Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(15):4463-4477. doi:10.7150/jca.60008 This issue Cite
Research Paper
MST4 negatively regulates the EMT, invasion and metastasis of HCC cells by inactivating PI3K/AKT/Snail1 axis
Mei-Juan Dian1,2*, Jing Li3*, Xiao-Ling Zhang4*, Zi-Jian Li1, Ying Zhou1, Wei Zhou1, Qiu-Ling Zhong5, Wen-Qian Pang1, Xiao-Lin Lin1, Tao Liu1, Yi-An Liu1, Yong-Long Li2, Liu-Xin Han6, Wen-Tao Zhao7, Jun-Shuang Jia1, Sheng-Jun Xiao8, Dong Xiao1,2 ![]() , Jia-Wei Xia6
, Jia-Wei Xia6 ![]() , Wei-Chao Hao9
, Wei-Chao Hao9 ![]()
1. Cancer Research Institute, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
2. Institute of Comparative Medicine & Laboratory Animal Center, Southern Medical University, Guangzhou 510515, China.
3. Radiotherapy Center, the First People's Hospital of Chenzhou, Xiangnan University, Chenzhou 423000, China.
4. Department of Physiology, Faculty of Basic Medical Sciences, Guilin Medical University, Guilin 541004, China.
5. State Key Laboratory of Organ Failure Research, Key Laboratory of Mental Health of the Ministry of Education, Guangdong-Hong Kong-Macao Greater Bay Area Center for Brain Science and Brain-Inspired Intelligence, Guangdong Province Key Laboratory of Psychiatric Disorders, Collaborative Innovation Center for Brain Science, Department of Neurobiology, School of Basic Medical Sciences, Southern Medical University, Guangzhou 510515, China.
6. The third people's hospital of Kunming (The Sixth Affiliated Hospital of Dali University), Kunming 650041, China.
7. Department of Gastrointestinal Oncology, The Third Affiliated Hospital of Kunming Medical University (Yunnan Cancer Hospital, Yunnan Cancer Center), Kunming 650118, China.
8. Department of Pathology, the Second Affiliated Hospital, Guilin Medical University, Guilin 541199, China.
9. Department of Oncology, The First Affiliation Hospital of Guangdong Pharmaceutical University, Guangzhou 510062, China.
*These authors contributed equally to this work.
Received 2021-3-2; Accepted 2021-5-8; Published 2021-5-27
Abstract

Background: Hepatocellular carcinoma (HCC) is one of the most common cancers worldwide and has a poor prognosis due to the high incidence of invasion and metastasis-related progression. However, the underlying mechanism remains elusive, and valuable biomarkers for predicting invasion, metastasis, and poor prognosis of HCC patients are still lacking.
Methods: Immunohistochemistry (IHC) was performed on HCC tissues (n = 325), and the correlations between MST4 expression of the clinical HCC tissues, the clinicopathologic features, and survival were further evaluated. The effects of MST4 on HCC cell migratory and invasive properties in vitro were evaluated by Transwell and Boyden assays. The intrahepatic metastasis mouse model was established to evaluate the HCC metastasis in vivo. The PI3K inhibitor, LY294002, and a specific siRNA against Snail1 were used to investigate the roles of PI3K/AKT pathway and Snail1 in MST4-regulated EMT, migration, and invasion of HCC cells, respectively.
Results: In this study, by comprehensively analyzing our clinical data, we discovered that low MST4 expression is highly associated with the advanced progression of HCC and serves as a prognostic biomarker for HCC patients of clinical-stage III-IV. Functional studies indicate that MST4 inactivation induces epithelial-to-mesenchymal transition (EMT) of HCC cells, promotes their migratory and invasive potential in vitro, and facilitates their intrahepatic metastasis in vivo, whereas MST4 overexpression exhibits the opposite phenotypes. Mechanistically, MST4 inactivation elevates the expression and nuclear translocation of Snail1, a key EMT transcription factor (EMT-TF), through the PI3K/AKT signaling pathway, thus inducing the EMT phenotype of HCC cells, and enhancing their invasive and metastatic potential. Moreover, a negative correlation between MST4 and p-AKT, Snail1, and Ki67 and a positive correlation between MST4 and E-cadherin were determined in clinical HCC samples.
Conclusions: Our findings indicate that MST4 suppresses EMT, invasion, and metastasis of HCC cells by modulating the PI3K/AKT/Snail1 axis, suggesting that MST4 may be a potential prognostic biomarker for aggressive and metastatic HCC.
Keywords: MST4, Hepatocellular carcinoma, Metastasis, Prognosis, EMT, PI3K/AKT, Snail1
Introduction
Hepatocellular carcinoma (HCC) is ranked as the sixth most common cancer and the fourth leading cause of cancer death [1]. Due to the metastasis-related high intrahepatic recurrence rate, the long-term survival of patients with HCC is very poor [2]. However, no effective treatment options to reduce the risk of metastasis-associated intrahepatic recurrence are available [3]. Surgical resection and ablation are the frontline treatment choices for patients with early HCC [4, 5]. Unfortunately, 70% of the cases would have tumor recurrence within the remaining liver at 5 years [6]. Liver transplantation is an effective treatment to prevent intrahepatic recurrence, but the source of liver donors is limited [1, 3, 6]. Sorafenib, a targeted therapeutic drug, is recommended for patients with advanced or intermediate-stage HCC [7]. It is a multi-kinase inhibitor that blocks Raf signaling, VEGF, PDGF, and c-Kit, and has anti-proliferative and anti-angiogenic effects [8-11]. Although it has been shown to be beneficial for HCC patients' survival [12, 13], it has also been proven to not prevent HCC tumor intrahepatic recurrence [14]. Therefore, it is urgent to understand the cellular and molecular mechanism of HCC metastasis and to find potential molecules that can be used to predict the aggressive invasion, metastasis, and poor prognosis of HCC, so as to provide opportunities to prevent intrahepatic recurrence of this malignancy.
Metastasis is a complex multi-step process that involves the dissemination of cancer cells from the primary tumor; intravasation into the circulatory system; and subsequent colonization in the distant tissue to form metastatic foci [15]. Intrahepatic metastasis (IM) is the most common hematogenous metastasis of HCC, which is usually caused by tumor cell dispersal via the portal vein [16]. Epithelial-mesenchymal transition (EMT) is a process of cell transdifferentiation in which cells lose their epithelial characteristics and acquire mesenchymal phenotype [17, 18]. For HCC progression, EMT is usually a crucial prerequisite for its invasive and metastatic potential [17]. Therefore, investigation of potential key molecules involved in EMT properties may contribute to develop new therapeutic strategies against HCC invasion and metastasis.
Mammalian sterile-20-like kinase 4 (MST4) is a member of the germinal center kinase (GCK) group III family of kinases. Studies have shown that MST4 affects a variety of physiological functions, including Golgi reorientation, autophagy, intestinal epithelial cell brush border formation, gastric parietal cell polarized epithelial secretion, and inflammatory response [19-22]. The functional role of MST4 in cancers has not been fully elucidated and remains controversial. It is reported that MST4 facilitates the growth of prostate cancer, pancreatic cancer, and glioblastoma [23, 24]. In breast cancer and gastric cancer, the expression of MST4 is associated with metastasis and poor prognosis [25, 26]. Another study showed that MST4 can inhibit gastric carcinogenesis by phosphorylating and inactivating YAP oncoprotein [27]. In choriocarcinoma, MST4 is shown to inhibit cell migration and invasion by suppressing the TGF-β1-induced EMT program [28]. In our recent work, we found that MST4 is down-regulated in HCC, and that it suppresses HCC cell proliferation and cell cycle progression by inactivating PI3K/AKT pathway [29]. However, as the major cause of HCC progression and poor prognosis, the role of MST4 in the regulation of invasion and metastasis of HCC is worth further exploration.
In this study, we found that the expression of MST4 is negatively correlated with the progression and poor prognosis of HCC. Functional inactivation of MST4 can promote the migration, invasion, and metastasis of HCC cells, whereas the overexpression of MST4 has the opposite effect. Mechanistically, we found that MST4 inhibits the motility and invasive potential of HCC cells by inactivating PI3K/AKT signaling pathway, which leads to the decrease of Snail1 expression, thus blocking the EMT phenotype. Collectively, our results revealed that MST4 is an important prognostic biomarker for aggressive invasion and IM in patients with HCC, and provides new insights into the role and molecular mechanism of MST4 in HCC progression.
Materials and methods
Clinical specimens
A total of 335 formalin-fixed paraffin-embedded HCC biopsies were obtained from the Department of Pathology, the Second Affiliated Hospital of Guilin Medical College, China. 85 HCC patients who had complete follow-up data were enrolled for survival analysis. All HCC patients enrolled in this study were histopathologically and clinically diagnosed. None of these patients received preoperative radiotherapy or chemotherapy. The clinical information of 10 SHC patients is presented in Table S1. Retrieval of tissue and clinical data was performed according to the regulations of the institutional review boards (IRB) of the Second Affiliated Hospital of Guilin Medical University and data safety laws with specific regard to ethical standards and patient confidentiality. The patients whose tissues were used provided written informed consent. Ethical approval was given by the Medical Ethics Committee of Southern Medical University. The study is compliant with all relevant ethical regulations involving human participants.
Histological analysis and immunohistochemistry (IHC)
Histological analysis and immunohistochemical staining were performed as described previously [30]. Formalin-fixed, paraffin-embedded tumor tissues from patients with HCC were collected. The paraffin sections were deparaffinized in xylene and rehydrated in ethanol with decreasing concentrations. The antigen retrieval was performed in a steam pressure cooker with citrate buffered saline (pH 6.0) for 3 minutes. Endogenous peroxidase was blocked by incubation with 3% hydrogen peroxide for 30 minutes. After incubation with 5% normal goat serum for 30 minutes at room temperature to block unspecific labeling, sections were incubated overnight in humidified chamber at 4 °C with primary antibodies (Table S2). The slides were then stained with a corresponding secondary antibody at 37 °C for 20 minutes. Finally, the sections were stained with diaminobenzidine (DAB) and counterstained with hematoxylin. Histopathology and immunohistochemistry analyses were performed independently by two pathologists without knowledge of the backgrounds of the patients. Staining intensity and percentage of staining-positive cells were evaluated for the IHC scores. Staining intensity was classified as 0 (negative), 1 (weak), 2 (moderate) and 3 (strong). Percentage of stained cells were classified as 0 (<5 %), 1 (≥ 5%, <25 %), 2 (≥25, <75%) and 3 (≥75%). The final IHC score is score of intensity multiplied by score of percentage. The scores 0, 1, 2, 3, 4 were defined as low, and the scores 6 and 9 were defined as high. Slides with conflicting evaluations were reassessed, and a consensus was reached.
Cell lines and cell culture
HCC cell lines (Bel-7402, Bel-7404 and Huh7) and HEK-293T cells were purchased from the Cell Bank of Shanghai Institutes for Biological Sciences (SIBS), Chinese Academy of Sciences (CAS) (Shanghai, China) or ATCC. All cells were cultured in Dulbecco's modified Eagle medium (DMEM) (10-013-CVR, Corning) supplemented with 10% fetal bovine serum (ST30-3302, PAN) at 37 °C under 5% CO2 in a humidified chamber.
Plasmid constructs and interfering RNAs
The pcDNA3.1-MST4-WT and pcDNA3.1-MST4-T178A (dnMST4: a dominant negative mutant of MST4-WT) were kindly provided by Prof. Hans Clevers [19]. The lentiviral pLV-MST4 and pLV-dnMST4 plasmids were generated by subcloning the MST4-WT fragments (from pcDNA3.1-MST4-WT) and MST4-T178A fragments (from pcDNA3.1-MST4-T178A) into pCDH-EF1-GFP-Puro (System Biosciences, Palo Alto, CA, #CD550A-1) lentiviral vectors. The lentiviral packaging plasmids psPAX2 and pMD2.G were kindly provided by Prof. Didier Trono (University of Geneva, Geneva, Switzerland). For RNA interference in HCC cells, specific siRNAs were used for Snail1 (Snail1 siRNA (antisense): UGGCACUGGUACUUCUUGACAUCUGTT.
Virus packaging and infection
The pLV-MST4, pLV-dnMST4 or pCDH-EF1-GFP-puro empty vector was respectively co-transfected into HEK-293T cells with lentiviral packaging plasmids (psPAX2 and pMD2.G) to produce the lentiviruses. The viral supernatants were harvested at 72 hours after transfection, and subsequently used to infect the indicated cells to generate vector-, MST4- or dnMST4-expressing cancer cell lines, respectively.
Western blot assay
Western blotting was performed as previously described [29]. Briefly, the same amount of total denatured protein (20 μg) was isolated by 10% SDS-PAGE and transferred to PVDF membranes (P2120-2, Millipore) using BIO-RAD system. After blocking for nonspecific binding using 5% skim milk, the membranes were then incubated with primary antibodies (Table S3) overnight at 4 °C, followed by incubation with an HRP-labeled goat anti-rabbit IgG or anti-mouse IgG for one hour at room temperature. After washing four times in TBST, protein bands were visualized on a chemiluminescence imaging system (ChemiDoc XRS+, Bio-Rad).
Transwell migration and invasion assay
Approximately 1×105 HCC cells were suspended in serum-free medium and added in either top-chambers that were not coated with Matrigel for migration assay or Matrigel-coated chambers with 8 μm pores (3422, Corning Costar) for invasion assay. The culture medium containing 10% FBS was added to the bottom chamber. After incubation at 37 °C for eighteen hours, HCC cells may migrate to the bottom chamber through the membrane. Membranes were washed three times with 1×PBS, fixed in 4% paraformaldehyde, and stained with crystal violet staining solution (1.09218, Sigma). The number of migrated or invaded cells through the membrane was separately counted under the microscope from 10 random fields of view.
Sphere formation assay
The HCC cells (1×105) were cultured in sphere medium and plated in 6-well ultra-low cluster plates. The sphere medium was composed of DMEM:F-12 serum-free medium supplemented with 20 ng/ml rhEGF (Peprotech), 20 ng/ml rbFGF (Peprotech), 1% B-27 (17504044, Gibco), and 2 mg/ml heparin (H3149, Sigma) [31].The HCC cells were cultured in sphere medium at 37 °C in 5% CO2, and counted floating spheres after ten days. The representative images were taken with Nikon microscopes. Every experiment was carried out multiple independent experiments.
Immunofluorescence (IF) staining
IF staining was performed according to the protocol of a standard method described previously [32]. The slides were counterstained with DAPI (Sigma) for five minutes to visualize the nuclei and imaged with a confocal laser-scanning microscope (Nikon A1). The primary antibodies used for IF staining were listed in Table S2.
PI3K inhibitor treatment
For long-term inhibitor treatment, 10 μM PI3K inhibitor LY294002 (Catalog # HY-10108; MedChemExpress (MCE) was added in the cultured cells every 3 days due to the half-life of LY294002.
In vivo metastasis analysis in nude mice
BALB/c nude mice (3-4 weeks) were purchased from the Animal Center of Southern Medical University and raised in a pathogen-free environment. For in vivo metastasis assays, the vector- or dnMST4-expressing Bel-7402 cells (1.0×106) were subcapsularly transplanted into the liver of nude mice, respectively. All animals were sacrificed after transplantation, and liver were collected for metastasis analysis. This study was carried out in accordance with the Guide for the Care and Use of Laboratory Animals of the Southern Medical University. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Southern Medical University. Subcapsular cell transplantation was performed under pentobarbital sodium anesthesia to minimize mice suffering.
Statistical analysis
The data were presented as mean ±SEM from three independent experiments. Statistical analyses were conducted using the SPSS 20.0 software package and Graphpad 8.0 software. A two-tailed Student's t test was used for comparisons of two independent groups. The χ2 test was used to analyze the association between clinicopathological characteristics and MST4 expression. Overall survival (OS) was measured from the onset of treatment to the date of death or the survival status at the last date of follow-up. OS probabilities were estimated by the Kaplan-Meier method and the significance of differences was assessed by the log-rank test. Values are statistically significant at *P<0.05, **P<0.01 and ***P<0.001.
Results
Low MST4 expression correlates with HCC progression and poor prognosis
Our recent work showed the expression of MST4 in HCC is lower than that in paracancerous liver tissues, and MST4 has an anti-tumorigenic effect on HCC [29]. For patients, HCC with high invasiveness and metastasis usually leads to rapid progression and indicates a poor prognosis. Higher AJCC T-stage based on larger tumor size, microvascular invasion, portal or hepatic vein invasion, and adjacent organ invasion. N-stage and M-stage define regional lymph nodes and distant metastasis, respectively. To determine the clinical relevance of MST4 in HCC progression, we examined the expression of MST4 in 325 paraffin-embedded human HCC specimens by immunohistochemical analysis. Our data showed the frequency of MST4 expression was significantly higher in T1-2 stage (99/225, 44%) than in T3-4 stage (28/100, 28%), and in N0 stage (124/305, 41%) than in N1 stage (3/20, 15%) of HCC. A similar trend was found in comparison MST4 expression in M0 and M1 stages (despite this not being statistically different) (Figure 1A, 1B and Table 1).
Low MST4 expression correlates with HCC progression and poor prognosis. (A) Representative images of MST4 immunohistochemical (IHC) staining in HCC specimens (n=325) with different TNM stages. (B) The percentages of cases with high or low expression of MST4 according to different TNM stages of HCC. (C) Representative images of MST4 IHC staining in HCC specimens (n=325) with different clinical stages and histological grades. (D) The percentages of cases with high or low expression of MST4 according to different clinical stages and histological grades of HCC. (E) Kaplan-Meier survival curves of overall survival (OS) in 85 HCC patients with high MST4 expression (red line) and low MST4 expression (green line) (Log-rank test; P=0.021). (F) Univariate and multivariate analyses of prognostic indicators in HCC (n=85). (G) The correlation of MST4 expression with survival time for HCC patients stratified by clinical stage I-II (upper panel) (Log-rank test; P=0.725) and clinical stage III-IV (lower panel) (Log-rank test; P=0.005). Data are mean ± SEM (*P< 0.05, **P <0.01, ***P <0.001).
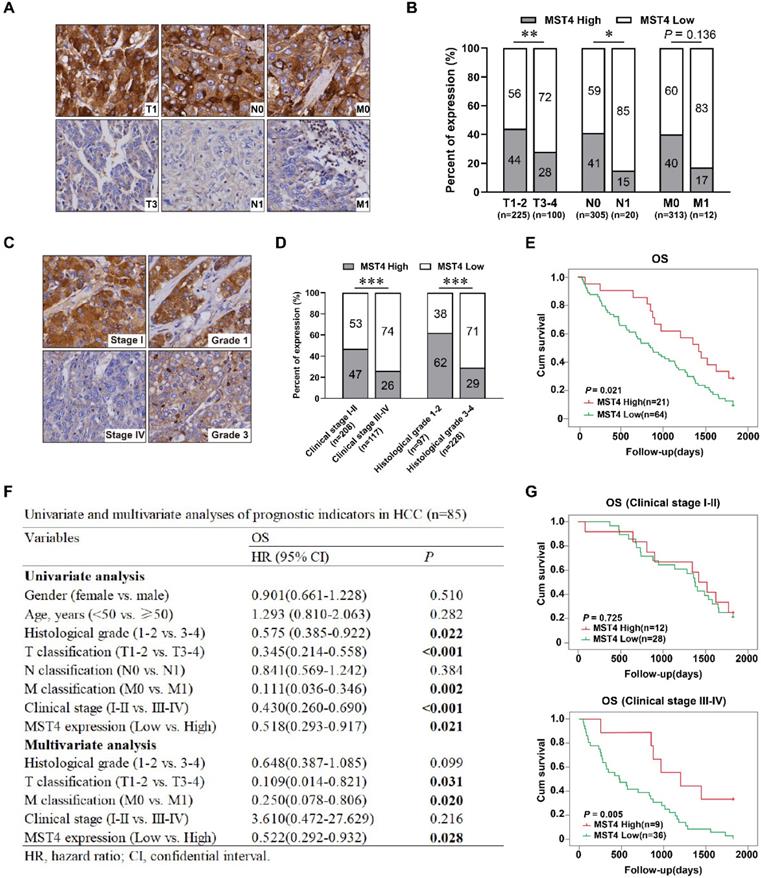
Correlation between the clinicopathological features and MST4 expression in 325 HCC tissue specimens
| Variables | Case No. | MST4 expression | χ2 | P | |
|---|---|---|---|---|---|
| (n) | High (n, %) | Low (n, %) | |||
| Gender | |||||
| Female | 60 | 20 (33.3) | 40 (66.7) | 0.745 | 0.38 |
| Male | 265 | 107 (40.4) | 158 (59.6) | ||
| Age (years) | |||||
| <50 | 158 | 57 (36.1) | 101 (63.9) | 0.931 | 0.307 |
| ≥50 | 167 | 70 (41.9) | 97 (58.1) | ||
| Histological grade | |||||
| I-II | 97 | 60 (61.9) | 37 (38.1) | 28.79 | <0.001 |
| III-IV | 228 | 67 (29.4) | 161 (70.6) | ||
| T classification | |||||
| T1-T2 | 225 | 99 (44.0) | 126 (56.0) | 6.788 | 0.007 |
| T3-T4 | 100 | 28 (28.0) | 72 (72.0) | ||
| N classification | |||||
| N0 | 305 | 124 (40.7) | 181 (59.3) | 5.189 | 0.031 |
| N1 | 20 | 3 (15.0) | 17 (85.0) | ||
| M classification | |||||
| M0 | 313 | 125 (39.9) | 188 (60.1) | 2.629 | 0.136 |
| M1 | 12 | 2 (16.7) | 10 (83.3) | ||
| Clinical stage | |||||
| I-II | 208 | 97 (46.6) | 111 (53.4) | 13 | <0.001 |
| III-IV | 117 | 30 (25.6) | 87 (74.4) | ||
T: tumor size; N: lymph node metastasis; M: distant metastasis.
The clinical stage according to the AJCC TNM staging system is of great significance for predicting the prognosis of HCC patients and determine the optimal clinical treatment. The histological grade is assigned by the degrees of HCC differentiation (according to the Edmondson Steiner grading system), and serves as an independent prognostic indicator for HCC patients. Our study demonstrated a significantly higher frequency of MST4 expression in HCC patients with low clinical stage (stage I-II; 97/208, 47%) than those with high clinical stage (stage III-IV; 30/117, 26%), and in patients with well or moderately differentiated HCC (histological grade 1-2; 60/97, 62%) than those with poorly differentiated HCC (histological grade 3-4; 67/228; 29%) (Figure 1C, 1D and Table 1), implicating MST4 may serve as a prognostic indicator for HCC.
Further we studied the prognostic value of MST4 based on clinical follow-up data. Kaplan-Meier survival analysis showed that HCC patients with high MST4 expression has a notably longer overall survival (OS) than those with low MST4 expression (P=0.021) (Figure 1E). Univariate and multivariate analyses revealed that MST4 expression is an independent prognostic factor for OS (P = 0.028, HR = 0.522) in HCC patients (Figure 1F). Interestingly, for patients with stage III-IV, the OS of the MST4 high group was significantly longer than that of the MST4 low group (P=0.005) (Figure 1G), whereas for patients with stage I-II, the differences in OS between the high group and the low group were not statistically significant (Figure 1G). Overall, these results suggested that low MST4 expression is closely correlated with the progression and poor prognosis of HCC, and can be used as a new prognostic biomarker for patients with advanced HCC (stage III-IV).
MST4 inactivation promotes the migration, invasion, and metastasis of HCC cells
To determine the role of MST4 in the invasion and metastasis of HCC cells, we performed Transwell migration assays using the dominant-negative mutant MST4 (dnMST4) and the wild-type MST4 stably transfected HCC cells. Our data showed that dnMST4 expression significantly increased the cellular motility of HCC cells (Figure 2A and 2B), whereas forced MST4 expression lowered this ability (Figure 2C and 2D). The invasive potential of tumor cells is a prerequisite for metastasis. Once tumor cells penetrate the basement membrane or extracellular matrix, they journey to new locations directly or through the circulatory system [17]. The Boyden assays showed that dnMST4 expression significantly enhanced the invasive potential of HCC cells (Figure 2A and 2B), whereas forced MST4 expression reduced this potential (Figure 2C and 2D).
MST4 negatively regulates HCC cell migration, invasion, and intrahepatic metastasis (IM) in vitro and in vivo. (A) Representative photographs of the migratory and invasive properties of dnMST4-expressing Bel-7402 and Bel-7404 cells. (B) Quantification of migratory and invasive properties of dnMST4-expressing Bel-7402 and Bel-7404 cells. (C) Representative images of the migratory and invasive properties of MST4-overexpressing Bel-7402 and Bel-7404 cells. (D) Quantification of migratory and invasive properties of MST4-overexpressing HCC cells. (E) Representative pictures of the livers from nude mice after subcapsular liver transplantation of vector-expressing (LV-con) or dnMST4-expressing (LV-dnMST4) Bel-7402 cells (7 mice/group). Photographs were recorded under natural light (upper panel) or under fluorescent stereomicroscope (lower panel). (F) The diameter of the primary tumors in the livers of nude mice. (G) The number of IM nodules in the livers of nude mice. (H) Representative H&E staining of liver tissue samples from nude mice after subcapsular liver transplantation of vector- or dnMST4-expressing Bel-7402 cells. (a and b) primary tumor, (c) satellite nodules near the primary tumor lesion, (d) vascular invasion, (e) lymph node metastasis, and (f) peripheral nerve invasion. P: primary tumor; M: metastatic nodule. (I) Survival curve of nude mice transplanted with vector- (bule line) or dnMST4-expressing (red line) Bel-7402 cells into hepatic subcapsular space. Data are mean ± SEM (*P< 0.05, **P <0.01, ***P <0.001).
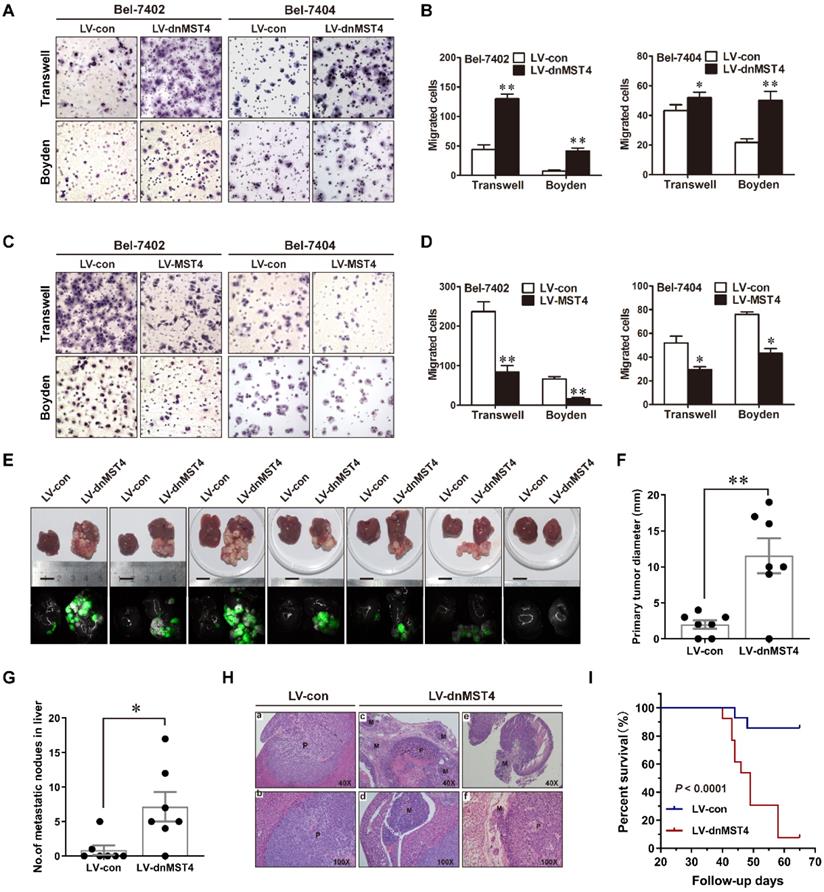
To further explore the effect of MST4 on the HCC cell metastatic potential in vivo, we established orthotopically metastatic models of human HCC in nude mice implanted of dnMST4 and control Bel-7402 cells, respectively (Figure 2E). The orthotopically implanted tumor in the dnMST4 group showed a remarkable larger volume compared to that of the control group (Figure 2F), demonstrating that functional inactivation of MST4 effectively promoted tumor growth. As expected, the number of metastatic nodules in the liver were dramatically increased in the dnMST4 group compared with the control group after in situ growth for 6 weeks (Figure 2G). Additionally, hematoxylin and eosin (HE) staining indicated satellite intrahepatic metastatic nodules, tumor thrombus, lymph node involvement, and perineural invasion are detected in the dnMST4 group (Figure 2H), indicating that MST4 functionally loss strengthened the intrahepatic metastatic potential of HCC cells. Given that metastasis is the main cause of death in cancer patients, we asked whether MST4 functionally loss could have an effect on overall survival (OS). The follow-up data revealed that the OS of mice bearing dnMST4 Bel-7402 cells (n=13) was significantly shorter than those bearing control cells (n=14) (Figure 2I), further suggesting the prognostic value of MST4 for HCC. Collectively, these observations indicate that MST4 inactivation promotes the migration, invasion, and metastasis of HCC cells, whereas MST4 overexpression exhibits the opposite phenotypes.
MST4 repressed the EMT phenotype of HCC cells
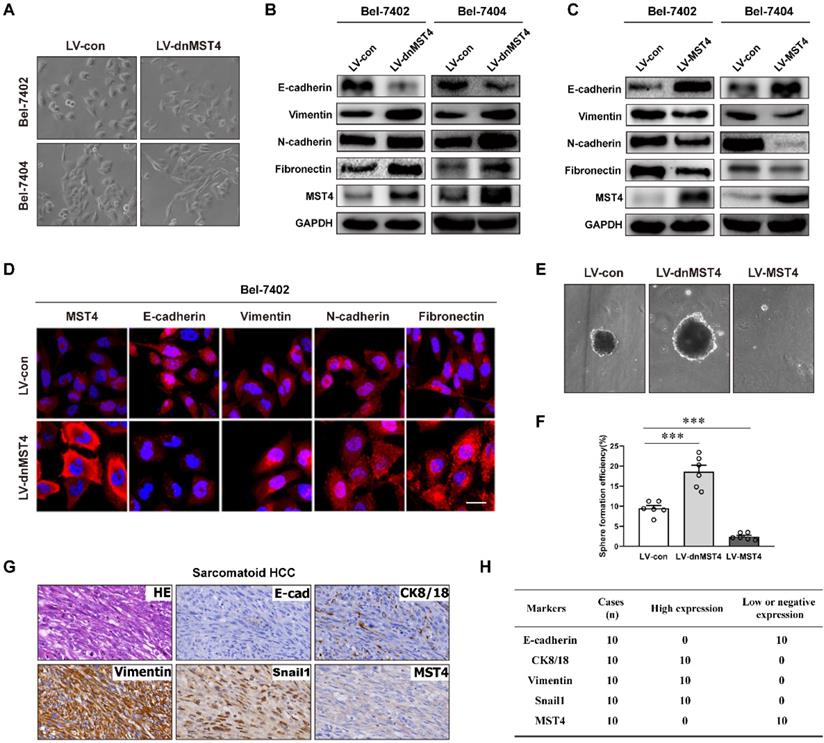
It is widely embraced the existence of EMT is essential to enhance tumor progression and metastasis [15, 17, 33-36]. Intriguingly, we noticed that the expression of dnMST4 causes significant morphological changes from cobblestone-like to the spindle-shaped appearance in both Bel-7402 and Bel-7404 cells (Figure 3A), indicating that MST4 functionally loss may facilitate the EMT process of HCC cells. EMT markers were then probed by western blot, including epithelial protein E-cadherin and mesenchymal markers N-cadherin, vimentin, and fibronectin. The dnMST4-expression HCC cells showed a typical EMT phenotype, including downregulation of E-cadherin and upregulation of N-cadherin, vimentin, and fibronectin (Figure 3B). Correspondingly, the MST4 overexpression leads to a reversed EMT phenotype, including the increase of E‑cadherin and the decreases of N-cadherin, vimentin, and fibronectin (Figure 3C). The phenotype was further confirmed by immunofluorescent staining (Figure 3D). It is known that cancer cells undergo EMT can also acquire stemness properties [37], so we investigated whether the MST4 functionally inactivation promotes the stemness of HCC cells. As expected, the expression of dnMST4 significantly increased the sphere-forming efficiency (SFE), which can be used to evaluate the population of cancer stem-like cells (CSLCs) [38], whereas the forced expression of MST4 reduced that (Figure 3E and 3F). Sarcomatoid hepatocellular carcinoma (SHC) is a rare type of HCC with positive expression of vimentin and predominantly composed of anaplastic spindle-like cells [39-41]. CK8 positive staining is used for differentiating SHC from real sarcomas [40]. EMT is implicated in the development of this cancer [42]. Immunohistochemical analysis showed that the expression of MST4 was negative or low in all 10 cases of SHC (Figure 3G and 3 H), which indicated a negative correlation between the EMT phenotype and MST4 expression in HCC cells. Collectively, our results supported the inhibitory potential of MST4 expression against the EMT phenotype of HCC cells.
MST4 represses EMT in HCC cells. (A) Phase-contrast micrograph of the morphology of vector- or dnMST4-expressing Bel-7402 cells and Bel-7404 cells. (B) Western blot analysis for the detection of the indicated protein expression in dnMST4-expressing Bel-7402 cells and Bel-7404 cells. (C) Western blot analysis for the detection of the indicated protein expression in MST4-overexpressing Bel-7402 cells and Bel-7404 cells. (D) Immunofluorescence images showing the expression of EMT marker proteins of dnMST4-expressing Bel-7402 cells. Blue: DAPI. Scale bar, 20 µm. (E) Representative pictures of tumorsphere in dnMST4-expressing and MST4-overexpressing Huh7 cells. (F) The number of tumorspheres in dnMST4-expressing and MST4-overexpressing Huh7 cells. (G) Representative pictures of H&E and IHC staining for EMT markers in SHC specimens. (H) The number of cases with high or low indicated protein expression in SHC specimens. Data are mean ± SEM (*P< 0.05, **P <0.01, ***P <0.001).
MST4 inactivation induces the EMT phenotype of HCC cells, and promotes their invasive potential through activation of PI3K/AKT/Snail1 signaling pathway
Next, we investigated the mechanism by which MST4 suppressed the EMT phenotype and the invasive and metastatic potential of HCC cells. In our previous study, we have unraveled an inhibitory effect of MST4 on PI3K/AKT pathway [29]. Here, we investigated whether the inactivation of PI3K/AKT pathway is essential for the suppressed EMT phenotype by MST4 high expression in HCC cells. To this end, we employed a PI3K/AKT inhibitor LY294002, in dnMST4-expressing cells and found that inhibition of the PI3K/AKT pathway could eliminate the up-regulation of vimentin and fibronectin, and down-regulation of E-cadherin induced by dnMST4 expression (Figure 4A). This finding suggested that PI3K/AKT pathway plays a vital role in the MST4-mediated repression of EMT phenotype of HCC cells. In addition, we examined the migration and invasion capability of Bel-7402 cells, and observed that PI3K/AKT inactivation abolished the cell migration and invasion promoted by dnMST4 expression (Figure 4B and 4C), suggesting the necessary role of PI3K/AKT inactivation in MST4 inhibiting the invasive and metastatic potential of HCC cells.
MST4 suppresses EMT and invasion of HCC cells through inactivation of PI3K/AKT/Snail1 signaling axis. (A) Western blot analysis for indicated protein levels in dnMST4-expressing Bel-7402 cells treated with or without LY294002 for 24 hours. (B) Representative images of migratory and invasive properties of dnMST4-expressing Bel-7402 cells treated with or without LY294002 for 24 hours. (C) Quantification of migratory and invasive properties of dnMST4-expressing Bel-7402 cells treated with or without LY294002 for 24 hours. (D) Immunoblot analysis of Snail1 protein levels in dnMST4- and LV-MST4-expressing Bel-7402 cells. (E) Representative immunofluorescence images for Snail1 levels in vector- and dnMST4-expressing Bel-7402 cells and Bel-7404 cells. Scale bar, 20 µm. (F) Immunoblot analysis of the expression of Snail1, E-cadherin and vimentin in vector- and dnMST4-expressing Bel-7402 cells with or without Snail1 knockdown. (G) Phase-contrast micrographs of the cell morphology of vector- and dnMST4-expressing HCC cells with or without Snail1 knockdown. (H) Representative pictures of the migration and invasion in vector- and dnMST4-expressing Bel-7402 cells with or without Snail1 knockdown. (I) Quantification of the migration and invasion in vector- and dnMST4-expressing Bel-7402 cells with or without Snail1 knockdown. (J) Western blot analysis of Snail1 expression in dnMST4-expressing Bel-7402 cells with or without LY294002. (K) Representative immunofluorescence images for Snail1 in dnMST4-expressing Bel-7402 cells and Bel-7404 cells with or without LY294002. Scale bar, 20 µm. Data are mean ± SEM (*P< 0.05, **P <0.01, ***P <0.001).
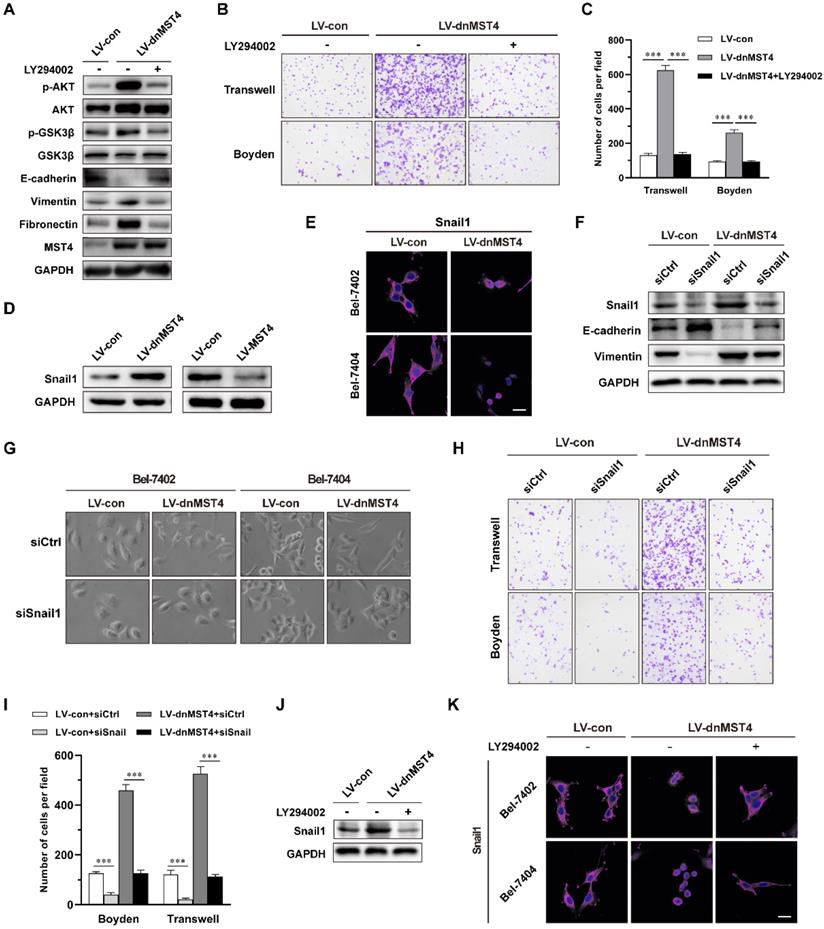
The transcription factor Snail1 is a critical inducer of EMT and plays an important role in cancer invasion and metastasis [43]. The activation of AKT can inhibit GSK3β activation, thus stabilize Snail1 and facilitate Snail1 nuclear translocation, promoting EMT and metastasis [44, 45]. As shown in Figure 4D, a significant increase in the level of Snail1 was observed in dnMST4-expressing Bel-7402 cells, whereas the MST4 overexpression led to a remarkably reduced level. We also noticed that the expression of dnMST4 caused the translocation of Snail1 into the nucleus (Figure 4E). Next, we examined whether the inhibition of Snail1 is necessary for MST4 to inhibit EMT phenotype, migratory and invasive potential of HCC cells. After the introduction of siRNAs specific for Snail1 into dnMST4-expressing Bel-7402 cells, we analyzed the expression of epithelial and mesenchymal markers. Our results showed that silencing of Snail1 significantly reversed the EMT phenotype induced by dnMST4 expression in Bel-7402 cells, as evidenced by the enhanced expression of the epithelial marker (E-cadherin), and the reduced expression of the mesenchymal marker (vimentin) (Figure 4F). Furthermore, we observed that siSnail1 abolished the spindle-like, fibroblastic morphology and enforced invasive potential promoted by dnMST4 expression in HCC cells (Figure 4G, 4H and 4I). To further elucidate whether the regulatory effect of MST4 on Snail1 is dependent on PI3K/AKT pathway, we applied LY294002 in dnMST4 cells and detected the expression of Snail1. The dnMST4-enforced expression of Snail1 was abolished (Figure 4J) and the location of Snail1 was reversed into cytoplasm (Figure 4K), demonstrating that the effect of MST4 on Snail1 is dependent on PI3K/AKT pathway. Collectively, these results suggested that MST4 inactivation can induce the EMT phenotype of HCC cells, and promote their invasive potential through activation of PI3K/AKT/Snail1 signaling pathway.
High MST4 expression correlates with the inactivation of PI3K/AKT/Snail signaling and reduced EMT phenotype in HCC patients
We investigated the expression of MST4, PI3K/AKT/Snail1 pathway components, EMT marker E-cadherin, and proliferation marker Ki67 by immunohistochemical staining in the 325 cases of primary HCC. Representative cases of immunohistochemical staining are shown in Figure 5A. Our results showed that in HCC patients with high MST4 expression, the levels of p-AKT, nuclear Snail1, and Ki67 are down-regulated, while the expression of E-cadherin is up-regulated in these patients (Figure 5A, 5B and Table 2). On the other hand, in patients with low expression of MST4, the levels of p-AKT, nuclear Snail1, and Ki67 were up-regulated, and the typical EMT phenotype characterized by downregulation of E-cadherin were observed.
The expression of MST4, p-AKT, Snail1, E-cadherin and Ki67 in human clinical HCC tissue specimens. (A-B) Representative images (A) of immunohistochemical staining for MST4, p-AKT, Snail1, E-cadherin and Ki67 in HCC tissues, and statistical analysis across 325 HCC specimens (B). (C) A proposed model of MST4 regulating the PI3K/AKT/Snail1 signaling pathway to modulate EMT, invasion and metastasis in HCC cells.
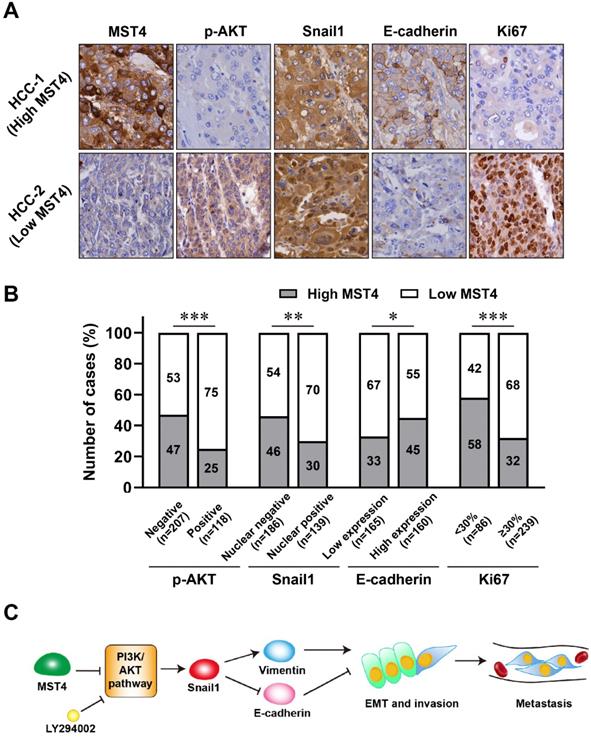
Correlation between MST4 expression and p-AkT level, Snail1 expression, E-cadherin expression & Ki67 in 325 HCC tissue specimens
| Markers | Case No. | MST4 expression | χ2 | P | |
|---|---|---|---|---|---|
| (n) | High (n, %) | Low (n, %) | |||
| p-AkT | |||||
| Negative | 207 | 98 (47.34) | 109 (52.66) | 15.421 | <0.001 |
| Positive | 118 | 29 (24.58) | 89 (75.42) | ||
| Snail1 | |||||
| Nuclear negative | 186 | 85 (45.70) | 101 (54.30) | 7.373 | 0.003 |
| Nuclear positive | 139 | 42 (30.22) | 97 (69.78) | ||
| E-cadherin | |||||
| Low | 165 | 55 (33.33) | 110 (66.67) | 4.167 | 0.041 |
| High | 160 | 72 (45.00) | 88 (55.00) | ||
| Ki67 index (%) | |||||
| <30% | 86 | 50 (58.14) | 36 (41.86) | 16.778 | <0.001 |
| ≥30% | 239 | 77 (32.22) | 162 (67.78) | ||
Discussion
HCC with a high tendency for invasion and metastasis often indicates poor survival [46]. As such, there is an urgent need to identify the key indicators to predict the risk of aggressive invasion and metastasis, with the aim of identifying new therapeutic targets. EMT phenotype is closely related to high invasion, intrahepatic metastasis (IM), and poor prognosis in patients with HCC [47]. Here, we disclosed that MST4, a member of the GCKIII kinase family, can suppress the EMT, invasive and metastatic potential of HCC cells by inactivating PI3K/AKT/Snail1 axis, and can act as a potential biomarker for the HCC progression and prognosis.
In clinics, early HCC can be cured by surgical resection. However, recurrence after radical resection often leads to treatment failure and affects the long-term survival of HCC patients [48]. Postoperative recurrent HCC (RHCC) can be caused by IM of the primary tumor, with a high incidence and poor prognosis [49, 50]. The highly invasive properties of HCC cells into macro- and microvascular vessels can give rise to IM with high frequency [51]. Therefore, it is necessary to identify the HCC patients with high invasive and IM potential to increase the chances of curative interventions. In our study, we found that MST4 was robustly down-regulated in HCC with advanced TNM stage, clinical stage, and histological grade. Kaplan-Meier analysis revealed that low MST4 expression correlated with poor prognosis in HCC patients. Strikingly, further subgroup analysis of the patients' clinical stages showed that there was no correlation between the expression of MST4 and OS in patients with I-II stage, whereas in patients with stage III-IV, the OS of patients with low expression of MST4 was significantly worse than that of patients with high expression. By using an orthotopically metastatic model of human HCC in nude mice, we showed that the functional inactivation of MST4 can promote the IM of HCC cells, demonstrating an inhibitory role of MST4 on HCC progression. These findings suggest that MST4 is a potential prognostic indicator for patients with HCC and can be used to predict the potential for HCC aggressive invasion, metastasis, and poor prognosis.
The occurrence of tumor metastasis requires the infiltrating of tumor cells from primary focus into adjacent tissues, trans-endothelial migration into vessels, and survival in the circulation known as anchorage-independent growth [52, 53]. Our previous study has shown that MST4 can limit the unanchored growth of HCC cells [29], implicating its potential inhibitory effect on metastasis. In this study, we found that MST4 suppressed the migration and invasion of HCC cells in vitro, which represent the initial steps associated with the metastasis [53]. Additionally, we observed that the MST4 functionally inactivation promoted the intrahepatic dissemination, lymph node localization, vasculature transmigration, and perineural infiltration of HCC cells in mouse models, and contributed to a worse prognosis. These observations indicate that MST4 plays an important role in blocking the invasive and metastatic progression of HCC. However, the specific mechanism of each step in this process may be complex and different, which is worthy of our further exploration.
EMT is a process of cell de-differentiation from epithelial into mesenchymal phenotype. In HCC, EMT can reduce the cell contacts and gain increased motility, thus causing cell spreading [46]. Multiple studies clarified the correlation between EMT and a high tendency to migrate, invade, and metastasize. An early study of 72 HCC patients with chronic liver disease showed that the E-cadherin expression in metastatic HCC was lower than that in non-metastatic HCC [54]. Another immunohistochemical study established the decreased expression of E-cadherin was significantly correlated with IM and capsular invasion in HCC [55, 56]. Our clinical data showed that MST4 was inversely correlated with the histological grade of HCC, which was based on the degree of differentiation [55], indicating a potential relevance of MST4 in the EMT of HCC. Further immunohistochemical analysis revealed that MST4 expression was positively correlated with E-cadherin and reversely correlated with E-cadherin transcriptional repressor Snail1. In our 10 cases of SHC, which expressed EMT-related markers Vimentin and Snail1 and lacked E-cadherin expression, loss of MST4 is observed. These results confirmed a significant negative correlation between MST4 expression and EMT status in HCC. In addition, our mechanistic study uncovered that inhibition of MST4 promoted the typical EMT phenotype, while MST4 overexpression reduced this phenotype. Above all, our data demonstrate that MST4 may affect the HCC aggressiveness and metastasis by altering the EMT status.
Another crucial finding of our work is that the inactivation of PI3K/AKT signaling is the cause of MST4-mediated inhibition of EMT, invasion, and metastasis in HCC. The PI3K/AKT signaling plays an important role in regulating cell proliferation and maintaining multiple biological characteristics of malignant cells [57, 58]. An early study has established that PI3K/AKT activation is essential for the EMT and cell migration in tumor cells induced by TGF-β1 [59]. Another finding reported that PI3K/AKT can sustain NF-kB activation in the absence of TGF-β1, thus inducing EMT and promoting tumor formation and metastasis [60]. Our previous study illustrated that MST4 inhibits HCC cell proliferation by inactivating the PI3K/AKT signaling pathway [29]. Here, our data further confirmed that the PI3K inhibitor can reverse the EMT phenotypic change and the increased capabilities of migration and invasion of HCC cells caused by the MST4 functionally inactivation. Therefore, we can conclude that the inhibition of PI3K/AKT signaling pathway is also essential for MST4 to inhibit the EMT phenotype and capabilities of invasion and metastasis of HCC cells.
Snail1 is a zinc-finger protein that binds to E-box, an E-cadherin promoter region, and represses the expression of the adhesion molecule, thereby breaking the cell-cell tightly contacts and triggering the EMT phenotypic change [45, 61]. The nuclear localization of Snail1 facilitates the expression of mesenchymal markers, such as vimentin and fibronectin, completes the EMT and increases migration, invasion, and metastasis of tumor cells [62-64]. PI3K/AKT signaling pathway can regulate Snail1 intracellular expression through multiple mechanisms [45]. For instance, PI3K/AKT can phosphorylate GSK-3β at serine of the 9th residue, leading to its ubiquitination and degradation, thereby blocking the phosphorylation of Snail1 mediated by GSK-3β and increasing its stability and location in the nucleus [44]. From our results, the phosphorylation levels of AKT and GSK-3β were upregulated by MST4 inactivation, indicating Snail1 may function as a downstream target of MST4 in HCC. By using siRNA against Snail1, we confirmed that the reduced expression of Snail1 is responsible for the attenuated EMT, migration and invasion of HCC cells triggered by MST4. Furthermore, using the PI3K inhibitor, we proved that the PI3K/AKT signaling pathway is the hinge between MST4 and Snail1. Therefore, we suggest that through inactivation of PI3K/AKT signaling pathway, MST4 suppressed Snail1 expression, which led to the upregulation of epithelial marker E-cadherin and downregulation of mesenchymal markers vimentin and fibronectin, thus inhibiting the potential of EMT, invasion, and metastasis of HCC cells (Figure 5C).
Based on our mechanistic studies, we further detected the relative expression levels of MST4, PI3K/AKT/Snail1 pathway components, as well as the EMT marker E-cadherin in HCC patients. We found that the frequency of PI3K/AKT/Snail1 pathway inactivation and elevated level of the epithelial marker E-cadherin was higher in HCC patients with high MST4 expression. In contrast, patients with low MST4 expression had a higher frequency of PI3K/AKT/Snail1 axis activation and EMT progression characterized by E-cadherin down-regulation. Taken together, our findings suggest that MST4 serves as a tumor suppressor on HCC cell aggressive invasion and metastasis, and its low expression is a potential biomarker for the HCC poor prognosis.
Supplementary Material
Supplementary tables.
Acknowledgements
Data availability
All data generated and analyzed in the current study are included in this article and its supplementary information.
Author Contributions
The study concept and design were performed by DX, MD, and WH. MD, WH, JL, ZL, XL, YZ, WZ, TL, YL, YL, WP, and QZ conducted experiments. MD, DX, and WH performed development of methodology and writing, review and revision of the paper. LH, JJ, WZ and JX were responsible for the statistical analysis. XZ, SX, JX and XL collected the clinical specimens and clinical data. XZ and SX performed immunohistochemical staining for clinical specimens and pathological analysis. XL, JX and JJ provided technical assistance and expertise in the mouse model preparation. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 81872209, 81672689, 81372896 and 81172587, to D. Xiao; Grant No. 81600488 and 81870602, to X.-L. Lin; Grant No. 81702778, to J.S. Jia; Grant No. 82060500, 81760491 and 81560441, to S.J. Xiao; Grant No. 81660485, to X.L. Zhang; Grant No. 82060425, to W.T. Zhao), the Natural Science Foundation of Guangdong Province of China (Grant No. 2014A030313294 to D. Xiao), the Science and Technology Planning Project of Guangdong Province of China (Grant No. 2017A010105017, 2013B060300013 and 2009B060300008, to D. Xiao; Grant No. 2017A030303018, to J.S. Jia; Grant No. 2015A030302024, to X.-L. Lin), the China Postdoctoral Science Foundation (Grant No. 2015M572338, 2016T90792, 2017M622740 and 2018T110884, to X.-L. Lin; Grant No. 2020M673595XB, to W.T. Zhao), the Basic Research Foundation of Yunnan Province Local Universities (Grant No. 202001BA070001-063, to J.W. Xia; Grant No. 202001BA070001-043, to L.X. Han), the Applied Basic Research Foundation of Yunnan Province Science and Technology Department & Kunming Medical University of China (Grant No. 2018FE001(-249), to W.T. Zhao; Grant No. 2018FE001(-243), to X.D. Xiang), the Science and Technology Planning Project of Kunming City of China (Grant No. 2019-1-S-25318000001329, to J.W. Xia), the Guangxi Natural Science Foundation of China (Grant No. 2020GXNSFAA159131, to X.L. Zhang) the Doctoral Science Startup Foundation of the First People's Hospital of Chenzhou of China (Grant No. BSQDJ-023, to J. Li) and the Medical Scientific Research Foundation of Guangdong Province of China (Grant No. A2017420, to J.S. Jia).
Ethics approval
Retrieval of tissue and clinical data was performed according to the regulations of the institutional review boards (IRB) of the Second Affiliated Hospital of Guilin Medical University and data safety laws with specific regard to ethical standards and patient confidentiality. The patients whose tissues were used provided written informed consent. Ethical approval was given by the Medical Ethics Committee of Southern Medical University. The study is compliant with all relevant ethical regulations involving human participants. This study was carried out in accordance with the Guide for the Care and Use of Laboratory Animals of the Southern Medical University. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Southern Medical University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Villanueva A. Hepatocellular Carcinoma. N Engl J Med. 2019;380:1450-62
2. Altekruse SF, McGlynn KA, Dickie LA, Kleiner DE. Hepatocellular carcinoma confirmation, treatment, and survival in surveillance, epidemiology, and end results registries, 1992-2008. Hepatology. 2012;55:476-82
3. Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301-14
4. Marrero JA, Kulik LM, Sirlin CB, Zhu AX, Finn RS, Abecassis MM. et al. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology. 2018;68:723-50
5. Roayaie S, Jibara G, Tabrizian P, Park JW, Yang J, Yan L. et al. The role of hepatic resection in the treatment of hepatocellular cancer. Hepatology. 2015;62:440-51
6. Ishizawa T, Hasegawa K, Aoki T, Takahashi M, Inoue Y, Sano K. et al. Neither multiple tumors nor portal hypertension are surgical contraindications for hepatocellular carcinoma. Gastroenterology. 2008;134:1908-16
7. Nicole D. Ferrante AP, and Amit G. Singal. Update on the Diagnosis and Treatment of Hepatocellular Carcinoma. Gastroenterology & Hepatology. 2020;16:11
8. Saeed A, Park R, Sun W. The integration of immune checkpoint inhibitors with VEGF targeted agents in advanced gastric and gastroesophageal adenocarcinoma: a review on the rationale and results of early phase trials. J Hematol Oncol. 2021;14:13
9. Kardas G, Daszyńska-Kardas A, Marynowski M, Brząkalska O, Kuna P, Panek M. Role of Platelet-Derived Growth Factor (PDGF) in Asthma as an Immunoregulatory Factor Mediating Airway Remodeling and Possible Pharmacological Target. Front Pharmacol. 2020;11:47
10. Proietti I, Skroza N, Bernardini N, Tolino E, Balduzzi V, Marchesiello A. et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers (Basel). 2020 12
11. Abbaspour Babaei M, Kamalidehghan B, Saleem M, Huri HZ, Ahmadipour F. Receptor tyrosine kinase (c-Kit) inhibitors: a potential therapeutic target in cancer cells. Drug Des Devel Ther. 2016;10:2443-59
12. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS. et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10:25-34
13. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90
14. Bruix J, Takayama T, Mazzaferro V, Chau GY, Yang J, Kudo M. et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2015;16:1344-54
15. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell. 2017;168:670-91
16. Yamanaka N, Okamoto E, Fujihara S, Kato T, Fujimoto J, Oriyama T. et al. Do the tumor cells of hepatocellular carcinomas dislodge into the portal venous stream during hepatic resection? Cancer. 1992;70:2263-7
17. Bakir B, Chiarella AM, Pitarresi JR, Rustgi AK. EMT, MET, Plasticity, and Tumor Metastasis. Trends Cell Biol. 2020;30:764-76
18. Liu X, You J, Peng X, Wang Q, Li C, Jiang N. et al. Mammalian Ste20-like kinase 4 inhibits the inflammatory response in Aspergillus fumigatus keratitis. Int Immunopharmacol. 2020;88:107021
19. ten Klooster JP, Jansen M, Yuan J, Oorschot V, Begthel H, Di Giacomo V. et al. Mst4 and Ezrin induce brush borders downstream of the Lkb1/Strad/Mo25 polarization complex. Dev Cell. 2009;16:551-62
20. Dent P, Booth L, Poklepovic A, Martinez J, Hoff DV, Hancock JF. Neratinib degrades MST4 via autophagy that reduces membrane stiffness and is essential for the inactivation of PI3K, ERK1/2, and YAP/TAZ signaling. J Cell Physiol. 2020;235:7889-99
21. Yuan X, Yao PY, Jiang J, Zhang Y, Su Z, Yao W. et al. MST4 kinase phosphorylates ACAP4 protein to orchestrate apical membrane remodeling during gastric acid secretion. J Biol Chem. 2017;292:16174-87
22. Mardakheh FK, Self A, Marshall CJ. RHO binding to FAM65A regulates Golgi reorientation during cell migration. J Cell Sci. 2016;129:4466-79
23. Sung V, Luo W, Qian D, Lee I, Jallal B, Gishizky M. The Ste20 kinase MST4 plays a role in prostate cancer progression. Cancer Res. 2003;63:3356-63
24. Chen M, Zhang H, Shi Z, Li Y, Zhang X, Gao Z. et al. The MST4-MOB4 complex disrupts the MST1-MOB1 complex in the Hippo-YAP pathway and plays a pro-oncogenic role in pancreatic cancer. J Biol Chem. 2018;293:14455-69
25. Li T, Deng L, He X, Jiang G, Hu F, Ye S. et al. MST4 Predicts Poor Prognosis And Promotes Metastasis By Facilitating Epithelial-Mesenchymal Transition In Gastric Cancer. Cancer Manag Res. 2019;11:9353-69
26. Madsen CD, Hooper S, Tozluoglu M, Bruckbauer A, Fletcher G, Erler JT. et al. STRIPAK components determine mode of cancer cell migration and metastasis. Nat Cell Biol. 2015;17:68-80
27. An L, Nie P, Chen M, Tang Y, Zhang H, Guan J. et al. MST4 kinase suppresses gastric tumorigenesis by limiting YAP activation via a non-canonical pathway. J Exp Med. 2020 217
28. Yu H, Zhang W, Han P, Yang B, Feng X, Zhou P. et al. MST4 Regulates Epithelial-Mesenchymal Transition of Choriocarcinoma by Mediating TGF-β1 Expression. Onco Targets Ther. 2020;13:11935-46
29. Hao WC, Zhong QL, Pang WQ, Dian MJ, Li J, Han LX. et al. MST4 inhibits human hepatocellular carcinoma cell proliferation and induces cell cycle arrest via suppression of PI3K/AKT pathway. J Cancer. 2020;11:5106-17
30. Essig K, Hu D, Guimaraes JC, Alterauge D, Edelmann S, Raj T. et al. Roquin Suppresses the PI3K-mTOR Signaling Pathway to Inhibit T Helper Cell Differentiation and Conversion of Treg to Tfr Cells. Immunity. 2017;47:1067-82.e12
31. Williams JP, Wu J, Johansson G, Rizvi TA, Miller SC, Geiger H. et al. Nf1 mutation expands an EGFR-dependent peripheral nerve progenitor that confers neurofibroma tumorigenic potential. Cell Stem Cell. 2008;3:658-69
32. Shi JW, Zhang TT, Liu W, Yang J, Lin XL, Jia JS. et al. Direct conversion of pig fibroblasts to chondrocyte-like cells by c-Myc. Cell Death Discov. 2019;5:55
33. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20:69-84
34. Lu W, Kang Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell. 2019;49:361-74
35. Simeone P, Trerotola M, Franck J, Cardon T, Marchisio M, Fournier I. et al. The multiverse nature of epithelial to mesenchymal transition. Semin Cancer Biol. 2019;58:1-10
36. Williams ED, Gao D, Redfern A, Thompson EW. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat Rev Cancer. 2019;19:716-32
37. Lee SY, Jeong EK, Ju MK, Jeon HM, Kim MY, Kim CH. et al. Induction of metastasis, cancer stem cell phenotype, and oncogenic metabolism in cancer cells by ionizing radiation. Mol Cancer. 2017;16:10
38. Bahmad HF, Cheaito K, Chalhoub RM, Hadadeh O, Monzer A, Ballout F. et al. Sphere-Formation Assay: Three-Dimensional in vitro Culturing of Prostate Cancer Stem/Progenitor Sphere-Forming Cells. Front Oncol. 2018;8:347
39. Liao SH, Su TH, Jeng YM, Liang PC, Chen DS, Chen CH. et al. Clinical Manifestations and Outcomes of Patients with Sarcomatoid Hepatocellular Carcinoma. Hepatology. 2019;69:209-21
40. Yen CH, Lai CC, Liao CC, Wang SF, Liao YJ, Tung CY. et al. Characterization of a new murine cell line of sarcomatoid hepatocellular carcinoma and its application for biomarker/therapy development. Sci Rep. 2017;7:3052
41. Wang QB, Cui BK, Weng JM, Wu QL, Qiu JL, Lin XJ. Clinicopathological characteristics and outcome of primary sarcomatoid carcinoma and carcinosarcoma of the liver. J Gastrointest Surg. 2012;16:1715-26
42. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442-54
43. Dong J, Zhai B, Sun W, Hu F, Cheng H, Xu J. Activation of phosphatidylinositol 3-kinase/AKT/snail signaling pathway contributes to epithelial-mesenchymal transition-induced multi-drug resistance to sorafenib in hepatocellular carcinoma cells. PLoS One. 2017;12:e0185088
44. Lee YJ, Han HJ. Troglitazone ameliorates high glucose-induced EMT and dysfunction of SGLTs through PI3K/Akt, GSK-3β, Snail1, and β-catenin in renal proximal tubule cells. Am J Physiol Renal Physiol. 2010;298:F1263-75
45. Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr. 2015;9:317-24
46. Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. 2016;65:798-808
47. van Zijl F, Zulehner G, Petz M, Schneller D, Kornauth C, Hau M. et al. Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol. 2009;5:1169-79
48. Feo F, Pascale RM. Multifocal hepatocellular carcinoma: intrahepatic metastasis or multicentric carcinogenesis? Ann Transl Med. 2015;3:4
49. Portolani N, Coniglio A, Ghidoni S, Giovanelli M, Benetti A, Tiberio GA. et al. Early and late recurrence after liver resection for hepatocellular carcinoma: prognostic and therapeutic implications. Ann Surg. 2006;243:229-35
50. Wang B, Xia CY, Lau WY, Lu XY, Dong H, Yu WL. et al. Determination of clonal origin of recurrent hepatocellular carcinoma for personalized therapy and outcomes evaluation: a new strategy for hepatic surgery. J Am Coll Surg. 2013;217:1054-62
51. Zimmermann A. Invasion Patterns and Metastatic Patterns of Hepatocellular Carcinoma.: In: Tumors and Tumor-Like Lesions of the Hepatobiliary Tract. Springer, Cham.; 2017
52. Nakanishi K, Sakamoto M, Yamasaki S, Todo S, Hirohashi S. Akt phosphorylation is a risk factor for early disease recurrence and poor prognosis in hepatocellular carcinoma. Cancer. 2005;103:307-12
53. van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res. 2011;728:23-34
54. Fransvea E, Angelotti U, Antonaci S, Giannelli G. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology. 2008;47:1557-66
55. Board tWCoTE. WHO Classification of Tumours. 2011/10/07 ed. 2011
56. Zhai B, Yan HX, Liu SQ, Chen L, Wu MC, Wang HY. Reduced expression of E-cadherin/catenin complex in hepatocellular carcinomas. World J Gastroenterol. 2008;14:5665-73
57. Corpuz AD, Ramos JW, Matter ML. PTRH2: an adhesion regulated molecular switch at the nexus of life, death, and differentiation. Cell Death Discov. 2020;6:124
58. Figueiredo AM, Villacampa P, Diéguez-Hurtado R, José Lozano J, Kobialka P, Cortazar AR. et al. Phosphoinositide 3-Kinase-Regulated Pericyte Maturation Governs Vascular Remodeling. Circulation. 2020;142:688-704
59. Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J Biol Chem. 2000;275:36803-10
60. Maier HJ, Schmidt-Strassburger U, Huber MA, Wiedemann EM, Beug H, Wirth T. NF-kappaB promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010;295:214-28
61. Villarejo A, Cortés-Cabrera A, Molina-Ortíz P, Portillo F, Cano A. Differential role of Snail1 and Snail2 zinc fingers in E-cadherin repression and epithelial to mesenchymal transition. J Biol Chem. 2014;289:930-41
62. Cano A, Pérez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG. et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76-83
63. Kaufhold S, Bonavida B. Central role of Snail1 in the regulation of EMT and resistance in cancer: a target for therapeutic intervention. J Exp Clin Cancer Res. 2014;33:62
64. Qiao B, Johnson NW, Gao J. Epithelial-mesenchymal transition in oral squamous cell carcinoma triggered by transforming growth factor-beta1 is Snail family-dependent and correlates with matrix metalloproteinase-2 and -9 expressions. Int J Oncol. 2010;37:663-8
Author contact
![]() Corresponding authors: Dong Xiao, E-mail: xiaodongedu.cn, xiao_dcom; Jia-Wei Xia, E-mail: 1115076481com; Wei-Chao Hao, E-mail: haoweichaocom.
Corresponding authors: Dong Xiao, E-mail: xiaodongedu.cn, xiao_dcom; Jia-Wei Xia, E-mail: 1115076481com; Wei-Chao Hao, E-mail: haoweichaocom.