERβ has not been yet identified [93]. In addition, in vivo experimentation suggests 27-HC treatment promotes metastasis phenomena due to LXR agonist function, inducing EMTs [92, 94]. Moreover, exposure to 27-HC in breast carcinoma MCF7 cells showed lower expression of E-cadherin and β-catenin, indicating a role of 27-HC during the development of EMTs [95]. Elevated 27-HC concentrations have been reported in plasma and breast cancer tissues on in vivo models [94, 96].
Taking into account 27-HC could bind to the LXR, promoting EMTs, a condition associated with the worst prognosis in BC patients [94, 96-98], LXR activation of tumor immune cells triggers CD8+ T cells suppression activity and, inflammasome formation [94, 99]. Likewise, overexpression of CYP27A1 in tumor cells and TAMs promote a tumor microenvironment rich in 27-HC, which has been associated with aggressive characteristics of ER+ in post-menopausal women [92]. 27-HC also promotes cell growth and proliferation in the MCF7 cells (ER+) [100]. A report of proteomics in patients with TNBC versus luminal type A breast cancer, suggests LXRα levels do not show significant differences among both molecular subtypes [101]. However, a slight increase in CYP7B1 expression was found in patients with luminal type A cancer, CYP7B1 is an enzyme inducer of 27-HC catabolism. Then, the efficient degradation of 27-HC in luminal type A is considered of better prognosis in this histological subtype [101]. Moreover, the analysis of CYP7B1 expression in 406 ER+ tumors versus 63 normal breast tissue samples (The Cancer Genome Atlas) showed a CYP7B1 diminution by 50 % in ER+ tumors compared to controls, possibly the increase of 27-HC registered in ER+ tumors is related to an alteration of 27-HC catabolism (Figure 2) [100].
Likewise, in the characterization of 22 BC specimens for oxysterol content to evaluate intra- and inter-tumor variation, authors reported significant differences of 27-HC concentrations among ER+ versus ER- [102]. Evidence suggests the heterogeneity in 27-HC content in the cancer tissues, authors propose a possible association due to differential invasion of macrophages, fibroblast and/or adipocytes, capable of synthesizing oxysterols [102]. In a recent report, serum oxysterols were determined in 58 patients with primary breast carcinoma in different tumor stages before treatment, reporting lower 27-HC levels in small tumors, in contrast with higher tumor burden [103]. In a prospective study, evaluation of oxysterols-profile in 29 patients before and after treatment with Tamoxifen (Tam) and aromatase inhibitors (AI) showed increased concentrations of 27-HC under AI response and not significant changes in Tam-treatment [104]. In another analysis, samples from 24 patients diagnosed with primary breast carcinoma before and after surgical tumor removal were characterized, 27-HC serum levels were lower after surgery, and patients with ERα+ tumor and treated with Tam showed a reduction of 27-HC with respect to ER- tumors [87, 105].
However, studies in humans have not been conclusive. In the evaluation of 58 patients with ERα+ BC and 18 cancer-free subjects, the tumor content of 27-HC was elevated by 2.3-fold compared to controls [100]. Likewise, results of the analysis of the nested case-control study of European Investigation into Cancer and Nutrition cohort (530 incident invasive BC cases/1036 controls), suggested that higher serum 27-HC levels were associated with lower BC risk among postmenopausal women; in addition, an association between 27-HC and BC risk among premenopausal women was not registered [93]. On the other hand, a report of 42 BC patients treated with atorvastatin within a phase II clinical trial, reported a decreased serum 27-HC and CYP27A1 expression in tumors; however, these changes were not directly correlated with anti-proliferative responses [106].
Another member of the oxysterol family, 25-hydroxysterol (25-HC) promoted the activation of the pro-tumoral ERα cascade in ER+ cellular models (MCF-7 and BG-1 cells) [107]. In line with this notion, in gastric cancer models 25-HC promoted cell invasion associated with the upregulation of matrix metalloproteinases-expression in vitro and in vivo [104, 108]. Higher serum levels of 25-HC associated with hormone receptor-positive metastatic disease have been reported [104], however, this phenomenon was not replicated by other authors in a larger population [103].
Likewise, oxysterols have multiple functions in shaping the immunological landscape, considering active cholesterol metabolism shows an essential role in promoting cancer progression. For instance, similar to cancer cells, activated T cells also undergo rapid proliferation and, therefore are dependent on elevated cholesterol metabolism. Whereas SREBP2 signaling is essential for CD8 T cell proliferation and for its effector function [109], LXR signaling negatively regulates T cell activation [109]. Therefore, high levels of oxysterols in the tumor microenvironment could inhibit T cell anti-tumoral immunity via LXR activation, whereas upregulation of intrinsic cholesterol synthesis de novo or endocytosis in T cells could boost T-cell antitumoral functions.
Epoxide-cholesterol pathway
Other modifications occur on cholesterol structure, oxygenation reactions could be generated by enzymatic and auto-oxidation phenomena, this last process is promoted by reactive oxygen species or indirectly through lipid peroxidation [113, 114]. For instance, cholesterol-5,6-epoxide (5,6-EC) is derived from cholesterol-autoxidation [115], although specifically, enzymatic systems can catalyze the synthesis of 5,6-EC [113]. Tam-treatment and ERα modulators promote oxidative stress and inhibit cholesterol-5,6-epoxide hydrolases, triggering the accumulation of 5,6-ECs, these metabolites are associated with differentiation and apoptosis of BC cells [116]. Evidence suggests a multifaceted role of epoxides in neoplastic pathology, being 5,6-EC the main metabolite (Figure 3). Therefore, their analysis is a critical condition.
Representation of metabolism of cholesterol epoxides. 5,6α-EC and 5,6β-EC are metabolites produced by oxidation of cholesterol, then specifically 5,6α-EC is hydrated by Cholesterol epoxide hydrolase (ChEH), originating cholestan-3β,5α,6β-triol (CT). ChEH is a heterodimeric complex integrated by 3β-hydroxysteroid-∆8-∆7-isomerase (D8D7I) and 3β-hydroxysteroid-∆7-reductase (DHCR7). Later, CT is metabolized into 6-oxo-cholestan-3β, 5α-diol (OCDO) by 11-β-hydroxysteroid dehydrogenase of type 2 (11HSD2), OCDO is a promoter of cancer cell proliferation. OCDO is an agonist of the glucocorticoid receptor (GR) and it is translocated to the nucleus. Glucocorticoid induces the expression of genes associated with protection against cell apoptosis. Dendrogenin A (DDA) pathway is represented, which is synthesized from 5,6α-EC and histamine (His); DDA could induce autophagy in cancer cells. Adapted from: [147, 155, 157].
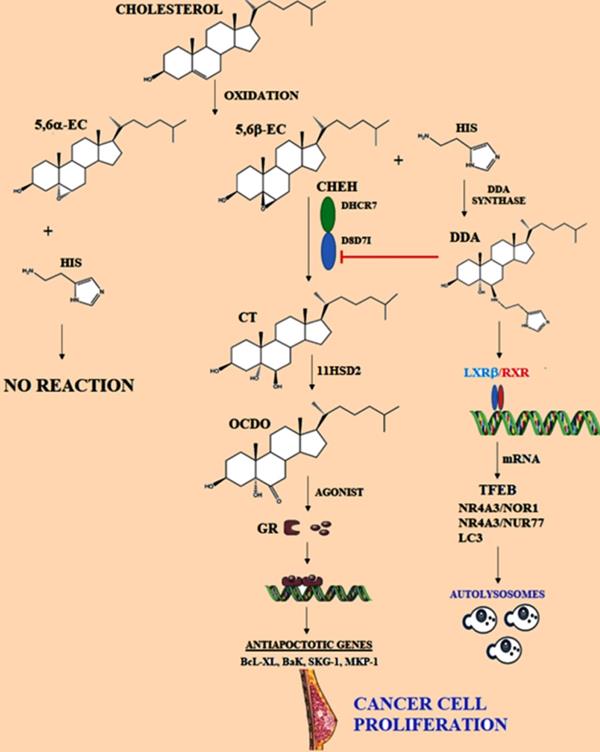
6-oxo-cholestan-3β,5α-diol (OCDO)
5,6-EC is a highly stable metabolite, allowing the synthesis of cholestane-3β,5α-triol (CT) by 5,6-epoxide hydrolase (ChEH) activity [117]. In cancer cells, using CT as a substrate, the 11-β-hydroxysteroid dehydrogenase of type 2 (11HSD2) catalyzes the synthesis of 6-oxo-cholestan-3β,5α-diol (OCDO) [117, 118] (Figure 3). OCDO could promote breast tumor development through the interaction with the glucocorticoid receptor (GR) regardless of their ERα expression status, then promoting the activation of cellular-proliferation genes. Importantly, 11HSD2 is described as a regulator of BC cell proliferation both in vitro and in vivo through OCDO synthesis. Likewise, 11HSD2 is described to regulate glucocorticoid metabolism by converting active cortisol into inactive cortisone [119].
As a cortisol agonist, OCDO causes GR-translocation to the nucleus for the regulation of GR-dependent transcription [120], such as the targets cyclin-dependent kinases (CDKs), CDK4, CDK6, and cyclin D3. Although OCDO does not stimulate the transcription of SGK1 or MKP1/DUSP1 genes, which play an important role in the negative regulation of cellular proliferation, as has been reported with cortisol; however, OCDO increases MMP1 gene expression through the GR. In BC cell lines, since low OCDO concentrations (≤10 µM), treatment induces proliferation, growth, and cellular invasion, as well as tumor growth in murine models. Besides, in the absence of the GR, OCDO does not induce proliferation and invasion in cellular and in vivo models [104]. Therefore, OCDO proliferative effects through the GR, and the identification of the enzymes regulating its synthesis could be determinant targets implicated in the biology of BC. Synthesis of glucocorticoids (GCs) is increased under stressful conditions, leading to regulation of inflammatory and immune responses, as well as cellular proliferation and apoptosis [121-124]. However, the correlation between GR expression and the prognosis of breast cancer or its malignancy is still debated [125-131].
GR is considered a mediator of the GCs acute-release associated with stress conditions. In vitro evidence and clinical studies, suggest that stress phenomenon contributes to cancer progression [132, 133]. Taking into account GR was found significantly expressed in breast cells and described to be activated by stress-induced GCs, GR might play a role in cancer development associated with stressful conditions [134]. In breast cancer cells, GCs induce the expression of genes connected with protection against cell apoptosis, such as Bcl-xL, Bak, SGK-1, and MKP-1 [126, 127, 135-137]. Specifically, in breast cancer cell lines, GR exerts anti-apoptotic actions through the activation of NF-κB signaling [138-140]. GR survival effect is mediated by its interaction with the AP-1 transcription factor family [141]. Finally, GR also disrupts p53 cell survival regulation [142], as we have analyzed previously, p53 mutant is correlated in preclinical cancer models with invasion, migration and metastasis [76, 77]. Therefore, the signaling pathways associated with GCs and the GR could trigger several hits in cancer development processes.
Cholestane-3β,5α,6β-triol (CT)
Considering 5,6-EC diastereoisomers can be hydrated by the ChEH enzyme to generate CT (Figure 3), ChEH has been reported to preferentially binds 5,6α-EC than 5,6β-EC during synthesizes of CT [143, 144]. Indeed, CT was reported to be involved in carcinogenesis indirectly through the stimulation of oxidative processes [145] and thus, inhibition of ChEH may contribute to chemopreventive action of Tam and other ChEH inhibitors. Likewise, the metabolite 7-KC is described as an inhibitor of ChEH [146], then 7-KC may contribute to the accumulation of 5,6-EC. In this sense, in a 58 patient study with primary breast carcinoma, the low levels of CT were related with early stages of the disease, and a positive association was reported as dependent on the patients age. Authors have proposed a correlation among higher CT levels and a shorter disease-free survival compared with patients with lower levels [103]. However, further exploration of the implications and the metabolic fate of CT during oncogenesis are necessary.
Dendrogenin A (DDA) pathway
On the other hand, throughout the metabolism of 5,6α-EC, the conjugation of this metabolite with histamine generates dendrogenin A (DDA), the first endogenous steroidal-alkaloid identified in mammals [147]. Anti-cancer effect of DDA has been established in several mice and human cancers such as breast, melanoma, and acute myeloid leukemia [145]. Indeed, treatment of cancer cells with DDA induced cancer cell re-differentiation, blockade of OCDO synthesis, and lethal autophagy in tumors [117]. Evidence suggests DDA as the most potent inhibitor of ChEH, the critical enzyme for CT synthesis [8, 104], then cell differentiation and death could be induced under DDA treatment [148, 149] (Figure 3). Specifically, DDA inhibits the D8D7I subunit (3β-hydroxysterol-Δ8,7- isomerase) of ChEH leading to accumulation of 5,6-ECs [150]. This accumulation shows a chemopreventive function and therapeutic effects of drugs such as Tam. Likewise, anti-cancer effects of DDA have been associated with increased tumor infiltration of T lymphocytes and CD11c+ dendritic cells. Therefore, ChEH inhibition could represent a new target involved in the anti-cancer function of DDA [147]. In murine models treated with grafts of tumors B16F10 and TS/A, DDA promoted inhibition of tumor size, also increased the survival time [147]. Importantly, differences of five-fold lower levels of DDA in BC tissues compared with normal matched tissues were reported [147].
On the other hand, TFEB is a master transcription factor, controlling genes involved in autophagy and lysosome biogenesis, evidence suggests LXRβ and LXRβ-agonists could act as TFEB repressors, and notably, DDA decreases LXRβ-binding to TFEB enhancer and, this phenomenon could stimulate TFEB expression [151-154] (Figure 3). Evidence revealed that LXR through DDA-binding could be a new target in autophagy and lysosome biogenesis. Reports suggest that DDA treatment in B16F10 cells increased the mRNA levels of ABCG5, NOR1, Nur77, LC3A, and LC3B [155-157], proteins involucrated in the formation of autophagosomes.
Proliferating cancer cells require high levels of cholesterol for membrane biogenesis and other functional requirements; therefore cholesterol metabolism substantially contributes to cancer progression, including cell proliferation, migration, and invasion. For instance, the cholesterol-derived metabolite 6-oxo-cholestan-3β,5α-diol, which is enriched in patients with breast cancer, binds glucocorticoid receptors and subsequently promotes tumor growth [104]. In general, cholesterol metabolism substantially contributes to cancer progression, including cell proliferation, migration and invasion [158-161].
Discussion
Cancer cells are exposed to highly fluctuating metabolic conditions, signaling metabolites, stromal biomolecules, and chemotherapeutic agents, which together generate a microenvironment that induces changes in phenotype [97, 98]. Cellular adaptations may involve genetic alterations, and importantly entail transcriptional or epigenetic changes modifying cholesterol pathways [162]. For instance, in the characterization of a genome-wide transcriptional survey in 995 breast tissue samples, 215 long non-coding RNAs (lncRNAs) showed a differential expression in mammary tumors compared to controls. Although several molecular processes were identified, such as activation of PI3K pathway, fibroblast growth factor, TGF-β, and activation of EGFR-dependent pathway [163], lncRNAs associated with cholesterol metabolism did not were reported for the authors [163]. However, in a subsequent analysis performed by our group on these 215 lncRNA, we evidenced an altered expression of some lncRNAs associated with the lipid metabolism, such as HOTAIR and H19. In a previous report, HOTAIR could ameliorate ox-LDL-induced inflammatory response in macrophages via inhibition of NF-κB pathway [164]. In contrast, silencing of H-19 could inhibit the adipogenesis and inflammation response in ox-LDL-treated macrophages [165]. Possibly these lncRNAs perform critical roles in compensate inflammatory conditions on BC tissues, promoting survival. lncRNA are only one epigenetic factor that could contribute to the metabolic adaptation of tumor cells. And possibly under hypercholesterolemic conditions, several mechanisms that are performed in metabolic tissues such as hepatic or in foam cells during meta-inflammation and development of atherosclerotic plaque, are shared by breast cancer cells.
The role of cholesterol-dyslipidemias on BC risk is a phenomenon that has been strengthened, wherein the function of cholesterol-derived metabolites and alteration of cholesterol-targets induce mechanisms of cellular adaptation during BC development. For instance, bile acids, oxysterols, and epoxy-cholesterols are secondary products of cholesterol metabolism, however, in the description of these metabolites, new critical biological properties have been found, through the regulation of nuclear receptors such as LXR, FXR, ERα, ROR, GR or through G-protein coupled receptors [94, 104, 155, 166-168], which supports a new feature of these metabolites in physiological functions, and pathological disorders. Although activation of LXR, ERα, and GR promotes phenomena such as proliferation, cell invasion, and tumor growth, it is important to determine which are the cellular signals that regulate the synthesis of 27-HC, CT, OCDO, and DDA during carcinogenesis, considering the highly complex regulation of cholesterol pathways, as well as their location within the different regions of the tumor.
In addition, it is necessary characterize the mechanisms associated with dyslipidemias and their direct effects on intracellular concentration of oxysterols and epoxy-cholesterols in tumors. For instance, DDA was confirmed as a steroidal-alkaloid metabolite, identified in mammalian tissues including breast, brain, liver, spleen, and blood [147]; however the diminution of DDA concentrations in tumors compared to normal tissues [147, 157] has not been explained in detail. In this case, has not been considered if the expression of the enzymes such as CHEH, 11HSD2 or DDA synthase is dependent on the SREBP2 sensor, YAP/TAZ or mutant p53. Moreover, changes in cholesterol concentration in the endoplasmic reticulum or several metabolites such as mevalonate and geranyl-geranyl pyrophosphate could be critical modulators.
In breast cancer development, extracellular cholesterol levels as well as intracellular cholesterol content should play an important role in cellular proliferation. An antagonistic role of HDL with LDL and even VLDL has been reported in the processes of cell growth, invasion, and metastasis. Thus, when intracellular cholesterol levels exceed a management threshold, the description of mechanisms that initiate and enhance processes such as EMTs, growth, migration, invasion, metastasis or resistance to chemotherapy, is a critical phenomenon that has not been elucidated in detail. The complex interactions among cholesterol pathways and the epigenetic regulation could explain several processes involved in the tumor development, as has been proposed for several lncRNAs. And how tumor cells express proteins differently to acquire various phenotypes that contribute to cellular heterogeneity in the primary tumor, possibly, the effect of cholesterol derived-metabolites could explain these phenomena in the complex cellular communication network.
Furthermore, subtle modulation between various phenotypes (EMT and MET) not only allows tumor cells to survive and proliferate in secondary sites, since by acquiring this cellular plasticity they could increase their tolerance to chemotherapeutic treatments. Moreover, this epithelial-mesenchymal plasticity that plays vital roles during normal development and tissue function [169] maintaining the cellular homeostasis, could be modified by cholesterol dyslipidemias. Therefore, regulation of this phenomenon will lead to new anti-tumoral treatments considering the cellular plasticity as a target.
Dyslipidemias and cholesterol-metabolism could impact neoplastic processes, resulting in the modulation of the microenvironment that promotes cellular oncogenic transformation, then tumor development. Indeed, the behavior of cells in the artery wall and the process of atherogenesis promoted by LDL and VLDL could share many similarities with the progression of the neoplastic processes [170]. Although is not described the role of critical targets such as transporters ABCA, ABCG or SRB1 during this oncogenic transformation. Therefore, discovering of cellular mechanisms associated with cholesterol metabolism and its derivatives could facilitate the understanding of the pathology and evolution of the disease, in order to design strategies to prevent the development of breast cancer related to interventions in the lifestyle of patients with MetS, as well as in the development of effective therapies.
Acknowledgements
A.G.-O. is a medical degree student and performed her social service in Biochemistry Department, Facultad de Medicina Mexicali. The authors recognize the administrative support of Josue Villegas Sandoval and Mónica López Valladares.
Funding
This research was supported by Fondo Sectorial de Investigación para la Educación CB 2017-2018 (A1-S-28653/SEP/CONACYT); 21a. Convocatoria Interna de Apoyo a Proyectos de Investigación (Coordinación General de Posgrado e Investigación/UABC) and Convocatoria 2019 Apoyos para Adquisición y Mantenimiento de Infraestructura en Instituciones y Laboratorios de Investigación Especializada (CONACYT, 300015). Convocatoria PRODEP-Apoyos Posdoctorales 2019. 511-6/2019.-15015.
Author Contributions
Conceived the idea: V.G.-G. and O.G.-H. Wrote the manuscript: V.G.-G., O.G.-H., A.G.-O., G.N.H-A and G.H.-U. Figures preparation: V.G.-G., O.G.-H., A.G.-O., G.N.H-A and G.H.-U. Critically reviewed manuscript and figures: V.G.-G., O.G.-H. and A.G.-O. All authors approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yamagishi K, Iso H. The criteria for metabolic syndrome and the national health screening and education system in Japan. Epidemiology and health. 2017;39:e2017003
2. Huang PL. A comprehensive definition for metabolic syndrome. Disease models & mechanisms. 2009;2:231-7
3. Chen Y, Wen YY, Li ZR, Luo DL, Zhang XH. The molecular mechanisms between metabolic syndrome and breast cancer. Biochemical and biophysical research communications. 2016;471:391-5
4. Calip GS, Malone KE, Gralow JR, Stergachis A, Hubbard RA, Boudreau DM. Metabolic syndrome and outcomes following early-stage breast cancer. Breast Cancer Research and Treatment. 2014;148:363-77
5. Dibaba DT, Ogunsina K, Braithwaite D, Akinyemiju T. Metabolic syndrome and risk of breast cancer mortality by menopause, obesity, and subtype. Breast Cancer Research and Treatment. 2019;174:209-18
6. Acosta-Montano P, Rodriguez-Velazquez E, Ibarra-Lopez E, Frayde-Gomez H, Mas-Oliva J, Delgado-Coello B. et al. Fatty Acid and Lipopolysaccharide Effect on Beta Cells Proteostasis and its Impact on Insulin Secretion. Cells. 2019 8
7. Melvin JC, Seth D, Holmberg L, Garmo H, Hammar N, Jungner I. et al. Lipid profiles and risk of breast and ovarian cancer in the swedish AMORIS study. Cancer Epidemiology Biomarkers and Prevention. 2012;21:1381-4
8. Raza U, Asif MR, Rehman AB, Sheikh A. Hyperlipidemia and hyper glycaemia in Breast Cancer Patients is related to disease stage. Pakistan Journal of Medical Sciences. 2018;34:209-14
9. Garcia-Gonzalez V, Diaz-Villanueva JF, Galindo-Hernandez O, Martinez-Navarro I, Hurtado-Ureta G, Perez-Arias AA. Ceramide Metabolism Balance, a Multifaceted Factor in Critical Steps of Breast Cancer Development. Int J Mol Sci. 2018 19
10. Schug ZT, Gottlieb E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta. 2009;1788:2022-31
11. Zhao W, Prijic S, Urban BC, Tisza MJ, Zuo Y, Li L. et al. Candidate Antimetastasis Drugs Suppress the Metastatic Capacity of Breast Cancer Cells by Reducing Membrane Fluidity. Cancer research. 2016;76:2037-49
12. Carracedo A, Cantley LC, Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nature reviews Cancer. 2013;13:227-32
13. Rohrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nature reviews Cancer. 2016;16:732-49
14. Martinez-Navarro I, Diaz-Molina R, Pulido-Capiz A, Mas-Oliva J, Luna-Reyes I, Rodriguez-Velazquez E. et al. Lipid Modulation in the Formation of beta-Sheet Structures. Implications for De Novo Design of Human Islet Amyloid Polypeptide and the Impact on beta-Cell Homeostasis. Biomolecules. 2020 10
15. Seiler A, Chen MA, Brown RL, Fagundes CP. Obesity, Dietary Factors, Nutrition, and Breast Cancer Risk. Current breast cancer reports. 2018;10:14-27
16. Ehmsen S, Pedersen MH, Wang G, Terp MG, Arslanagic A, Hood BL. et al. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Reports. 2019;27:3927-38.e6
17. Borgquist S, Tamimi RM, Chen WY, Garber JE, Eliassen AH, Ahern TP. Statin Use and Breast Cancer Risk in the Nurses' Health Study. Cancer Epidemiol Biomarkers Prev. 2016;25:201-6
18. McDougall JA, Malone KE, Daling JR, Cushing-Haugen KL, Porter PL, Li CI. Long-term statin use and risk of ductal and lobular breast cancer among women 55 to 74 years of age. Cancer Epidemiol Biomarkers Prev. 2013;22:1529-37
19. Van Wyhe RD, Rahal OM, Woodward WA. Effect of statins on breast cancer recurrence and mortality: a review. Breast cancer. 2017;9:559-65
20. Argolo DF, Hudis CA, Iyengar NM. The Impact of Obesity on Breast Cancer. Current oncology reports. 2018;20:47
21. Chan DSM, Vieira AR, Aune D, Bandera EV, Greenwood DC, McTiernan A. et al. Body mass index and survival in women with breast cancer-systematic literature review and meta-analysis of 82 follow-up studies. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2014;25:1901-14
22. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11:85-97
23. Naik A, Monjazeb AM, Decock J. The Obesity Paradox in Cancer, Tumor Immunology, and Immunotherapy: Potential Therapeutic Implications in Triple Negative Breast Cancer. Front Immunol. 2019;10:1940
24. Duggan C, Irwin ML, Xiao L, Henderson KD, Smith AW, Baumgartner RN. et al. Associations of insulin resistance and adiponectin with mortality in women with breast cancer. Journal of Clinical Oncology. 2011;29:32-9
25. Caan BJ, Kwan ML, Hartzell G, Castillo A, Slattery ML, Sternfeld B. et al. Pre-diagnosis body mass index, post-diagnosis weight change, and prognosis among women with early stage breast cancer. Cancer Causes and Control. 2008;19:1319-28
26. Esposito K, Chiodini P, Colao A, Lenzi A, Giugliano D. Metabolic syndrome and risk of cancer: A systematic review and meta-analysis. Diabetes Care. 2012;35:2402-11
27. Dibaba DT, Braithwaite D, Akinyemiju T. Metabolic syndrome and the risk of breast cancer and subtypes by race, menopause and BMI. Cancers. 2018 10
28. Simon MS, Beebe-Dimmer JL, Hastert TA, Manson JAE, Cespedes Feliciano EM, Neuhouser ML. et al. Cardiometabolic risk factors and survival after breast cancer in the Women's Health Initiative. Cancer. 2018;124:1798-807
29. Dieli-Conwright CM, Sweeney FC, Courneya KS, Tripathy D, Sami N, Lee K. et al. Hispanic ethnicity as a moderator of the effects of aerobic and resistance exercise in survivors of breast cancer. Cancer. 2019;125:910-20
30. Dieli-Conwright CM, Wong L, Waliany S, Bernstein L, Salehian B, Mortimer JE. An observational study to examine changes in metabolic syndrome components in patients with breast cancer receiving neoadjuvant or adjuvant chemotherapy. Cancer. 2016;122:2646-53
31. Samadi S, Ghayour-Mobarhan M, Mohammadpour A, Farjami Z, Tabadkani M, Hosseinnia M. et al. High-density lipoprotein functionality and breast cancer: A potential therapeutic target. Journal of Cellular Biochemistry. 2019;120:5756-65
32. Lu CW, Lo YH, Chen CH, Lin CY, Tsai CH, Chen PJ. et al. VLDL and LDL, but not HDL, promote breast cancer cell proliferation, metastasis and angiogenesis. Cancer Letters. 2017;388:130-8
33. Wolfe AR, Atkinson RL, Reddy JP, Debeb BG, Larson R, Li L. et al. High-density and very-low-density lipoprotein have opposing roles in regulating tumor-initiating cells and sensitivity to radiation in inflammatory breast cancer. International Journal of Radiation Oncology Biology Physics. 2015;91:1072-80
34. Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. The Framingham study. The American Journal of Medicine. 1977;62:707-14
35. Cooke AL, Morris J, Melchior JT, Street SE, Gray Jerome W, Huang R. et al. A thumbwheel mechanism for APOA1 activation of LCAT activity in HDL. Journal of Lipid Research. 2018;59:1244-55
36. Mendoza-Espinosa P, Montalvan-Sorrosa D, García-González V, Moreno A, Castillo R, Mas-Oliva J. Microenvironmentally controlled secondary structure motifs of apolipoprotein A-I derived peptides. Molecular and Cellular Biochemistry. 2014;393:99-109
37. Garcia-Gonzalez V, Delgado-Coello B, Perez-Torres A, Mas-Oliva J. Reality of a Vaccine in the Prevention and Treatment of Atherosclerosis. Archives of medical research. 2015;46:427-37
38. Llanos AA, Makambi KH, Tucker CA, Wallington SF, Shields PG, Adams-Campbell LL. Cholesterol, lipoproteins, and breast cancer risk in African American women. Ethnicity & disease. 2012;22:281-7
39. Chang SJ, Hou MF, Tsai SM, Wu SH, Ann Hou L, Ma H. et al. The association between lipid profiles and breast cancer among Taiwanese women. Clinical Chemistry and Laboratory Medicine. 2007;45:1219-23
40. Kucharska-Newton AM, Rosamond WD, Mink PJ, Alberg AJ, Shahar E, Folsom AR. HDL-Cholesterol and Incidence of Breast Cancer in the ARIC Cohort Study. Annals of Epidemiology. 2008;18:671-7
41. Zhou P, Li B, Liu B, Chen T, Xiao J. Prognostic role of serum total cholesterol and high-density lipoprotein cholesterol in cancer survivors: A systematic review and meta-analysis. Clin Chim Acta. 2018;477:94-104
42. Flote VG, Vettukattil R, Bathen TF, Egeland T, McTiernan A, Frydenberg H. et al. Lipoprotein subfractions by nuclear magnetic resonance are associated with tumor characteristics in breast cancer. Lipids in Health and Disease. 2016 15
43. Kim Y, Park SK, Han W, Kim DH, Hong YC, Ha EH. et al. Serum high-density lipoprotein cholesterol and breast cancer risk by menopausal status, body mass index, and hormonal receptor in Korea. Cancer Epidemiol Biomarkers Prev. 2009;18:508-15
44. Touvier M, Fassier P, His M, Norat T, Chan DS, Blacher J. et al. Cholesterol and breast cancer risk: a systematic review and meta-analysis of prospective studies. The British journal of nutrition. 2015;114:347-57
45. Li X, Tang H, Wang J, Xie X, Liu P, Kong Y. et al. The effect of preoperative serum triglycerides and high-density lipoprotein-cholesterol levels on the prognosis of breast cancer. Breast. 2017;32:1-6
46. Pan B, Ren H, He Y, Lv X, Ma Y, Li J. et al. HDL of patients with type 2 diabetes mellitus elevates the capability of promoting breast cancer metastasis. Clinical Cancer Research. 2012;18:1246-56
47. Balci H, Genc H, Papila C, Can G, Papila B, Yanardag H. et al. Serum Lipid Hydroperoxide Levels and Paraoxonase Activity in Patients With Lung, Breast, and Colorectal Cancer. Journal of Clinical Laboratory Analysis. 2012;26:155-60
48. Gallagher EJ, Zelenko Z, Neel BA, Antoniou IM, Rajan L, Kase N. et al. Elevated tumor LDLR expression accelerates LDL cholesterol-mediated breast cancer growth in mouse models of hyperlipidemia. Oncogene. 2017;36:6462-71
49. O'Neill S, Bohl M, Gregersen S, Hermansen K, O'Driscoll L. Blood-Based Biomarkers for Metabolic Syndrome. Trends Endocrinol Metab. 2016;27:363-74
50. Ray KK, Corral P, Morales E, Nicholls SJ. Pharmacological lipid-modification therapies for prevention of ischaemic heart disease: current and future options. Lancet. 2019;394:697-708
51. Beckwitt CH, Brufsky A, Oltvai ZN, Wells A. Statin drugs to reduce breast cancer recurrence and mortality. Breast Cancer Res. 2018;20:144
52. Rodrigues dos Santos C, Fonseca I, Dias S, de Almeida JCM. Plasma level of LDL-cholesterol at diagnosis is a predictor factor of breast tumor progression. BMC Cancer. 2014 14
53. Li X, Liu ZL, Wu YT, Wu H, Dai W, Arshad B. et al. Status of lipid and lipoprotein in female breast cancer patients at initial diagnosis and during chemotherapy. Lipids in Health and Disease. 2018 17
54. Gupta RK, Patel AK, Kumari R, Chugh S, Shrivastav C, Mehra S. et al. Interactions between oxidative stress, lipid profile and antioxidants in breast cancer: a case control study. Asian Pac J Cancer Prev. 2012;13:6295-8
55. Borgquist S, Butt T, Almgren P, Shiffman D, Stocks T, Orho-Melander M. et al. Apolipoproteins, lipids and risk of cancer. International Journal of Cancer. 2016;138:2648-56
56. Nowak C, Arnlov J. A Mendelian randomization study of the effects of blood lipids on breast cancer risk. Nat Commun. 2018;9:3957
57. Deedwania P, Murphy SA, Scheen A, Badariene J, Pineda AL, Honarpour N. et al. Efficacy and Safety of PCSK9 Inhibition With Evolocumab in Reducing Cardiovascular Events in Patients With Metabolic Syndrome Receiving Statin Therapy: Secondary Analysis From the FOURIER Randomized Clinical Trial. JAMA Cardiol. 2020
58. Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, Liebow A, Bettencourt BR, Sutherland JE. et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet. 2014;383:60-8
59. Alikhani N, Ferguson RD, Novosyadlyy R, Gallagher EJ, Scheinman EJ, Yakar S. et al. Mammary tumor growth and pulmonary metastasis are enhanced in a hyperlipidemic mouse model. Oncogene. 2013;32:961-7
60. Kim EJ, Choi MR, Park H, Kim M, Hong JE, Lee JY. et al. Dietary fat increases solid tumor growth and metastasis of 4T1 murine mammary carcinoma cells and mortality in obesity-resistant BALB/c mice. Breast Cancer Res. 2011;13:R78
61. Antalis CJ, Uchida A, Buhman KK, Siddiqui RA. Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clinical and Experimental Metastasis. 2011;28:733-41
62. Kang DW, Choi KY, Min do S. Functional regulation of phospholipase D expression in cancer and inflammation. J Biol Chem. 2014;289:22575-82
63. dos Santos CR, Domingues G, Matias I, Matos J, Fonseca I, de Almeida JM. et al. LDL-cholesterol signaling induces breast cancer proliferation and invasion. Lipids Health Dis. 2014;13:16
64. Roche J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers. 2018 10
65. Borgquist S, Bjarnadottir O, Kimbung S, Ahern TP. Statins: a role in breast cancer therapy? J Intern Med. 2018;284:346-57
66. Madison BB. Srebp2: A master regulator of sterol and fatty acid synthesis. J Lipid Res. 2016;57:333-5
67. Yang HX, Zhang M, Long SY, Tuo QH, Tian Y, Chen JX. et al. Cholesterol in LDL receptor recycling and degradation. Clin Chim Acta. 2020;500:81-6
68. Jie Z, Xie Z, Xu W, Zhao X, Jin G, Sun X. et al. SREBP-2 aggravates breast cancer associated osteolysis by promoting osteoclastogenesis and breast cancer metastasis. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2019;1865:115-25
69. Blake ML, Tometsko M, Miller R, Jones JC, Dougall WC. RANK expression on breast cancer cells promotes skeletal metastasis. Clinical and Experimental Metastasis. 2014;31:233-45
70. Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nature reviews Cancer. 2002;2:584-93
71. Futakuchi M, Fukamachi K, Suzui M. Heterogeneity of tumor cells in the bone microenvironment: Mechanisms and therapeutic targets for bone metastasis of prostate or breast cancer. Advanced drug delivery reviews. 2016;99:206-11
72. Zhao B, Ye X, Yu J, Li L, Li W, Li S. et al. TEAD mediates YAP-dependent gene induction and growth control. Genes and Development. 2008;22:1962-71
73. Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S. et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nature Cell Biology. 2014;16:357-66
74. Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM. et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861-72
75. Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT. et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847-60
76. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B. et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137:87-98
77. Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S. et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327-41
78. Freed-Pastor WA, Mizuno H, Zhao X, Langerød A, Moon SH, Rodriguez-Barrueco R. et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244-58
79. Widenmaier SB, Snyder NA, Nguyen TB, Arduini A, Lee GY, Arruda AP. et al. NRF1 Is an ER Membrane Sensor that Is Central to Cholesterol Homeostasis. Cell. 2017;171:1094.e15-109.e15
80. Oh DH, Rigas D, Cho A, Chan JY. Deficiency in the nuclear-related factor erythroid 2 transcription factor (Nrf1) leads to genetic instability. FEBS Journal. 2012;279:4121-30
81. Levy D, Correa de Melo T, Ohira BY, Fidelis ML, Ruiz JLM, Rodrigues A. et al. Oxysterols selectively promote short-term apoptosis in tumor cell lines. Biochemical and Biophysical Research Communications. 2018;505:1043-9
82. Brzeska M, Szymczyk K, Szterk A. Current Knowledge about Oxysterols: A Review. Journal of food science. 2016;81:R2299-R308
83. Zmyslowski A, Szterk A. Current knowledge on the mechanism of atherosclerosis and pro-atherosclerotic properties of oxysterols. Lipids Health Dis. 2017;16:188
84. Vine DF, Mamo CL, Beilin LJ, Mori TA, Croft KD. Dietary oxysterols are incorporated in plasma triglyceride-rich lipoproteins, increase their susceptibility to oxidation and increase aortic cholesterol concentration of rabbits. J Lipid Res. 1998;39:1995-2004
85. Rasmiena AA, Barlow CK, Ng TW, Tull D, Meikle PJ. High density lipoprotein efficiently accepts surface but not internal oxidised lipids from oxidised low density lipoprotein. Biochim Biophys Acta. 2016;1861:69-77
86. Munir MT, Ponce C, Powell CA, Tarafdar K, Yanagita T, Choudhury M. et al. The contribution of cholesterol and epigenetic changes to the pathophysiology of breast cancer. J Steroid Biochem Mol Biol. 2018;183:1-9
87. Kloudova A, Guengerich FP, Soucek P. The Role of Oxysterols in Human Cancer. Trends Endocrinol Metab. 2017;28:485-96
88. Silva J, Beckedorf A, Bieberich E. Osteoblast-derived oxysterol is a migration-inducing factor for human breast cancer cells. J Biol Chem. 2003;278:25376-85
89. Silva J, Dasgupta S, Wang G, Krishnamurthy K, Ritter E, Bieberich E. Lipids isolated from bone induce the migration of human breast cancer cells. J Lipid Res. 2006;47:724-33
90. Moresco MA, Raccosta L, Corna G, Maggioni D, Soncini M, Bicciato S. et al. Enzymatic Inactivation of Oxysterols in Breast Tumor Cells Constraints Metastasis Formation by Reprogramming the Metastatic Lung Microenvironment. Frontiers in Immunology. 2018 9
91. Marwarha G, Raza S, Hammer K, Ghribi O. 27-hydroxycholesterol: A novel player in molecular carcinogenesis of breast and prostate cancer. Chem Phys Lipids. 2017;207:108-26
92. Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK. et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342:1094-8
93. Lu DL, Le Cornet C, Sookthai D, Johnson TS, Kaaks R, Fortner RT. Circulating 27-Hydroxycholesterol and Breast Cancer Risk: Results From the EPIC-Heidelberg Cohort. J Natl Cancer Inst. 2019;111:365-71
94. Baek AE, Yu YRA, He S, Wardell SE, Chang CY, Kwon S. et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nature Communications. 2017 8
95. Torres CG, Ramirez ME, Cruz P, Epunan MJ, Valladares LE, Sierralta WD. 27-hydroxycholesterol induces the transition of MCF7 cells into a mesenchymal phenotype. Oncol Rep. 2011;26:389-97
96. Kuzu OF, Noory MA, Robertson GP. The Role of Cholesterol in Cancer. Cancer research. 2016;76:2063-70
97. Galindo-Hernandez O, Serna-Marquez N, Castillo-Sanchez R, Salazar EP. Extracellular vesicles from MDA-MB-231 breast cancer cells stimulated with linoleic acid promote an EMT-like process in MCF10A cells. Prostaglandins Leukotrienes and Essential Fatty Acids. 2014;91:299-310
98. Galindo-Hernandez O, Gonzales-Vazquez C, Cortes-Reynosa P, Reyes-Uribe E, Chavez-Ocaña S, Reyes-Hernandez O. et al. Extracellular vesicles from women with breast cancer promote an epithelial-mesenchymal transition-like process in mammary epithelial cells MCF10A. Tumor Biology. 2015;36:9649-59
99. Vejux A, Lizard G. Cytotoxic effects of oxysterols associated with human diseases: Induction of cell death (apoptosis and/or oncosis), oxidative and inflammatory activities, and phospholipidosis. Molecular aspects of medicine. 2009;30:153-70
100. Wu Q, Ishikawa T, Sirianni R, Tang H, McDonald JG, Yuhanna IS. et al. 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Reports. 2013;5:637-45
101. Dessi S, Batetta B, Pulisci D, Spano O, Cherchi R, Lanfranco G. et al. Altered pattern of lipid metabolism in patients with lung cancer. Oncology. 1992;49:436-41
102. Solheim S, Hutchinson SA, Lundanes E, Wilson SR, Thorne JL, Roberg-Larsen H. Fast liquid chromatography-mass spectrometry reveals side chain oxysterol heterogeneity in breast cancer tumour samples. J Steroid Biochem Mol Biol. 2019;192:105309
103. Kloudova-Spalenkova A, Ueng YF, Wei S, Kopeckova K, Peter Guengerich F, Soucek P. Plasma oxysterol levels in luminal subtype breast cancer patients are associated with clinical data. J Steroid Biochem Mol Biol. 2020;197:105566
104. Voisin M, de Medina P, Mallinger A, Dalenc F, Huc-Claustre E, Leignadier J. et al. Identification of a tumor-promoter cholesterol metabolite in human breast cancers acting through the glucocorticoid receptor. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:E9346-E55
105. Soucek P, Vrana D, Ueng YF, Wei S, Kozevnikovova R, Guengerich FP. Selective changes in cholesterol metabolite levels in plasma of breast cancer patients after tumor removal. Clinical chemistry and laboratory medicine. 2018;56:e78-e81
106. Kimbung S, Chang C-y, Bendahl P-O, Dubois L, Thompson JW, McDonnell DP. et al. Impact of 27-hydroxylase (CYP27A1) and 27-hydroxycholesterol in breast cancer. Endocrine-Related Cancer. 2017:339-49
107. Lappano R, Recchia AG, de Francesco EM, Angelone T, Cerra MC, Picard D. et al. The cholesterol metabolite 25-Hydroxycholesterol activates estrogen receptor α-Mediated signaling in cancer cells and in cardiomyocytes. PLoS ONE. 2011 6
108. Wang S, Yao Y, Rao C, Zheng G, Chen W. 25-HC decreases the sensitivity of human gastric cancer cells to 5-fluorouracil and promotes cells invasion via the TLR2/NF-κB signaling pathway. International Journal of Oncology. 2019;54:966-80
109. Kidani Y, Elsaesser H, Hock MB, Vergnes L, Williams KJ, Argus JP. et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat Immunol. 2013;14:489-99
110. Sallam T, Jones MC, Gilliland T, Zhang L, Wu X, Eskin A. et al. Feedback modulation of cholesterol metabolism by the lipid-responsive non-coding RNA LeXis. Nature. 2016;534:124-8
111. Favero GM, Paz JL, Otake AH, Maria DA, Caldini EG, de Medeiros RSS. et al. Cell internalization of 7-ketocholesterol-containing nanoemulsion through LDL receptor reduces melanoma growth in vitro and in vivo: A preliminary report. Oncotarget. 2018;9:14160-74
112. McDonnell DP, Park S, Goulet MT, Jasper J, Wardell SE, Chang CY. et al. Obesity, cholesterol metabolism, and breast cancer pathogenesis. Cancer research. 2014;74:4976-82
113. Silvente-Poirot S, Poirot M. Cholesterol epoxide hydrolase and cancer. Curr Opin Pharmacol. 2012;12:696-703
114. Zerbinati C, Iuliano L. Cholesterol and related sterols autoxidation. Free Radic Biol Med. 2017;111:151-5
115. Schroepfer GJ Jr. Oxysterols: modulators of cholesterol metabolism and other processes. Physiol Rev. 2000;80:361-554
116. Leignadier J, Dalenc F, Poirot M, Silvente-Poirot S. Improving the efficacy of hormone therapy in breast cancer: The role of cholesterol metabolism in SERM-mediated autophagy, cell differentiation and death. Biochemical pharmacology. 2017;144:18-28
117. Poirot M, Soules R, Mallinger A, Dalenc F, Silvente-Poirot S. Chemistry, biochemistry, metabolic fate and mechanism of action of 6-oxo-cholestan-3beta,5alpha-diol (OCDO), a tumor promoter and cholesterol metabolite. Biochimie. 2018;153:139-49
118. Aringer L, Eneroth P. Formation and metabolism in vitro of 5,6-epoxides of cholesterol and beta-sitosterol. J Lipid Res. 1974;15:389-98
119. Chapman K, Holmes M, Seckl J. 11beta-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. 2013;93:1139-206
120. Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM. et al. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol. 2013;380:41-54
121. Kricker A, Price M, Butow P, Goumas C, Armes JE, Armstrong BK. Effects of life event stress and social support on the odds of a >or= 2 cm breast cancer. Cancer Causes Control. 2009;20:437-47
122. Michael YL, Carlson NE, Chlebowski RT, Aickin M, Weihs KL, Ockene JK. et al. Influence of stressors on breast cancer incidence in the Women's Health Initiative. Health Psychol. 2009;28:137-46
123. Hermes GL, Delgado B, Tretiakova M, Cavigelli SA, Krausz T, Conzen SD. et al. Social isolation dysregulates endocrine and behavioral stress while increasing malignant burden of spontaneous mammary tumors. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:22393-8
124. Baschant U, Tuckermann J. The role of the glucocorticoid receptor in inflammation and immunity. J Steroid Biochem Mol Biol. 2010;120:69-75
125. Moran TJ, Gray S, Mikosz CA, Conzen SD. The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer research. 2000;60:867-72
126. Schorr K, Furth PA. Induction of bcl-xL expression in mammary epithelial cells is glucocorticoid-dependent but not signal transducer and activator of transcription 5-dependent. Cancer research. 2000;60:5950-3
127. Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD. Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem. 2001;276:16649-54
128. Lippman M, Bolan G, Huff K. The effects of glucocorticoids and progesterone on hormone-responsive human breast cancer in long-term tissue culture. Cancer research. 1976;36:4602-9
129. Osborne CK, Monaco ME, Kahn CR, Huff K, Bronzert D, Lippman ME. Direct inhibition of growth and antagonism of insulin action by glucocorticoids in human breast cancer cells in culture. Cancer research. 1979;39:2422-8
130. Huff KK, Knabbe C, Lindsey R, Kaufman D, Bronzert D, Lippman ME. et al. Multihormonal regulation of insulin-like growth factor-I-related protein in MCF-7 human breast cancer cells. Mol Endocrinol. 1988;2:200-8
131. Harris RA, Hiles ID, Page MJ, O'Hare MJ. The induction of apoptosis in human mammary luminal epithelial cells by expression of activated c-neu and its abrogation by glucocorticoids. Br J Cancer. 1995;72:386-92
132. Lillberg K, Verkasalo PK, Kaprio J, Teppo L, Helenius H, Koskenvuo M. Stressful life events and risk of breast cancer in 10,808 women: a cohort study. Am J Epidemiol. 2003;157:415-23
133. Reiche EM, Nunes SO, Morimoto HK. Stress, depression, the immune system, and cancer. Lancet Oncol. 2004;5:617-25
134. Madden E, Logue SE, Healy SJ, Manie S, Samali A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biology of the cell. 2019;111:1-17
135. Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer research. 2004;64:1757-64
136. Wu W, Pew T, Zou M, Pang D, Conzen SD. Glucocorticoid receptor-induced MAPK phosphatase-1 (MPK-1) expression inhibits paclitaxel-associated MAPK activation and contributes to breast cancer cell survival. J Biol Chem. 2005;280:4117-24
137. Messmer UK, Winkel G, Briner VA, Pfeilschifter J. Suppression of apoptosis by glucocorticoids in glomerular endothelial cells: effects on proapoptotic pathways. Br J Pharmacol. 2000;129:1673-83
138. De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488-522
139. Lin A, Karin M. NF-kappaB in cancer: a marked target. Semin Cancer Biol. 2003;13:107-14
140. Herr I, Gassler N, Friess H, Buchler MW. Regulation of differential pro- and anti-apoptotic signaling by glucocorticoids. Apoptosis. 2007;12:271-91
141. Feng Z, Marti A, Jehn B, Altermatt HJ, Chicaiza G, Jaggi R. Glucocorticoid and progesterone inhibit involution and programmed cell death in the mouse mammary gland. J Cell Biol. 1995;131:1095-103
142. Vilasco M, Communal L, Mourra N, Courtin A, Forgez P, Gompel A. Glucocorticoid receptor and breast cancer. Breast Cancer Res Treat. 2011;130:1-10
143. Sevanian A, McLeod LL. Catalytic properties and inhibition of hepatic cholesterol-epoxide hydrolase. J Biol Chem. 1986;261:54-9
144. Nashed NT, Michaud DP, Levin W, Jerina DM. Properties of liver microsomal cholesterol 5,6-oxide hydrolase. Arch Biochem Biophys. 1985;241:149-62
145. Cheng YW, Kang JJ, Shih YL, Lo YL, Wang CF. Cholesterol-3-beta, 5-alpha, 6-beta-triol induced genotoxicity through reactive oxygen species formation. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2005;43:617-22
146. de Medina P, Paillasse MR, Segala G, Poirot M, Silvente-Poirot S. Identification and pharmacological characterization of cholesterol-5,6-epoxide hydrolase as a target for tamoxifen and AEBS ligands. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:13520-5
147. de Medina P, Paillasse MR, Segala G, Voisin M, Mhamdi L, Dalenc F. et al. Dendrogenin A arises from cholesterol and histamine metabolism and shows cell differentiation and anti-tumour properties. Nat Commun. 2013;4:1840
148. Payre B, de Medina P, Boubekeur N, Mhamdi L, Bertrand-Michel J, Terce F. et al. Microsomal antiestrogen-binding site ligands induce growth control and differentiation of human breast cancer cells through the modulation of cholesterol metabolism. Mol Cancer Ther. 2008;7:3707-18
149. de Medina P, Payre B, Boubekeur N, Bertrand-Michel J, Terce F, Silvente-Poirot S. et al. Ligands of the antiestrogen-binding site induce active cell death and autophagy in human breast cancer cells through the modulation of cholesterol metabolism. Cell Death Differ. 2009;16:1372-84
150. Kotsopoulos J. BRCA Mutations and Breast Cancer Prevention. Cancers. 2018;10:524 -
151. Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S. et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109
152. Jakobsson T, Treuter E, Gustafsson JA, Steffensen KR. Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol Sci. 2012;33:394-404
153. Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129:2475-81
154. Viennois E, Mouzat K, Dufour J, Morel L, Lobaccaro JM, Baron S. Selective liver X receptor modulators (SLiMs): what use in human health? Mol Cell Endocrinol. 2012;351:129-41
155. Segala G, David M, de Medina P, Poirot MC, Serhan N, Vergez F. et al. Dendrogenin A drives LXR to trigger lethal autophagy in cancers. Nat Commun. 2017;8:1903
156. Silvente-Poirot S, Segala G, Poirot MC, Poirot M. Ligand-dependent transcriptional induction of lethal autophagy: A new perspective for cancer treatment. Autophagy. 2018;14:555-7
157. Poirot M, Silvente-Poirot S. The tumor-suppressor cholesterol metabolite, dendrogenin A, is a new class of LXR modulator activating lethal autophagy in cancers. Biochemical pharmacology. 2018;153:75-81
158. Chimento A, Casaburi I, Avena P, Trotta F, De Luca A, Rago V. et al. Cholesterol and Its Metabolites in Tumor Growth: Therapeutic Potential of Statins in Cancer Treatment. Front Endocrinol (Lausanne). 2018;9:807
159. Ding X, Zhang W, Li S, Yang H. The role of cholesterol metabolism in cancer. Am J Cancer Res. 2019;9:219-27
160. Wang Y, Liu C, Hu L. Cholesterol regulates cell proliferation and apoptosis of colorectal cancer by modulating miR-33a-PIM3 pathway. Biochemical and biophysical research communications. 2019;511:685-92
161. Liu Z, Liu X, Liu S, Cao Q. Cholesterol promotes the migration and invasion of renal carcinoma cells by regulating the KLF5/miR-27a/FBXW7 pathway. Biochemical and biophysical research communications. 2018;502:69-75
162. Li D, Cheng M, Niu Y, Chi X, Liu X, Fan J. et al. Identification of a novel human long non-coding RNA that regulates hepatic lipid metabolism by inhibiting SREBP-1c. International Journal of Biological Sciences. 2017;13:349-57
163. Van Grembergen O, Bizet M, De Bony EJ, Calonne E, Putmans P, Brohée S. et al. Portraying breast cancers with long noncoding RNAs. Science Advances. 2016 2
164. Pang JL, Wang JW, Hu PY, Jiang JS, Yu C. HOTAIR alleviates ox-LDL-induced inflammatory response in Raw264.7 cells via inhibiting NF-κB pathway. European review for medical and pharmacological sciences. 2018;22:6991-8
165. Han Y, Ma J, Wang J, Wang L. Silencing of H19 inhibits the adipogenesis and inflammation response in ox-LDL-treated Raw264.7 cells by up-regulating miR-130b. Molecular Immunology. 2018;93:107-14
166. Mutemberezi V, Guillemot-Legris O, Muccioli GG. Oxysterols: From cholesterol metabolites to key mediators. Prog Lipid Res. 2016;64:152-69
167. Hannedouche S, Zhang J, Yi T, Shen W, Nguyen D, Pereira JP. et al. Oxysterols direct immune cell migration via EBI2. Nature. 2011;475:524-7
168. Theofilopoulos S, Arenas E. Liver X receptors and cholesterol metabolism: role in ventral midbrain development and neurodegeneration. F1000Prime Rep. 2015;7:37
169. Plygawko AT, Kan S, Campbell K. Epithelial-mesenchymal plasticity: emerging parallels between tissue morphogenesis and cancer metastasis. Philos Trans R Soc Lond B Biol Sci. 2020;375:20200087
170. Tapia-Vieyra JV, Delgado-Coello B, Mas-Oliva J. Atherosclerosis and Cancer; A Resemblance with Far-reaching Implications. Archives of medical research. 2017;48:12-26
Author contact
![]() Corresponding author: Departamento de Bioquímica, Facultad de Medicina Mexicali, Universidad Autónoma de Baja California, 21000 Mexicali, México. Tel.: +52-1-686-557-1622 (ext. 121); Fax: +52-1-686-557-1622 (ext. 109). E-mail: vgarcia62edu.mx (V. García-González).
Corresponding author: Departamento de Bioquímica, Facultad de Medicina Mexicali, Universidad Autónoma de Baja California, 21000 Mexicali, México. Tel.: +52-1-686-557-1622 (ext. 121); Fax: +52-1-686-557-1622 (ext. 109). E-mail: vgarcia62edu.mx (V. García-González).