Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(15):4672-4685. doi:10.7150/jca.53954 This issue Cite
Review
Hindsight: Review of Preclinical Disease Models for the Development of New Treatments for Uveal Melanoma
Caoimhe Goldrick1, Letizia Palanga1, Bobby Tang2, Grace Mealy1, John Crown1,3, Noel Horgan2, Susan Kennedy1,2, Naomi Walsh1 ![]()
1. National Institute for Cellular Biotechnology, School of Biotechnology, Dublin City University, Dublin, Ireland.
2. Royal Victoria Eye and Ear Research Foundation, Royal Victoria Eye and Ear Hospital, Adelaide Road, Dublin, Ireland.
3. Department of Medical Oncology, St. Vincent's University Hospital, Dublin, Ireland.
Received 2020-9-30; Accepted 2021-5-19; Published 2021-6-4
Abstract

The molecular, histopathological, genomic and transcriptomic characteristics of uveal melanoma (UM) have identified four molecular subgroups with different clinical outcomes. Despite the improvements in UM classification and biological pathology, current treatments do not reduce the occurrence of metastasis. The development of effective adjuvant and metastatic therapies for UM has been slow and extremely limited. Preclinical models that closely resemble the molecular and genetic UM subgroups are essential for translating molecular findings into improved clinical treatment. In this review, we provide a retrospective view of the existing preclinical models used to study UM, and give an overview of their strengths and limitations. We review targeted therapy clinical trial data to evaluate the gap in the translation of preclinical findings to human studies. Reflecting on the current high attrition rates of clinical trials for UM, preclinical models that effectively recapitulate the human in vivo situation and/or accurately reflect the subtype classifications would enhance the translational impact of experimental data and have crucial implications for the advancement of personalised medicine.
Keywords: uveal melanoma, preclinical disease models, personalised medicine, cell lines, PDX, GEMM, zebrafish models
Introduction
Despite being a rare subtype of melanoma (approximately 3% of melanoma cases), uveal melanoma (UM) is the most common intraocular cancer in adults [1]. Approximately 50% of UM patients develop metastases within 10 years of diagnosis, with the liver being the most common site of metastasis, followed by lungs and bone [1]. Metastatic UM (mUM) patients have a median overall survival of 6 to 12 months [2,3], and while effective localised treatments exist to target the primary tumour, there are no current therapies available to cure patients at metastatic stages [4]. Therefore, generating preclinical models to better characterise the genetic landscape and evolution of UM is critical in order to identify new cancer biomarkers and therapies. This review will assess current preclinical models of UM and their merits in furthering the development of effective treatment strategies. In doing so, we provide a comprehensive overview of targeted therapies in clinical trials for UM and link preclinical research outcomes with clinical results.
Clinical epidemiology of uveal melanoma
Several variables may alter the incidence of UM, such as race, sex, and age. UM occurs most frequently in the Caucasian population, followed by Asian, Hispanic, and Black populations. This is likely to relate to physical features, including pale skin and light eye colour, which are known risk factors in the development of UM [5]. In Ireland, the age-adjusted incidence of uveal melanoma at 9.5 cases per million is one of the highest in the world [6]. UM most commonly arises in the choroid (90%), followed by the ciliary body (6%) and iris (4%) [7]. Tumours are histopathologically characterised by the morphology of the dominant cell type, including spindle, epithelioid, mixed, and necrotic cell types. Mixed cell type consists of varying amounts of spindle and epithelioid cells, while necrotic cell type, as the name suggests, is predominantly made up of dead cells of unidentifiable morphology [8]. Prognosis is determined by the analysis of tumour type, cellular morphology, mitotic figures, cytogenetic aberrations, tumour diameter, and the presence of metastases. Survival rates are highest for spindle cell-type tumours, followed by mixed, and necrotic, with tumours of epithelioid morphology having the poorest outcomes [2,9,10].
Genetic landscape of uveal melanoma
Understanding the molecular and genetic landscape of UM is vital in deciphering its pathogenesis and determining patient prognosis. While clinical features, such as tumour size, histopathological state, and location in the uvea can indicate the presence of metastatic risk, cytogenetic and molecular profiling, such as fluorescence in situ hybridisation (FISH) studies, can depict disease state accurately [11]. A patient's disease status can be classified as either low or high-risk. The former is a less aggressive form of UM, rarely leading to metastasis, while the latter features cancer stem-cell like cells and an aggressive, commonly metastatic disease [12].
Cytogenetic alterations involve losses relating to chromosomes 1p, 3, 6q and 8p, and amplifications relating to 6p and 8q are considered as UM biomarkers [13,14]. TCGA global and integrated molecular and genomic analyses classifies UM into four molecularly distinct, clinically relevant subtypes [15]. Monosomy 3 and 8q gain are particularly associated with a poor disease prognosis and metastatic state [13]. Conversely, disomy 3 is associated with low risk UM, and thus, low rates of metastasis.
An early event in primary UM resulting from genetic mutations is activation of G protein-coupled receptor signalling. This occurs through mutually exclusive mutations in GNAQ or GNA11, or via rare mutations in CYSLTR2, and/or PLCB4 [16,17]. GNAQ and GNA11 encode α subunits of Gαq/11 heterotrimeric G proteins, respectively. Abnormalities here are the most prominent initiating mutations in UM, arising in 90% of cases [18,19]. Such mutations alter Gln209 and Arg183, inhibiting GTPase activity, causing constitutive activation of G proteins [15]. Related downstream signalling pathways include MAPK, PI3K/Akt and Rho GTPase signalling, all of which are a large research focus in the field.
Although less prominent than GNAQ and GNA11 mutations, mutations in the cysteinyl-leukotriene receptor 2 (CYSLTR2) gene occur in 4% of primary UM, and also lead to the constitutive activation of Gαq proteins relating to CYSLTR2. The downstream effects of this abnormality were found to promote tumorigenesis in vivo [19]. Gain of function mutations in phospholipase β4 (PLCβ4) can also occur in UM, at a rate of 2.5-4% and similarly, enhances GPCR signalling [18]. PLCβ4 can act as a downstream target of Gα signalling, and namely the canonical target of GNAQ and GNA11 gene products. PLCβ4 hotspot mutation at amino acid residue, D630, specifically activates the PLC-PKC-MAPK signalling pathway downstream of GNAQ signalling, implying that this may be a pathway of particular importance in UM [18]. The MAPK signalling pathway is integral in growth regulation, stimulation of pro-proliferative and anti-apoptotic genes, indicating these mutations may confer a key role of Gαq signalling in the pathogenesis of UM [20].
The Breast Cancer 1 (BRCA1)-associated protein 1 (BAP1) is a tumour suppressor gene which is inactivated in 70% of metastatic UM. Consequently, BAP1 inactivation is associated with high risk UM [21]. BAP1 is found on chr 3p21.2, anomalies of which strongly correlate with monosomy 3 [22]. BAP1 promotes DNA repair, regulates transcription, cell differentiation, and cell death [23]. Owing to its role in DNA repair, loss of BAP1 function may contribute to the emergence of cytogenetic alterations later in UM progression.
Mutations in eukaryotic initiation factor 1A (EIF1AX) are thought to occur in cases of disomy 3, rarely presenting in monosomy 3 tumours, consolidating its relatively low metastatic risk [24-26]. This gene plays a role in initiation of translation, which is altered by arising mutations.
Mutations in splicing factor 3B subunit 1 (SF3B1) and serine and arginine rich splicing factor 2 (SRSF2) occur in intermediate risk tumours [22,27]. SF3B1 and SRSF2 encode spliceosomal proteins, mutations of which culminate in a large number of splice variants in UM. The precise effects of these abnormalities in UM remain elusive, however, in other cancers, these mutations can reduce DNA damage repair, and are therefore potential contributors to emerging cytogenetic mutations [28,29]. These aforementioned mutations of BAP1, EIF1AX, SF3B1 and SRSF2, tend to be mutually exclusive, providing effective molecular biomarkers of metastatic outlook using techniques such as gene expression profiling (GEP).
In vitro uveal melanoma cell line models
Established UM Cell Lines: 2D and 3D models
Experimental use of cell lines is a convenient and cost-effective method of carrying out preliminary research. Cell lines have been shown to accurately recapitulate the key genomic events of many types of cancer, including breast cancer and ovarian cancer [30,31]. Prior to the establishment of UM cell lines, cutaneous melanoma cell lines presented an attractive alternative. However, genetic discrepancies may have added to the lack of progression in positive research outcomes during this time, as numerous cell lines, such as OCM-1, OCM-3, and OCM-8, which were initially thought to be of UM origin have been found to be misidentified derivatives of melanoma cell lines [32,33]. Although a subtype of melanoma, UM presents a distinct genetic landscape to cutaneous melanoma. For example, while many cutaneous melanoma cell lines characteristically harbour a mutation in the BRAF gene, this aberration does not commonly arise in UM [34]. OCM-1, OCM-3 and OCM-8 each present with V600E mutation, with the addition of V599E in the case of OCM-1. Perhaps this discrepancy was, in part, responsible for lack of substantial progression in understanding UM pathogenesis and development of novel therapeutics prior to UM cell lines being readily available.
Therefore, accurately modelling the genetic landscape of UM is fundamental to advancements in preclinical research. In recent years, there has been a growing repertoire of established UM cell lines derived from primary and secondary patient tumours, or patient-derived xenografts (Table 1). Most of the established UM cell lines harbour at least one known UM driver mutation, and may mirror a proportion of the genetic landscape UM. Consideration of patient clinical and histopathological characteristics is also of importance in drawing connections with underlying genetic features and formulating a complete cell line profile [35]. Cell lines remain an established first line model in carrying out preliminary preclinical experimentation. In recent years, there have been tremendous initiatives to improve the library of UM cell lines in terms of capturing the mutational signature of UM. In general, most in vitro studies assessing the progression, development and treatment response of UM are performed using 2D UM cell lines. However, consideration needs to be given in the experimental design when using such models. In vitro, 2D culture of cells is restricted in its ability to mirror cellular interactions with the extracellular matrix, which is an important aspect in understanding tumorigenesis and cancer progression. The need for more complex cellular systems is required to further understand the development of this disease, and to accurately represent drug efficacy in UM patients.
Summarised established uveal melanoma cell lines
| Cell line [Refs] | Origin | Sex | Morphology | Pop. doubling time | GNAQ | GNA11 | BAP1 mut | BAP1 protein | EIF1AX | SF3B1 | Cytogenetics | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Chr 1 | Chr 3 | Chr 6 | Chr 8 | Chr 16 | Other | |||||||||||
| 92.1 [36,37] | Met | F | Mixed | 38 h | Q209L (626 A>T) | WT | WT | Y | c.17G/A | Yn(C1793 C>T) | Disomy 3 | 6p+ | 8q+ | der (X) t (X; 6)(q28; p11),+8 | ||
| MEL-202 [36-38] | Primary | F | Epithelioid | 43 h | Q209L (626 A>T) R210K (629 G>A) | WT | Y | Y | c.1793c>T | Disomy 3 | 6q-, 6p+ | 8q+41 | ||||
| MEL-285 [36,39] | Primary | F | WT | WT | WT | WT | Disomy 3, 3p26- | 6q- | Disomy 8p, 8q+ | |||||||
| MEL-270 [35,39] | Primary | M | Q209P (626 A>C) | WT | Y | Y | WT | WT | Disomy 3, 3p24-, 3q21.2-3q24 | 6p+ | Disomy 8q, 8+ | |||||
| MEL-290 [35,39] | Primary | F | Epithelioid | WT | WT | WT | WT | Disomy 3, 3p26 | Disomy 6 | Disomy 8 | ||||||
| OMM-1 [35,40] | Subcut. met. | M | Mixed | 34 h | WT | Q209L (626 A>T) | Y | Y | WT | WT | 3+ | 8p- | 4-, 7+, 9-, 11-, 12+, 15-, 17-, 20+, 21- | |||
| OMM-2.3 [35,40] | Liver met. | M | Q209P (626 A>C) | Y | Y | N/A | N/A | |||||||||
| OMM-2.5 [35,40] | Liver met. | M | Mixed | 50 h | Q209P (626 A>C) | Y | Y | |||||||||
| MM28 [41] | PDX liver met. | M | Mixed | 109 h | c.626A > T | c.1881C>A | N | 1p-, 1q+ | 3q- | 6q-, 6p+ | 8p-, 8q+ | 16- | ||||
| MM33 [41] | PDX skin met. | Spindle | 91 h | c.626 a > C | Y | c.22G/A | 1+ | 6q-, 6p+ | 8+ | 16+ | ||||||
| MM66 [41] | PDX liver met. | Mixed | 80 h | c.626A > T | N | Y | 1q+ | 6q- | 8+ | |||||||
| MP38 [41] | Primary | M | Spindle | 80 h | c.626 a > T | c.68-9_72 del | N | 3q- | 8+ | 16q- | ||||||
| MP41 [41] | PDX primary | F | Mixed | 41 h | c.626 a > A/T | N | Y | 1p-, 1q+ | 3- | 6q-, 6p+ | 8p-, 8q+ | 16- | ||||
| MP46 [41] | PDX primary | M | Mixed | 110 h | c.626 a > T | N | N | 1q+ | 6q-, 6p+ | 8p-, 8+ | 16q- | |||||
| MP65 [41] | Primary | F | Spindle | 120 h | c.626A > T | c.1717del | N | 1q+ | 6p+ | 8+ | ||||||
| UPMM-1 [42-44] | Primary | M | Mixed | 100-150 h | R183Q (548 G>A) | 3- | ||||||||||
| UPMM-2 [42-44] | Primary | Spindle | 150 h | Q209L (626 A>T) | 3- | |||||||||||
| UPMM-3 [42-44] | Primary | M | Epithelioid | 100 h | p.Gln209Pro (c.626A>C) | 3- | ||||||||||
| UPMM-4 [42-44] | Primary | M | 300 h | WT | WT | |||||||||||
| UPMD-1 [43] | Primary | 100 h | Q209L (626 A>T) | Disomy 3 | ||||||||||||
| UPMD-2 [43] | Primary | 150 h | Q209L (626 A>T) | Disomy 3 | ||||||||||||
| UMT2 [45] | M | 72 h | WT | Q209L (626 A>T) | Disomy 3 | |||||||||||
| UMT26 [45] | M | 2000-3000 h (3-4 months) | WT | Q209L (626 A>T) | ||||||||||||
| UMT33 [45] | F | 288 h | WT | Q209L (626 A>T) | ||||||||||||
| WM3618F [46] | Lymph Node Met. | F | Melanocytic | Y | Y | |||||||||||
| WM3772F [46] | Lung Met | F | Clumpy pigmented | Y | Y | |||||||||||
| MEL-20-06-039 [47] | Q209L (626 A>T) | |||||||||||||||
| MEL-20-06-045 [47] | Q209P (626 A>C)43,44 | |||||||||||||||
| MEL-20-07-070 [47] | Q209L (626 A>T) | |||||||||||||||
Other UM cell lines in existence for which such information is unavailable include: 92.2, BB90-MEL, OMM2, OMM2.2, OMM2.6, OMM3, MU2, MU8, EOM-3, EOM-29, C918, M619, MuM-2B, MKT-BR, YUGLIDE, YUCRENA.
Cell lines can also be used to generate 3D in vitro models, which offer a number of advantages over 2D cell culture, such as the establishment of living biobanks and genomic analysis. In comparison to 2D models, 3D cell culture models can provide a more realistic model of how therapeutic treatments take effect in cancer cells and allow for exploration of alternative treatments. Several studies have examined the use of 3D spheroids as models of UM, using established and primary UM cell lines. Recently, Aughton et al. assessed the ability of established UM cell lines (92.1, OMM-2.5, MM66, MP41, OMM-1, and MP46) and primary UM tissue to form spheroids. This study highlighted the utility of UM cell line spheroids in in vitro drug screening assays. Compared to 2D cell lines, the 3D cell line spheroids exhibited altered drug responses, with increased resistance to doxorubicin and higher sensitivity to treatment with selumetinib [48]. This study suggests the importance of 2D vs 3D culture drug efficacy studies, as drug penetration, different metabolic states, and cell cycle growth rate, are more likely to affect the predicted preclinical efficacy. Other studies have also shown the ability of UM cell lines to be cultured as 3D spheroids. Goncalves et al. embedded cell lines 92.1, MEL270 and MEL290 in collagen and demonstrated MEKi cytotoxicity [49]. The morphological nature of 3D cell culture makes it an excellent model for studying metastasis of UM also. Migration of UM cancer cells in 3D culture shows a more natural and realistic path than would be observed in 2D culture. Fornabaio et al. studied extravascular migratory metastasis and angiotropism in both cutaneous and uveal melanoma using 3D cell culture. While the most common metastatic path for tumour cells to spread occurs through intravascular or intralymphatic routes, metastasis may also occur through extravascular migratory metastasis, as with angiotropism. The UM cell lines OMM 2.3-GFP tagged and OMM 2.5-GFP tagged were added to a plate coated with basement membrane extract containing human endothelial HUVEC cells, which developed tubular structures. Real-time imaging of the 3D Matrigel-embedded UM cell line co-cultures found that both OMM-2.3-GFP and OMM-2.5-GFP showed migration of uveal melanoma cells along, and towards, endothelial cells, and subsequent attachment to the endothelial tubules. This provides evidence of angiotropism of uveal melanoma cells in endothelial tubules in 3D cultures [50]. Such studies suggest that 3D cell culture may be advantageous over 2D studies in some settings, and may provide a more accurate means to predicting in vivo tumour responses.
Mouse models of UM
Xenograft mouse models
Xenografts are a valued tool in cancer biology, providing an efficient model for exploring the in vivo tumour microenvironment, and drug screening. Such models can be generated using established cell lines, primary tumours, or secondary tumour metastases, such as from the liver, largely maintaining the genetic characteristics of the original patient tumour. Cells can be injected into selected compartments of the eye to mimic specific primary tumours, intravenously, or at secondary tumour sites to study metastases. This has been demonstrated in a growing number of studies, using a variety of established UM cell lines. For example, in assessing novel UM biomarkers, Barisione et al. carried out intrasplenic injection of either 92.1 or MEL270 cells into 6NU/NU and 9NOD/SCIDIL2Rγnull (NOG) mice [51]. Similarly, 92.1 or MEL202 have been inoculated subcutaneously into hIL2-NOG mice for use in exploring the in vivo efficacy of HER2 CAR-T cells in eradicating UM [52]. Metastatic cell line OMM-1 has been injected subcutaneously into NOD/SCID mice, and the resulting models used to evaluate the anti-tumour activity of niclosamide, preclinically [53].
However, the ability of cell line xenografts to recapitulate the genetic and molecular landscape of the patient tumour from which they were derived may be unsatisfactory, due to changes that occur through in vitro culturing. Established cell lines often lack some of the mutational burden present in the tumour of origin, such as cell lines MEL285 and MEL290 (Table 1). This discrepancy in recapturing the original genomic signature using cell line implants, gives way to an attractive alternative; patient-derived xenograft (PDX) models. These models have repeatedly been observed to retain the mutational burden, chromosomal abnormalities, and histopathological features of the original tumour. Successful generation of PDX models can also provide a means for establishing new cell lines from the animal (Table 2).
Summarised mouse xenograft models and GEMMs
| PDX Model | Phenotype | Limitations | Reference |
|---|---|---|---|
| MP34, MP38, MP41, MP42, MP46, MP47, MP55, MP71, MP77, and MP80 | Original chromosome 3 status maintained in xenograft in the case of disomy, heterozygosity and monosomy | Where original tumours were isodisomic for chromosome 3, corresponding xenografts harboured monosomy 3 anomaly | 41,54,58 |
| 6 Liver metastases successfully grafted 3 times | Key mutational and histological characteristics (CNV, immunohistochemical melanoma markers, ratio of Ki67 positive cells) of original tumour maintained in xenograft tumour | Orthotopic hepatic transplantation causes difficulties in monitoring tumour growth (CT scan is required) | 55 |
| ØPI-204 | Models human uveal malignant melanoma; transplanted cells retained morphological similarities with the primary tumour; immunohistochemically representative of malignant melanoma | Transplanted cells stained positive for vimentin, unlike the primary tumour | 65 |
| GEMM | |||
| Tyr:RAS+/Ink4a/Arf-/- | Spontaneous ocular and cutaneous melanomas, all choroidal origin; morphologically similar to human UM | No metastases; 50% of mice developed cutaneous melanoma | 61 |
| Tg (dct:Grm1) | Spontaneous choroidal and ciliary body uveal melanomas; cutaneous melanomas | Cutaneous melanoma present | 60 |
| Rosa26-LSL-GNA11Q209L/+;Tyr-CreERT2/+ Rosa26-LSL-GNA11Q209L; Bap1lox/lox; Tyr-CreERT2/+ | Spontaneous uveal, cutaneous and leptomeningeal melanomas; evidence of potential metastases in axillary lymph nodes and lungs | Cutaneous and leptomeningeal melanomas; no evidence of liver lesions | 60 |
| Accelerated rate of disease compared with above (Rosa26-LSL-GNA11Q209L/+;Tyr-CreERT2/+); Bap1 deletion contributed to growth of cutaneous melanoma and increased mortality rates | 60,66 | ||
| Rosa26-floxed stopGNAQQ209L/+; Mitf-cre/+ | Spontaneous uveal melanoma with occasional cutaneous,leptomeningeal and vestibular melanocytic lesions; evidence of lung metastasis | Cutaneous and leptomeningeal melanomas; no evidence of liver lesions | 64 |
| Dct-rtTA/+; tet-HA-GNAQQ209L/+; p16p19KO | Spontaneous cutaneous melanoma in >50% animals | No uveal lesions reported; cutaneous melanomas present | 58 |
Némati et al. established 16 PDX mouse models, using severe-combined immunodeficient (SCID) mice. Chromosome 3 status was examined as a method of assessing genomic correlation between xenograft tumours and patient tumours via single nucleotide polymorphism (SNP) array analyses. Results showed that in the case of disomic, heterozygous, and monosomic chromosome 3, status of xenografts corresponded to that of the tumour of origin. However, chromosome 3 isodisomic patient tumours gave rise to monosomic xenograft tumours [54].
Similarly, PDX models can be of use in studying metastasis in UM. Kageyama et al. generated an orthotopic PDX mouse model from UM liver metastases. In examining five key mutations in UM (GNAQ, GNA11, BAP1, SF3B1 and EIF1AX), the xenograft tumours matched the genomic signature of the original patient tumours in 83% of cases. Furthermore, features such as histological characteristics and DNA copy number variations of the tumour of origin were retained in the xenograft tumours [55].
UM PDX can be used as preclinical models in identifying novel therapeutic targets and advancing treatment strategies. Carita et al. demonstrated therapeutic efficacy for combination of PKC/mTOR inhibitors with PKC/p53-MDM2 inhibitors or mTORC1 inhibitors, both of which resulted in tumour regression in PDX mouse models by targeting the PKC pathway downstream of GNAQ and GNA11 driver mutations [56]. However, although PDX mouse models of UM are effective in modelling UM in vivo while maintaining many fundamental facets of the disease as it appears in humans, a consideration to this method is the need to suppress the mouse immune system to ensure the graft is accepted by the host. This limits the accuracy in studying the relationship between the tumour and its microenvironment in such models, as the involvement of the host immune response is diminished. This, consequently, hampers their use in studying immunotherapies. The cost and the time to initiate such models add additional limitations. Despite this, xenograft models present a useful tool in the preclinical setting for non-immunologic agents.
Genetically Engineered Mouse Models (GEMMs)
A major attraction to the use of genetically engineered mouse models (GEMMs) is that it encompasses native tumorigenesis in an immunocompetent host, mimicking the in vivo tumour microenvironment and stages of tumorigenesis. Fundamentally, this approach allows observation of specific effects of mutations on oncogenic signalling.
Prior to extensive study of the UM genome, some earlier GEMMs included tumours that originated in the retinal pigment epithelium, as opposed to the uveal tract [57]. Many of these earlier models are driven by molecular changes which do not occur in UM, therefore, limiting their clinical relevance (Table 2). Since the establishment of GNAQ/GNA11 as key oncogenic drivers in a large cohort of UM, genetically engineered overexpression of GNAQQ209L in mice deficient for p16Ink4a and p19Ink4b have been developed which demonstrate elevated YAP signalling downstream of oncogenic GNAQ. However, there was a high incidence of cutaneous melanoma, as opposed to UM in these models. While thought to underlie early events in UM tumorigenesis, GNAQ/GNA11 mutations alone are insufficient to recreate the UM phenotype in this model. In fact, uvea nevi which are not always precancerous are commonly present with GNAQ/GNA11 mutations [58,59]. In the case of GNA11 mutations, overexpression of GNA11Q209L at a conditional knock-in allele caused sporadic UM development in GEMMs. While the combination of BAP1 deletion with GNAQ11Q209L yielded larger cutaneous melanocytic lesions, there was no such observed effect in UM tumours [60]. This may indicate that other genetic cues may be required to replicate the aggressive phenotype associated with BAP1 anomalies in humans.
Tolleson et al. generated a Tyr:RAS+/Ink4a/Arf-/- transgenic mouse model, in which animals developed UM. In melanocytes, HRAS is driven under the control of tyrosinase promoter, in absence of Ink4a/Arf tumour suppressor. Spontaneous ocular and cutaneous melanomas occurred in 15.7% and 50.4% of cases, respectively. All UM tumours that developed were of choroidal origin, with none arising from retinal pigment epithelium. Morphologically, tumours resembled that of human UM tumours, having a spindle-like morphology. While many of the tumours extended to the subretinal space, no cases of metastasis arose in this model 61. However, the validity of this transgenic in modelling UM may raise concern, due to the co-occurrence of cutaneous melanoma in approximately 50% of cases. Furthermore, this model is based on overexpression of mutant HRAS, while RAS mutations are not thought to occur in UM [62].
Schiffner et al. developed a model of spontaneous UM using a Tg (dct:Grm1) transgenic mouse. Mouse dopachrome tautomerase (dct) promoter was cloned upstream of metabotropic glutamate receptor 1 (Grm1) transgene, to drive melanocyte-specific expression of Grm1. UM-like melanocytic tumours were observed intraocularly in the choroid and ciliary body, while cutaneous tumours occurred in the tails, ears, and anus. A range of tumour severity was seen in replicates of this model [63]. While this model gives rise to UM tumours, the evidence supporting an established role of Grm1, or elevated expression, is lacking. However, with further research, Grm1 may present a novel therapeutic target in the treatment of UM.
More recently, Huang et al. successfully replicated genetic mutations of UM in a model overexpressing oncogenic GNAQQ209L allele under the control of the Rosa26 promoter. After only 3 months, UM tumours occurred, driven by GNAQQ209L. Furthermore, most animals developed tumours in the lungs, which is a common site of metastases in human UM, aside from the liver. However, these lesions may have been of primary or metastatic origin. Yap activation, which occurs downstream of GNAQ, was detected in choroidal growths. Other results of this mutation were shown as central nervous system melanomas [64]. This study presents a strong oncogenic potential of GNAQQ209L in UM tumorigenesis.
Zebrafish Models of Uveal Melanoma
Zebrafish is a widely used invertebrate animal model in the preclinical setting, posing many beneficial characteristics, such as sharing large genomic similarities with humans. As much as 70% of human genes have similar zebrafish orthologs [67]. In fact, much of the genes associated with diseases in humans have corresponding zebrafish counterparts, including highly conserved tumour suppressor genes, cell-cycle genes, and oncogenic signalling, such as tp53 [68], cyclins, cdks and cdk inhibitors [69], and oncogenic Notch signalling [70], making for an extremely clinically relevant animal model. Furthermore, zebrafish models retain similarities with tumours on histological, genomic and gene expression levels [71]; therefore, presenting as a suitable animal model in the context of UM.
Alterations are primarily applied to the developing embryo, where ex-utero development allows for convenient manipulation. The highly adaptive, immature immune system of the early developing embryo permits the administration and integration of tumour grafts at these early embryonic stages [72]. This circumvents the issue of immunosuppression, required by most other xenograft models [73,74].
Additionally, the transparent nature of the fish embryos aids in tumour visualisation and live imaging of internal processes [75]. Xenograft zebrafish models, involving the transplantation of human tissue or cells into the recipient animal model, are an accessible, time and cost-effective method of performing in vivo screens for anti-cancer agents for UM. In a study by van der Ent et al., a panel of 5 UM cell lines were injected into 2-day old zebrafish embryo yolk sacs and fixed 6-days post-injection. Drug administration with individual treatment of either dasatinib, quisinostat or MLN-4924, was performed 1-day post injection in selected animals, to screen for therapeutic effects. The models displayed a phenotypic reflection of the cell lines' characteristics, consistent with clinical behaviour and underlying genetics of the UM patients of origin. OMM2.3, originating from a metastasis and harbouring the BAP1 mutation, showed increased onset of migrating tumour cells in zebrafish, compared to its primary tumour counterpart, MEL270. Src-inhibitor dasatinib caused reduced proliferation and migration in the high Src-expressing 92.1 cell line, while having little effect on low Src-expressing OMM-2.3 cells. Additionally, quinostat, a histone deacetylase inhibitor, and MLN-4924, a neddylation pathway inhibitor, prevented proliferation and migration in both 92.1 and OMM-2.3 cell lines [76]. Studies as such can provide a means of predicting patient response to treatment, identifying optimal therapeutics for UM cases of a specific disease class. This study effectively demonstrates the relevance of this model in drug screening and drug discovery.
Zebrafish UM xenografts are also beneficial in studying metastatic processes. Fornabaio et al., developed zebrafish xenografts from metastatic cell lines, OMM-2.3 and OMM-2.5, to investigate angiotropism and extravascular migratory metastasis in UM. Interactions between xenograft cancer cells and the external surfaces of vasculature were monitored via in vivo imaging techniques. Results of this study showed that UM cells migrated along vascular surfaces similarly for in vivo xenograft models, and in vitro endothelial tubule assays [50].
Zebrafish expressing human oncogenes can be generated using transgenesis, introducing a new phenotype to the developing zebrafish, which is also transmitted to offspring. Many transgenic zebrafish models have successfully been generated, for cancers including leukaemia and lymphoma, allowing for reverse genetic screening [77,78]. The first transgenic zebrafish model of UM was generated by Rose et al. This model displayed spontaneous development of UM from overexpression of a human UM oncogene, transgenically. Tg (mitfa:BRAF V600E);p53-/-;mitfa-/- transgenic embryos were injected with Tol2 transposase mRNA and transposon-based miniCoopR plasmid, overexpressing oncogenic GNA11 Q209L in rescued melanocytes [79]. HOXB7 overexpression was observed in the induction of UM in zebrafish melanocytes. There is no past evidence for a role of HOXB7 in UM tumorigenesis, but HOXB7 has been shown to be overexpressed in other cancers, including cutaneous melanoma [80], and could contribute to a pro-proliferative state in the presence of the tumour microenvironment. Furthermore, in the absence of melanocyte-restricted stable expression of the BRAFV600E transgene, no tumours were observed, which itself is a limitation to this model. In human UM, BRAF mutations are uncommon, meaning that this model may not reflect the genetics of human UM [81,82]. Another example of the use of transgenesis in zebrafish UM research, was that of Mouti et al. using a Tg (cryaa:Venus,mitfa:gnaq_Q209P);tp53 M214K transgenic zebrafish model. In this study, two transgenics were successfully generated, representing benign and malignant stages of UM development, and documenting the first in vivo characterisation of GNAQQ209P as a molecular driver in UM. GNAQQ209P, an oncogenic form of GNAQ, was expressed under the control of the mitfa promoter, which allowed selective targeting of melanocytes. This resulted in hyperproliferation of uveal melanocytes, and not cutaneous melanocytes, in the model, which reflects the clinical consequences of GNAQ mutations in human UM. Moreover, UM tumours derived from this model show that ERK1/2-MAPK signalling is weakly activated downstream of mutated GNAQ in UM, which is consistent with results from similar studies using established human UM cells. Interestingly, the addition of tp53 inactivation to this model was sufficient to introduce malignant transformation of uveal melanocytes, reflecting human UM, where cases harbouring GNAQ/11 are generally benign [83,84]. Similarly, a study by Perez et al. focused on zebrafish models expressing oncogenic forms of GNAQ and GNA11 (GNAQ/11Q209L). As in previous publications, combination of these driver mutations with mutant tp53 saw the development of ocular melanomas which were primarily choroidal, and shared molecular and histological characteristics with human UM. Expectedly, nuclear YAP localisation was noted, mirroring events in human UM of the same mutational burden. Tumours were also reported at cutaneous and internal sites in this model [85]. However, evidence of melanocytic alterations was also observed in the presence of either GNAQ/11Q209L mutations alone, therefore, indicating that such mutations may be sufficient to promote early tumorigenic processes in vivo. Despite such findings, it is likely that there is a plethora of mutational contributors that may depict the oncogenic strength of GNAQ/11Q209L. Ju et al. generated a transgenic zebrafish line expressing EGFP fusion protein of an activated zebrafish Smoothened (Smoa1-EGFP) using the Gal4VP16-UAS binary transgenic expression approach. While expression of Smao1-EGFP alone did not result in tumour formation, co-expression of constitutively active human AKT1, lead to ocular melanomas presenting in the retina, among other tumour types, suggesting a role for Smao1 in zebrafish tumorigenesis. Moreover, activation of the PI3K-AKT pathway was indicated by elevated levels of phosphorylated AKT [86]. Such findings propose that tumorigenesis can occur as a result of co-activation of hedgehog and AKT pathways in this model. This study demonstrates the utility of transgenic zebrafish models in examining the dynamics of oncogenic signalling pathways.
There is a clear potential for the use of transgenic zebrafish models in capturing the genetics of UM, and serving as an effective preclinical model of UM. Notably, cell lines from this model, as described by Mouti et al. were similar to human UM cell lines when comparing genetic and drug response characteristics, suggesting parallels between this animal model of disease, and the human correlate [83,84]. Such models are, therefore, useful in exploring novel genomic signatures in tumorigenesis, to further understand the complexity of the tumour microenvironment and its potential therapeutic targets. However, drug screening in zebrafish has several limitations. While there is a high degree of conservation between zebrafish and humans, differences remain, which could give rise to non-translatable chemical and drug screening hits.
Preclinical modelling and targeted therapy clinical trials in UM
Building on preclinical translational research to inform clinical studies is of high scientific relevance for the development of new therapeutic targets for UM. Preclinical models need to provide data demonstrating the efficacy of targeted therapeutics based on the (epi)/genomic and transcriptional landscape of the tumour. Barriers to the implementation and efficacy of clinical studies may relate to the (epi)/genetic and transcriptional heterogeneity of the original UM, confounded by the lack of appropriate preclinical models used to inform clinical studies.
Mutations commonly arising in GNAQ and GNA11 in UM can lead to increased activation of the Ras/Raf/MEK/ERK pathway, which is involved in the control of cell proliferation, survival, and differentiation [87]. Anomalies affecting this pathway are integral in the development of UM, therefore, targeting this pathway is a common point of therapeutic intervention. Table 3 outlines the results of completed targeted therapy clinical trials in UM and links associated preclinical studies and models. Clinical trials of MEK inhibitor selumetinib (phase III) in mUM, while proving safe, failed to demonstrate a significant improvement in progression-free survival (PFS) [88,89]. Despite a small number of experiments showing anti-tumour activity and reduction in tumour volume, selumetinib monotherapy, or in combination with dacarbazine (DTIC) or AKT inhibitor, did not cause a significant objective response (OR) in the chosen experimental models [90,91]. The MEK 1/2 inhibitor trametinib alone did not induce anti-proliferative effects in UM cell lines [92], in vitro studies in combination with ABT263 were synergistic, however this combination did not show anti-tumoral effects in PDX in vivo models [93]. Furthermore, phase II clinical trials of trametinib in combination with AKT inhibitor GSK2141795 did not improve PFS in advanced UM [94].
Completed clinical trials of targeted therapies and matched laboratory preclinical studies in uveal melanoma
| Targeted agent | Preclinical models | Preclinical results | Clinical Trial | Refs | ||||
|---|---|---|---|---|---|---|---|---|
| In vitro | In vivo | Trial | Study design | Results | Findings/Conclusion | |||
| Selumetinib (MEK 1/2 inhibitor) | Selumetinib and DTIC alone and in combination in 6 UM cell lines; MP38, MP41, MP46, MP65 (primary); MM28, MM66 (liver mets). Selumetinib and AKT inhibitor alone and in combination with Mel202, Mel270, Mel290, 92.1, OCM1A (primary); Omm1.3 (liver mets). In vivo: PDX-derived UM tumours | Selumetinib alone showed response in 3 out of 6 CL -Weak non-sig synergistic effect of combination in 5 of 6 cell lines -Combinatorial Selumetinib and AKTi treatments inhibited cell viability more effectively than monotherapy in GNAQ and BRAF-mutant cells | Selumetinib alone showed no sig TGI in 2 of 3 PDX models. OR = 18% DTIC alone showed no sig TGI in 2 of 3 PDX models OR = 15% Combination was not more effective than either drugs alone OR = 28% | Phase III SUMIT (n=129) NCT01974752 | mUM, no prior systemic therapy randomly assigned selumetinib (75 mg twice daily) plus DTIC (1,000 mg/m2 intravenously on day 1 of every 21-day cycle) (n=97) or placebo plus DTIC (n=32) | Selumetinib plus DTIC arm 85% achieved PFS event vs 75% placebo (median 2.8 v 1.8 mo) HR PFS 0.78 (95% CI, 0.48 to 1.27; two-sided P = 0.32). ORR 3% with selumetinib plus DTIC and 0% with placebo plus dacarbazine (two-sided P = 0.36) | Combination of selumetinib plus DTIC had a tolerable safety profile but did not significantly improve PFS compared with placebo plus DTIC. | 88-91 |
| Trametinib (MEK 1/2 inhibitor) | MET CL: MM28 (liver metastasis), MM66, OMM1 (subcutaneous met), OMM2.3 and OMM2.5 (liver metastasis) Primary CL: MP38, MP46, MP65 In vivo: Xenografted SCID female mice | 1/11 UM CL sensitive to trametinib Combination of trametinib and ABT263 (Bcl-2/XL/W inhibitor) displayed the best rank synergistic combination in 8 UM CL. | In vivo efficacy of ABT263 + trametinib in 6 UM PDX found trametinib alone had the best anti-tumour effect, but in combination with ABT263 did not show any additive effect. | Phase II (n=39) NCT01979523 | Advanced UM patients, no prior systemic or liver-directed therapy were randomized to one of two arms stratified by liver disease and LDH: trametinib 2mg daily (Arm A, 18 pts) or trametinib 1.5mg + GSK2141795 (AKT inhibitor) 50mg daily (Arm B, 21 pts). | Partial response was observed in each arm. No difference in median PFS between Arm B and Arm A (Median PFS trametinib + GSK2141795 was 7.9 wk). All pts had ≥1 adverse event (AE). | The addition of GSK2141795 to trametinib did not improve PFS. | 92-94 |
| AEB071 (PKC inhibitor) | Panel of 7 UM cell lines with GNAQ mutations. MET CL: OMM-1.3 OCM1. Cutaneous Melanoma CL: OCM3 Primary CL: C918 MEL285, MEL202, 92.1 In vivo: Allograft mouse model (GNAQQ209L were injected into the flanks of c57/Bl6 mouse) | AEB071 induced growth suppression of GNAQ mutant cells, with pronounced G1 arrest and induction of apoptosis in vitro. 7/11 UM CL sensitive/intermediately sensitive | The mice received 120 mg/kg AEB071 (n=9) or vehicle control (n=9) x3/day for 3 weeks by oral gavage. AEB071 significantly inhibits GNAQQ209L- mediated tumour growth in vivo in combination with MEK inhibitor. | Phase I (n=153) NCT01430416 Phase Ib/II (n=38) NCT01801358 NCT02273219 | mUM dose escalation and estimation of the maximum tolerated dose (MTD). 118 pts received AEB071 at total daily doses of 450-1400 mg either BID or TID. | Dose-limiting Toxicities (DLTs) were observed in 12 pts (11%) at doses ≥800 mg/day total. 55/118 (47%) pts achieved SD and progression-free survival (PFS) ranging from <1-57 weeks and with a median PFS of 15.4 weeks (95% CI 8.3-15.7). | Preliminary data suggests clinical activity of AEB071 and manageable toxicity at multiple dose levels, with evidence of PKC inhibition in patients with mUM. | 96,98,99 |
| Ulixertinib (BVD-523); (ERK inhibitor) | No uveal melanoma cell lines used RKO, SW480, HCT116, Colo205, HT-29 (colon/colorectal), A375 (cutaneous melanoma), MIAPaCa-2 (pancreas carcinoma), ZR-75-1 (breast ductal met site), AN3Ca (adenocarcinoma) and G-361 (malignant melanoma) In vivo: female athymic nude mice, xenograft model | In vitro ulixertinib treatment resulted in reduced proliferation and enhanced caspase activity in sensitive cells. | In vivo ulixertinib showed dose-dependent growth inhibition and tumor regression. ulixertinib yielded synergistic antiproliferative effects in a BRAFV600E-mutant melanoma cell line xenograft model when used in combination with BRAF inhibition. | Phase II NCT03417739 (n=13) | A phase II study to determine the efficacy and safety of ulixertinib in patients with mUM. | Median time to progression 2.0 months (90% CI: 1.8-3.6 mos.). Median survival time 6.9 months (90%CI: 3.2 to 8.3 mos.). | ERK inhibition with ulixertinib did not demonstrate activity in patients with mUM. The toxicities observed in the study were consistent with what would be expected with MAPK pathway inhibition. | 100,101 |
| Sunitinib; (RTK inhibitor) | No uveal melanoma cell lines Neuroblastoma cell lines: SK-N-BE(2), SH-SY5Y, LAN-5, NUB-7 cells, NB12, NB25, and NB88R2. In vivo: NB xenograft models, SK-N-BE(2) cells or NB12 cells injected subcutaneously into the groin fat pad of the NOD-SCID mice. | Neuroblastoma cell lines, SK-N-BE(2), NUB-7, SH-SY5Y, and LAN-5, were exposed to increasing concentrations of sunitinib for 72 hours and assayed. In vitro Sunitinib inhibits tumour cell proliferation and phosphorylation of VEGFRs. | Treatment with 20 mg/kg of sunitinib showed significant reduction (P < .05) in primary tumour growth. Sunitinib inhibits tumour growth, angiogenesis, and metastasis in vivo. | Phase II NCT01551459 (n=124) | Patients with mUM and no prior systemic therapy for advanced disease. They were randomized 1:1 to sunitinib (50mg daily for 28 days, followed by a 14-day break), or dacarbazine (1000 mg/m2 once every 21 days). | Overall response rates of 0% and 8% were observed in the sunitinib and dacarbazine arms; while stable disease was observed in 24% of pts on sunitinib, and 11% on DTIC. PFS and OS were not improved with sunitinib. | In these preliminary results, sunitinib did not exhibit significant clinical activity in mUM. | 103,104 |
| Cediranib (Multi-kinase inhibitor, VEGF) | No uveal melanoma cell lines Eight immortalized glioblastoma cell lines were used: SW1088, SW1783, U-87 MG, A172, SNB-19, GAMG, U251 and U373. In vivo: 6-to 8-week-old female BALB/c mice, injected with RENCA cells. | Cediranib IC50 of 1.71 ± 0.97 μM (range, 0.47-4.17 μM) Cediranib in vitro on glioblastoma cell lines inhibited cellular migration and invasion. | Mice were treated once daily with 5 mg/kg cediranib. Cediranib in vivo reduces primary tumour in mice in a dependent exposing time manner. | Phase II (n=24) NCT00243061 | mUM cediranib was given on a continuous, oral once daily schedule of 45 mg, on a 28-day cycle. | Of the 17 patients evaluable for response, there was stable disease in 8 patients, and progressive disease in 9 patients, with no objective responses seen. Only 2 patients had stable disease ≥6 months. | Although 2 patients had stable disease at 6 months, the short median time to progression and lack of any objective responses indicate that single agent cediranib at this dose and schedule is not sufficiently active to warrant study continuation. | 105-107 |
| Sorafenib (Multi-kinase inhibitor, VEGF inhibitor) | No uveal melanoma cell lines Paediatric hepatocellular carcinoma cell lines: HC-AFW1, Huh7, HUH6, HepT1. In vivo: Xenograft model of HC-AFW1 cells | Tumour cell proliferation in the HC-AFW1 cell line was effectively inhibited by sorafenib. | Sorafenib was administered orally/day with a dosage of 60 mg/kg body weight. Treating mice bearing HC-AFW1-derived tumours with sorafenib only led to a moderate tumor growth inhibition. | Phase II NCT00329641 (n=25) | Patients with mUM who had received 0-1 prior systemic therapy were enrolled. Treatment included up to 6 cycles of carboplatin (AUC = 6) and paclitaxel (225 mg/m2 administered on day 1 plus sorafenib (400 mg PO twice daily), followed by sorafenib monotherapy until disease progression. | (ORR = 0% [95% CI: 0-14%]) This study was terminated at the initial stage. Tumour regression <30% in 11 of 24 (45%) patients. The median PFS was 4 months and the 6-month PFS was 29%. The median OS was 11 months. | The overall efficacy of CP plus sorafenib in mUM did not warrant further clinical testing when assessed by ORR, although minor tumour responses and stable disease were observed in some patients. | 108-110 |
Abbreviations: CL, cell line; sig, significant; TGI, tumour growth inhibition; OR, overall response; ORR, objective response rate; met, metastatic; RTK, receptor tyrosine kinase; PFS, progression free survival; OS, overall survival.
Another example of indirect targeting of GNAQ and GNA11 signalling is via protein kinase C (PKC) inhibition. PKC family proteins are involved in a number of pathways, including those regulating cell proliferation, differentiation, survival and death [95]. PKC signalling alterations can be co-expressed with GNA11, GNAQ, and less commonly, PLCβ4 mutations, all of which signal upstream of PKC. Thus, inhibition of PKC is a rational point of therapeutic intervention. Currently AEB071 and LXS196, PKC inhibitors, are in clinical trials both alone and in combination with other drugs [96,97]. Preliminary data suggests clinical activity of AEB071 and manageable toxicity at multiple dose levels, with evidence of PKC inhibition in patients with UM [96]. Corresponding in vitro studies using primary and mUM and cutaneous melanoma cell lines, suggest that AEB071-induced growth suppression of GNAQ mutant cells is associated with pronounced G1 arrest, and induction of apoptosis [98]. While AEB071 inhibits PKC signalling in vivo in allograft mouse models, compensatory mechanisms prevent suppression of the MAP-kinase pathway. However, combinations of PKC and MEK inhibition were efficacious in vitro and in vivo causing marked tumor regression in a UM xenograft model [99].
A recent phase II clinical trial carried out by Buchbinder et al. investigated the anti-tumour capacity of ulixertinib (BVD-523) (ERK inhibitor) in mUM [100]. While preclinical in vitro studies by Germann et al. involving ulixertinib, exhibited anti-proliferative and pro-caspase activity, such studies were carried out on a variety of adenocarcinoma cell lines and malignant melanoma cell lines, and not UM cell lines [101]. Thus, the clinical translation of such models in the context of UM is unclear. Furthermore, a mouse model with a BRAF-mutated melanoma cell line xenograft was used as an in vivo model. Although ulixertinib demonstrated tumour growth inhibition and regression, as well as anti-proliferative synergism with BRAF inhibition, the absence of plausible UM models is questionable. Despite some parallels, in CM genetic aberrations and that of UM, there is a stark difference in each disease's genetic landscape, including the lack of BRAF mutations in UM, a hallmark of CM. Unfortunately, preliminary findings did not translate to the clinic. Progression as far as phase II revealed that ERK inhibition with ulixertinib was unsuccessful as a treatment for mUM, with no apparent improvement in PFS and OS observed [100]. This negative result is likely owing to the use of unsuitable preclinical UM models, which may have proved poor predictors of the true clinical outcome.
Receptor tyrosine kinases (RTKs) have also been implicated in tumorigenesis. RTKs are involved in the processes of cell-cell communication, cell growth and differentiation, among many, and are associated with growth and malignancy in cancers [102]. Sunitinib is a multi-targeted receptor targeted (RTK) kinase inhibitor against VEGFR, FMS-like tyrosine kinase 3 (FLT-3), C-KIT and platelet-derived growth factor receptor (PDGFR). A randomised phase II clinical trial combining sunitinib or DTIC was carried out on mUM patients, and found no improvement in PFS or OS, and lacked significant clinical activity [103]. The in vitro effects of sunitinib have been studied but in non-UM preclinical models. Studies in neuroblastoma cell lines and acral melanoma cell lines found sunitinib inhibited tumour cell proliferation and phosphorylation of VEGFRs. Similarly, in vivo sunitinib caused a reduction of tumour growth, angiogenesis, and metastasis of neuroblastoma models in vivo [104].
Vascular endothelial growth factor (VEGF) is another common target in cancer treatments. VEGF signalling promotes tumour survival as it plays a crucial role in tumour angiogenesis, and therefore, inhibitors of this pathway can have potent anti-tumour effects. Multi-kinase inhibitors such as cediranib and sorafenib, have been investigated as potential therapies for the treatment of UM. In a phase II clinical trial by McWhirter et al. cediranib was administered continuously on a 28-day cycle to mUM patients. While some patients had a stable disease following treatment, in the vast majority of cases, disease progression occurred over a short period of time. Moreover, the lack of OR observed in this study group indicates that cediranib may not be efficacious as a single agent treatment in UM [105]. Preclinical assessment was carried out using non-UM glioblastoma cell lines and mice with RENCA cell xenografts. In vitro studies showed that cediranib inhibits cellular migration and invasion, while in vivo, cediranib caused a significant reduction in tumour volume over a range of drug treatment periods [106,107]. Sorafenib has also been examined clinically as a mUM therapy. Phase II clinical trials were carried out on sorafenib in combination with carboplatin and paclitaxel in patients with mUM (SWOG S0512). Minor tumour responses and disease stability were observed in some patients, however, due to the poor OR, clinical examination was not further progressed using sorafenib and the combination [108]. Pre-clinical non-UM cell lines such as those derived from hepatocellular carcinoma demonstrated anti-cancer properties to sorafenib [109]. Sorafenib has been tested in a xenograft model in which uveal melanoma cell line 92.1 was dorsally injected subcutaneously; this study found that sorafenib inhibited of tumour growth (p≤0.0035) and fewer metastases after sorafenib treatment were observed (33% vs. 60%) [110]. However, this did not translate into clinical efficacy of sorafenib for the treatment of UM.
Conclusion
Preclinical modelling and drug screening in established cell lines and animal models are an integral part in bridging the translational gap (Figure 1), providing a roadmap for regulatory drug development and approval. While the current models in place for studying UM have proved vital in gaining a greater understanding of the disease, as with most preclinical models - they do not fully recapitulate the in vivo situation, and to date, no UM preclinical studies have predicted effective translation to the clinic. Therefore, better strategies are needed to improve the translational relevance of preclinical to clinical translational research in UM.
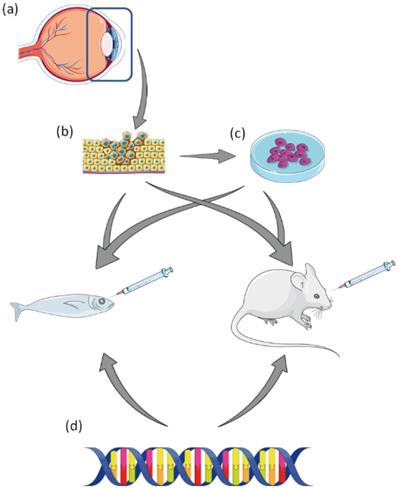
A summary of pre-clinical laboratory models of Uveal Melanoma. (a) Tumour resection of affected eye. (b) Resected tumour can be used to generate UM cell lines, or patient derived xenograft (PDX) in animal models. (c) Established UM cell lines can be used to generate xenograft animal models. (d) UM animal models can be created by genetic manipulation via transgenesis. This figure was created using Servier Medical Art templates, which have been modified. These images are licensed under a Creative Commons Attribution 3.0 Unported License; https://smart.servier.com.
Future directions
Advancements in in vitro cellular models such as 3D organoids have the potential to create a more accurate representation of how tumours grow and interact with surrounding cells and tissues. Preclinical models that reflect their original cancer help to make preclinical data more reproducible and translatable to the clinic. Therefore, utilisation of preclinical model systems which fully recapitulate the human in vivo situation may enhance the translational impact of experimental data, and inform future strategies for clinical impact.
Acknowledgements
This work was funded by a research grant from The Royal Victoria Eye and Ear Hospital Research Foundation (Annual Grant Scheme 2019) (BT/SK), IRC-Enterprise Partnership post-graduate fellowship (EPSPG/2020/401) (LP) and Cancer Clinical Research Trust (NW).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Singh AD, Topham A. Incidence of uveal melanoma in the United States: 1973-1997. Ophthalmology. 2003;110:956-61
2. Kaliki S, Shields CL, Shields JA. Uveal melanoma: Estimating prognosis. In: Indian Journal of Ophthalmology. 2015 p: 93-102
3. Rantala ES, Hernberg M, Kivelä TT. Overall survival after treatment for metastatic uveal melanoma: a systematic review and meta-analysis. Melanoma Res. 2019;29:561-8
4. Carvajal RD, Schwartz GK, Tezel T. et al. Metastatic disease from uveal melanoma: Treatment options and future prospects. Br. J. Ophthalmol. 2017;101:38-44
5. Weis E, Shah CP, Lajous M. et al. The association between host susceptibility factors and uveal melanoma: A meta-analysis. Arch Ophthalmol. 2006;124:54-60
6. Baily C, O'Neill V, Dunne M. et al. Uveal Melanoma in Ireland. Ocul Oncol Pathol. 2019;5:195-204
7. Shields CL, Furuta M, Thangappan A. et al. Metastasis of uveal melanoma millimeter-by-millimeter in 8033 consecutive eyes. Arch Ophthalmol. 2009;127:989-98
8. Rennie IG. The Ashton Lecture. Uveal melanoma: The past, the present and the future. In: Eye. 1997 p: 255-64
9. Paul E V, Parnell BL, Fraker M. Prognosis of malignant melanomas of the choroid and ciliary body. Int Ophthalmol Clin. 1962;2:387-402
10. Yousef YA, Alkilany M. Characterization, treatment, and outcome of uveal melanoma in the first two years of life. Hematol Oncol Stem Cell Ther. 2015;8:1-5
11. Patel KA, Edmondson ND, Talbot F. et al. Prediction of prognosis in patients with uveal melanoma using fluorescence in situ hybridisation. Br J Ophthalmol. 2001;85:1440-4
12. Onken MD, Worley LA, Ehlers JP. et al. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004;64:7205-9
13. Johansson P, Aoude LG, Wadt K. et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget. 2016;7:4624-31
14. Moore AR, Ceraudo E, Sher JJ. et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet. 2016;48:675-80
15. Van Raamsdonk CD, Bezrookove V, Green G. et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599-602
16. Van Raamsdonk CD, Griewank KG, Crosby MB. et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191-9
17. Robertson AG, Shih J, Yau C. et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell. 2017;32:204-220
18. Wei Z, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12:9-18
19. Szalai E, Wells JR, Ward L. et al. Uveal Melanoma Nuclear BRCA1-Associated Protein-1 Immunoreactivity Is an Indicator of Metastasis. Ophthalmology. 2018;125:203-9
20. Harbour JW. Genomic, Prognostic, and Cell-Signaling Advances in Uveal Melanoma. Am Soc Clin Oncol Educ B. 2013 p:388-91
21. Affar EB, Carbone M. BAP1 regulates different mechanisms of cell death. Cell Death Dis. 2018 9
22. Kilic E, Van Gils W, Lodder E. et al. Clinical and cytogenetic analyses in uveal melanoma. Investig Ophthalmol Vis Sci. 2006;47:3703-7
23. Shields CL, Say EAT, Hasanreisoglu M. et al. Cytogenetic Abnormalities in Uveal Melanoma Based on Tumor Features and Size in 1059 Patients: The 2016 W. Richard Green Lecture. Ophthalmology. 2017;124:609-18
24. Decatur CL, Ong E, Garg N. et al. Driver mutations in uveal melanoma associations with gene expression profile and patient outcomes. JAMA Ophthalmol. 2016;134:728-33
25. Shain AH, Bagger MM, Yu R. et al. The genetic evolution of metastatic uveal melanoma. Nat Genet. 2019;51:1123-30
26. Martin M, Maßhöfer L, Temming P. et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45:933-6
27. van Poppelen NM, Drabarek W, Smit KN. et al. SRSF2 mutations in uveal melanoma: A preference for in-frame deletions? Cancers (Basel). 2019;11:1200
28. Rahman MA, Lin KT, Bradley RK. et al. Recurrent SRSF2 mutations in MDS affect both splicing and NMD. Genes Dev. 2020;34:413-27
29. Te Raa GD, Derks IAM, Navrkalova V. et al. The impact of SF3B1 mutations in CLL on the DNA-damage response. Leukemia. 2015;29:1133-42
30. Jiang G, Zhang S, Yazdanparast A, Li M. et al. Comprehensive comparison of molecular portraits between cell lines and tumors in breast cancer. BMC Genomics. 2016;17:525
31. Tonin PN, Hudson TJ, Rodier F. et al. Microarray analysis of gene expression mirrors the biology of an ovarian cancer model. Oncogene. 2001;20:6617-26
32. Folberg R, Kadkol SHS, Frenkel S. et al. Authenticating cell lines in ophthalmic research laboratories. Investig Ophthalmol Vis Sci. 2008;49:4697-701
33. Korch C, Hall EM, Dirks WG. et al. Authentication of M14 melanoma cell line proves misidentification of MDA-MB-435 breast cancer cell line. Int J Cancer. 2018;142:561-72
34. Rimoldi D, Salvi S, Liénard D. et al. Lack of BRAF mutations in uveal melanoma. Cancer Res. 2003;63:5712-5
35. De Waard-Siebinga I, Blom DR, Griffioen M. et al. Establishment and characterization of an uveal-melanoma cell line. Int J Cancer. 1995;62:155-61
36. Jager MJ, Magner JAB, Ksander BR. et al. Uveal Melanoma Cell Lines: Where Do They Come From? Trans Am Ophthalmol Soc. 2016;114:1-16
37. Ksander BR, Rubsamen PE, Olsen KR. et al. Studies of tumor-infiltrating lymphocytes from a human choroidal melanoma. Investig Ophthalmol Vis Sci. 1991;32:3198-208
38. Verbik DJ, Murray TG, Tran JM. et al. Melanomas that develop within the eye inhibit lymphocyte proliferation. Int J Cancer. 1997;73:470-8
39. Chen PW, Murray TG, Uno T. et al. Expression of MAGE genes in ocular melanoma during progression from primary to metastatic disease. 1997;15:509-518.
40. Luyten GPM, Naus NC, Mooy CM. et al. Establishment and characterization of primary and metastatic uveal melanoma cell lines. Int J Cancer. 1996;66:380-7
41. Amirouchene-Angelozzi N, Nemati F, Gentien D. et al. Establishment of novel cell lines recapitulating the genetic landscape of uveal melanoma and preclinical validation of mTOR as a therapeutic target. Mol Oncol. 2014;8:1508-20
42. Nareyeck G, Zeschnigk M, Prescher G. et al. Establishment and characterization of two uveal melanoma cell lines derived from tumors with loss of one chromosome 3. Exp Eye Res. 2006;83:858-64
43. Nareyeck G, Zeschnigk M, Bornfeld N. et al. Novel cell lines derived by long-term culture of primary uveal melanomas. Ophthalmologica. 2009;223:196-201
44. Griewank KG, Yu X, Khalili J, Sozen MM. et al. Genetic and molecular characterization of uveal melanoma cell lines. Pigment Cell Melanoma Res. 2012;25:182-7
45. Suesskind D, Gauss S, Faust UEA. et al. Characterisation of novel uveal melanoma cell lines under serum-free conditions. Graefe's Arch Clin Exp Ophthalmol. 2013;251:2063-70
46. The Wistar Institute Cancer Centre. https://www.wistar.org/our-scientists/meenhard-herlyn
47. Burgess BL, Rao NP, Eskin A. et al. Characterization of three cell lines derived from fine needle biopsy of choroidal melanoma with metastatic outcome. Mol Vis. 2011;17:607-15
48. Aughton K, Shahidipour H, Djirackor L. et al. Characterization of Uveal Melanoma Cell Lines and Primary Tumor Samples in 3D Culture. Transl Vis Sci Technol. 2020;9:39
49. Gonçalves J, Emmons MF, Faião-Flores F. et al. Decitabine limits escape from MEK inhibition in uveal melanoma. Pigment Cell Melanoma Res. 2020;33:507-14
50. Fornabaio G, Barnhill RL, Lugassy C. et al. Angiotropism and extravascular migratory metastasis in cutaneous and uveal melanoma progression in a zebrafish model. Sci Rep. 2018;8:1-12
51. Barisione G, Fabbi M, Gino A. et al. Potential role of soluble c-met as a new Candidate biomarker of metastatic uveal melanoma. JAMA Ophthalmol. 2015;133:1013-21
52. Forsberg EMV, Lindberg MF, Jespersen H. et al. HER2 CAR-T cells eradicate uveal melanoma and T-cell therapy-resistant human melanoma in IL2 transgenic NOD/SCID IL2 receptor knockout mice. Cancer Res. 2019;79:899-904
53. Zhou J, Jin B, Jin Y. et al. The antihelminthic drug niclosamide effectively inhibits the malignant phenotypes of uveal melanoma in vitro and in vivo. Theranostics. 2017;7:1447-62
54. Némati F, Sastre-Garau X, Laurent C. et al. Establishment and characterization of a panel of human uveal melanoma xenografts derived from primary and/or metastatic tumors. Clin Cancer Res. 2010;16:2352-62
55. Kageyama K, Ohara M, Saito K. et al. Establishment of an orthotopic patient-derived xenograft mouse model using uveal melanoma hepatic metastasis. J Transl Med. 2017;15:145
56. Carita G, Frisch-Dit-Leitz E, Dahmani A. et al. Dual inhibition of protein kinase C and p53-MDM2 or PKC and mTORC1 are novel efficient therapeutic approaches for uveal melanoma. 2016;7:33542-56.
57. Mintz B, Klein-Szanto AJP. Malignancy of eye melanomas originating in the retinal pigment epithelium of transgenic mice after genetic ablation of choroidal melanocytes. Proc Natl Acad Sci USA. 1992;89:11421-5
58. Feng X, Degese MS, Iglesias-Bartolome R. et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a Trio-regulated Rho GTPase Signaling Circuitry. Cancer Cell. 2014;25:831-45
59. Vader MJC, Madigan MC, Versluis M. et al. GNAQ and GNA11 mutations and downstream YAP activation in choroidal nevi. Br J Cancer. 2017 117
60. Moore AR, Ran L, Guan Y. et al. GNA11 Q209L Mouse Model Reveals RasGRP3 as an Essential Signaling Node in Uveal Melanoma. Cell Rep. 2018;22:2455-68
61. Tolleson WH, Doss JC, Latendresse J. et al. Spontaneous uveal amelanotic melanoma in transgenic Tyr-RAS Ink4a/Arf-/- mice. Arch Ophthalmol. 2005;123:1088-94
62. Cruz F, Rubin BP, Wilson D. et al. Absence of BRAF and NRAS Mutations in Uveal Melanoma. Cancer Res. 2003;63:5761-6
63. Schiffner S, Braunger BM, de Jel MM. et al. Tg(Grm1) transgenic mice: A murine model that mimics spontaneous uveal melanoma in humans? Exp Eye Res. 2014;127:59-68
64. Huang JLY, Urtatiz O, Van Raamsdonk CD. Oncogenic G protein GNAQ induces uveal melanoma and intravasation in mice. Cancer Res. 2015;75:3384-97
65. Heegaard S, Spang-Thomsen M, Prause JU. Establishment and characterization of human uveal malignant melanoma xenografts in nude mice. Melanoma Res. 2003;13:247-51
66. Lafave LM, Béguelin W, Koche R. et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. 2015;21:1344-9
67. Howe K, Clark MD, Torroja CF. et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496:498-503
68. Berghmans S, Murphey RD, Wienholds E. et al. tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc Natl Acad Sci USA. 2005;102:407-12
69. Duffy KT, McAleer MF, Davidson WR. et al. Coordinate control of cell cycle regulatory genes in zebrafish development tested by cyclin D1 knockdown with morpholino phosphorodiamidates and hydroxyprolyl-phosphono peptide nucleic acids. Nucleic Acids Res. 2005;33:4914-21
70. Harrison NR, Laroche FJF, Gutierrez A. et al. Zebrafish models of human leukemia: Technological advances and mechanistic insights. Adv Exp Med Biol. 2016;916:335-69
71. White R, Rose K, Zon L. Zebrafish cancer: The state of the art and the path forward. Nat. Rev. Cancer. 2013;13:624-36
72. Xiao J, Glasgow E, Agarwal S. Zebrafish Xenografts for Drug Discovery and Personalized Medicine. Trends in Cancer. 2020;6:569-79
73. Jivrajani M, Shaikh MV, Shrivastava N. et al. An improved and versatile immunosuppression protocol for the development of tumor xenograft in mice. Anticancer Res. 2014;34:7177-83
74. Kan-Mitchell J, Mitchell MS, Liggett PE. Characterization of Uveal Melanoma Cell Lines that Grow as Xenografts in Rabbit Eyes. Invest Ophthalmol Vis Sci. 1989;30:829-34
75. Meyers JR. Zebrafish: Development of a Vertebrate Model Organism. Curr Protoc Essent Lab Tech. 2018 16
76. van der Ent W, Burrello C, Teunisse AFAS. et al. Modeling of human uveal melanoma in zebrafish xenograft embryos. Invest Ophthalmol Vis Sci. 2014;55:6612-22
77. Frazer JK, Meeker ND, Rudner L. et al. Heritable T-cell malignancy models established in a zebrafish phenotypic screen. Leukemia. 2009;23:1825-35
78. Langenau DM, Traver D, Ferrando AA. et al. Myc-induced T cell leukemia in transgenic zebrafish. Science. 2003;299:887-90
79. Rose KM. A Zebrafish Model of Uveal Melanoma. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. 2015
80. Maeda K, Hamada JI, Takahashi Y. et al. T. Altered expressions of HOX genes in human cutaneous malignant melanoma. Int J Cancer. 2005;114:436-41
81. Saldanha G, Purnell D, Fletcher A. et al. High BRAF mutation frequency does not characterize all melanocytic tumor types. Int J Cancer. 2004;111:705-10
82. Zuidervaart W, Van Nieuwpoort F, Stark M. et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br J Cancer. 2005;92:2032-8
83. Mouti MA. Engineering a Genetically Relevant Zebrafish Model of Human Uveal Melanoma. Doctoral dissertation, University of Manchester, Faculty of Life Sciences. 2016
84. Mouti MA, Dee C, Coupland SE. et al. Minimal contribution of ERK1/2-MAPK signalling towards the maintenance of oncogenic GNAQQ209P-driven uveal melanomas in zebrafish. Oncotarget. 2016;7:39654-70
85. Perez DE, Henle AM, Amsterdam A. et al. Uveal melanoma driver mutations in GNAQ/11 yield numerous changes in melanocyte biology. Pigment Cell Melanoma Res. 2018;31:604-13
86. Ju B, Spitsbergen J, Eden CJ. et al. Co-activation of hedgehog and AKT pathways promote tumorigenesis in zebrafish. Mol Cancer. 2009;8:40
87. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291-310
88. Carvajal RD, Schwartz GK, Mann H. et al. Study design and rationale for a randomised, placebo-controlled, double-blind study to assess the efficacy of selumetinib (AZD6244; ARRY-142886) in combination with dacarbazine in patients with metastatic uveal melanoma (SUMIT). BMC Cancer. 2015;15:467
89. Carvajal RD, Piperno-Neumann S, Kapiteijn E. et al. Selumetinib in Combination With Dacarbazine in Patients With Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J Clin Oncol. 2018;36:1232-9
90. Decaudin D, El Botty R, Diallo B. et al. Selumetinib-based therapy in uveal melanoma patient-derived xenografts. Oncotarget. 2018;9:21674-86
91. Ambrosini G, Musi E, Ho AL. et al. Inhibition of mutant GNAQ signaling in uveal melanoma induces AMPK-dependent autophagic cell death. Mol Cancer Ther. 2013;12:768-76
92. Park JJ, Stewart A, Irvine M. et al. Uveal melanoma cell lines depend on multiple signaling pathways for survival. Ann Oncol. 2019;30:v6-v7
93. Decaudin D, Frisch Dit Leitz E, Nemati F. et al. Preclinical evaluation of drug combinations identifies co-inhibition of Bcl-2/XL/W and MDM2 as a potential therapy in uveal melanoma. Eur J Cancer. 2020;126:93-103
94. Shoushtari AN, Kudchadkar RR, Panageas K. et al. A randomized phase 2 study of trametinib with or without GSK2141795 in patients with advanced uveal melanoma. J Clin Oncol. 2016;34:9511
95. Black AR, Black JD. Protein kinase C signaling and cell cycle regulation. Frontiers Immunol. 2012;3:423
96. Piperno-Neumann S, Kapiteijn E, Larkin JMG. et al. Phase I dose-escalation study of the protein kinase C (PKC) inhibitor AEB071 in patients with metastatic uveal melanoma. J Clin Oncol. 2014;32:9030-9030
97. Kapiteijn E, Carlino M, Boni V. et al. Abstract CT068: A Phase I trial of LXS196, a novel PKC inhibitor for metastatic uveal melanoma. In: Proceedings of the American Association for Cancer Research Annual Meeting 2019; Cancer Res. 2019;79(13 Suppl):Abstract nr CT068
98. Wu X, Li J, Zhu M. et al. Protein kinase C inhibitor AEB071 targets ocular melanoma harboring GNAQ mutations via effects on the PKC/Erk1/2 and PKC/NF-κB pathways. Mol Cancer Ther. 2012;11:1905-14
99. Chen X, Wu Q, Tan L. et al. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:4724-34
100. Buchbinder EI, Cohen JV, Haq R. et al. A phase II study of ERK inhibition by ulixertinib (BVD-523) in metastatic uveal melanoma. J Clin Oncol. 2020;38:10036
101. Germann UA, Furey BF, Markland W. et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol Cancer Ther. 2017;16:2351-63
102. Pottier C, Fresnais M, Gilon M. et al. Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers (Basel). 2020;12:731
103. Sacco JJ, Nathan PD, Danson S. et al. Sunitinib versus dacarbazine as first-line treatment in patients with metastatic uveal melanoma. J Clin Oncol. 2013;31:9031s
104. Zhang L, Smith KM, Chong AL. et al. In vivo antitumor and antimetastatic activity of Sunitinib in preclinical Neuroblastoma mouse model. Neoplasia. 2009;11:426-35
105. McWhirter E, Quirt I, Gajewski T. et al. A phase II study of cediranib, an oral VEGF inhibitor, in previously untreated patients with metastatic or recurrent malignant melanoma. Invest New Drugs. 2016;34:231-5
106. Medinger M, Esser N, Zirrgiebel U. et al. Antitumor and antiangiogenic activity of cediranib in a preclinical model of renal cell carcinoma. Anticancer Res. 2009;29:5065-76
107. Martinho O, Silva-Oliveira R, Miranda-Gonçalves V. et al. In vitro and In vivo Analysis of RTK Inhibitor Efficacy and Identification of its Novel Targets in Glioblastomas. Transl Oncol. 2013;6:187-96
108. Bhatia S, Moon J, Margolin KA. et al. Phase II Trial of Sorafenib in Combination with Carboplatin and Paclitaxel in Patients with Metastatic Uveal Melanoma: SWOG S0512. PLoS One. 2012 7
109. Nagel C, Armeanu-Ebinger S, Dewerth A. et al. Anti-tumor activity of sorafenib in a model of a pediatric hepatocellular carcinoma. Exp Cell Res. 2015;331:97-104
110. Mangiameli DP, Blansfield JA, Kachala S. et al. Combination therapy targeting the tumor microenvironment is effective in a model of human ocular melanoma. J Transl Med. 2007;5:38
Author contact
![]() Corresponding author: Naomi Walsh, National Institute for Cellular Biotechnology, School of Biotechnology, Dublin City University, Dublin, Ireland. Tel: + 35317005912; E-mail: naomi.walshie.
Corresponding author: Naomi Walsh, National Institute for Cellular Biotechnology, School of Biotechnology, Dublin City University, Dublin, Ireland. Tel: + 35317005912; E-mail: naomi.walshie.