Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(20):6198-6208. doi:10.7150/jca.61602 This issue Cite
Research Paper
Chidamide, a subtype-selective histone deacetylase inhibitor, enhances Bortezomib effects in multiple myeloma therapy
Yanjuan He1, Duanfeng Jiang2, Kaixuan Zhang1, Yinghong Zhu3, Jingyu Zhang3, Xuan Wu3, Jiliang Xia3, Yan Zhu1, Lang Zou1, Jian Hu1, Yajuan Cui4, Wen Zhou3, Fangping Chen1 ![]()
1. Department of Hematology, Xiangya Hospital, Central South University, Changsha, Hunan, China.
2. Department of Hematology, The 3rd Xiangya Hospital, Central South University, Changsha, China.
3. Cancer Research Institute, School of Basic Medical Sciences, Central South University, Changsha, China.
4. Department of Hematology, The Second Xiangya Hospital, Central South University, Changsha, China.
Received 2021-4-13; Accepted 2021-8-15; Published 2021-8-27
Abstract

Drug resistance is the major cause for disease relapse and patient death in multiple myeloma (MM). It is an urgent need to develop new therapies to overcome drug resistance in MM. Chidamide (CHI), a novel oral HDAC inhibitor targeting HDAC1, 2, 3 and 10, has shown potential therapeutic effect in MM. In this study, we determined that CHI exhibited significant anti-tumor effect on MM cells both in vitro and in vivo, which was positively correlated with the expression of HDAC1. Meanwhile, CHI enhanced Bortezomib (BTZ) effects synergistically in MM cells and a combination of CHI with BTZ induced myeloma cell apoptosis and G0/G1 arrest in vitro and in vivo. Mechanistically, the synergistic anti-tumor effect of CHI and BTZ was related with the increased production of reactive oxygen species (ROS) dependent DNA damage and the changes of cell apoptosis and cycle pathways. Our data indicate that CHI may be a suitable drug to sensitize BTZ in MM cells, which provides novel insight into the therapy for MM patients.
Keywords: Multiple myeloma, Chidamide, Bortezomib, Reactive oxygen species
Introduction
Multiple myeloma (MM) is a malignant disease characterized by proliferation of clonal plasma cells in the bone marrow and accompanied by detectable monoclonal immunoglobulins (M protein) in patients' serum or urine. Together with autologous stem cell transplantation (ASCT), multiple novel chemotherapeutic drugs such as proteasome inhibitors, immunomodulatory drugs, alkylating agents and corticoids have significantly increased response rates and patients' survival in MM in the past decades [1, 2]. However, MM is still a difficult-to-cure disease due to inevitable drug resistance, and development of novel therapies are urgently required.
The two major types of epigenetic modifications - DNA methylation and histone modification, both are important in the pathogenesis of MM. Though targeting DNA methylation has not yet been fully developed, the histone deacetylases inhibitors (HDACi) have shown promising therapeutic effects in MM [3]. Protein acetylation has various functions including modulation of gene expression, DNA replication and repair, cell cycle progression, cytoskeletal reorganization and protein chaperone activity [4]. Increased expression of HDACs has been identified in MM cells. High expression levels of Class I HDAC, particular HDAC1, are associated with poor prognosis in MM disease [5]. The HDACi as single agent induces MM cell death mainly through the inhibition of class I HDAC [6]. The class I specific HDACi, romidepsin, along with the pan-HDACi dacinostat (LAQ824), belinostat, vorinostat, and panobinostat showed clearly anti-myeloma cells in vitro and in vivo [7, 8]. Moreover, HDACi panobinostat, in combination with bortezomib and dexamethasone, has been approved as the first HDAC inhibitor to treat MM [9, 10].
Chidamide (CHI) (CS055/HBI-8000), discovered and developed by Chipscreen Biosciences, is a novel orally active HDACi with subtype selective activity against HDAC1, 2, 3 and 10 [11]. CHI was approved by the Chinese Food and Drug Administration (CFDA) for the treatment of recurrent or refractory peripheral T-cell lymphoma (PTCL) patients in December 2014. CHI was the first listed benzamide class of HDACi for global clinical trials in solid tumors including non-small cell lung cancer, breast cancer and prostate cancer in the United States and China. As an epigenetic modulator, CHI induced tumor cell growth arrest and apoptosis and enhanced cellular antitumor immunity [12-14]. CHI exhibited significant cytotoxicity against MM cells co-cultured with bone mesenchymal stromal cells and CHI-pretreated osteoclasts in recent studies [15-17].
The mechanism of proteasome inhibitor Bortezomib (BTZ), leading to disruption of intracellular protein metabolism, are well characterized [18, 19]. HDAC1 overexpression conferred resistance to BTZ in MM cells, and administration of the HDACi romidepsin restored sensitivity to BTZ in HDAC1-overexpressing cells both in vitro and in vivo [20]. Based on preclinical data, combining HDACi with proteasome inhibitors such as BTZ represent an attractive strategy for the treatment of patients with MM [8]. CHI, an improved and cheaper HDACi, has also achieved exciting result on PTCL therapy. Thus, it is meaningful to explore the single drug anti-myeloma of CHI or synergistic anti-myeloma activity of CHI with BTZ in MM.
In this study, we found that the expression level of HDAC1 was different in various MM cell lines and higher expression of HDAC1 in MM cell line resulted in more sensitivity to CHI. Meanwhile, the cell line resistant to BTZ had a higher expression level of HDAC1 and was more sensitive to CHI than wild type cell line. The effect of single drug CHI on MM cells, and the synergistic anti-myeloma effect of CHI and BTZ in vitro and in vivo was confirmed. Moreover, the potential mechanism of CHI synergized with BTZ was further explored in this study. Taken together, our findings will provide a novel and individuation therapeutic strategy for patients with MM.
Methods
Drugs and reagents
CHI was kindly provided by Shenzhen Chipscreen Biosciences, Ltd (Shenzhen, China). BTZ was obtained from BSP Pharmaceuticals S.p.A (Lazio, ITA), dissolved in saline solution as a stock solution for 5 mmol/L, aliquoted and stored at -80 °C. The following antibodies were used: rabbit anti-HDAC1 (catalog #AF0178), rabbit anti-cleaved Caspase-3 (catalog #AF7022), rabbit anti-cleaved PARP (catalog #AF7023), rabbit anti-cleaved Caspase-8 (cleaved-Asp384; catalog #AF5267), rabbit anti-γ-H2AX (Tyr143; catalog #AF8482), mouse monoclonal anti-β-actin (catalog #T0022) and goat anti-rabbit Alexa Fluor 594 (catalog #S0006) were purchased from Affinity Biosciences (Cincinnati, OH, USA). Rabbit anti-Ki67 (catalog #GB13030-2) was purchased from Servicebio (Woburn, MA). Matrigel (catalog #356237) was purchased from BD Biosciences Discovery Labware (Two Oak Park, Bedford, MA).
Cell culture
The human MM cell lines XG1, KMS-11, KMS-28, ANBL-6, RPMI-8226, ARP-1 and OCI-my5 were obtained from the Cancer Research Institute of Central South University. All MM cells were grown in RPMI 1640 (Gibco, USA) medium supplemented with 10% fetal bovine serum (Gibco, USA) at 37 °C in a 5% CO2 incubator.
Primary samples
Newly diagnosed or relapsed MM cases were defined according to the classification in the International Myeloma Working Group (IMWG) guidelines [21]. Two cases of newly diagnosed MM and one case of relapsed MM bone marrow samples were obtained from the Xiangya Hospital, Central South University. This study was approved by the Xiangya Hospital Ethics Review Board in accordance with the Declaration of Helsinki. Acquisition of bone marrow samples was performed with the informed consents of the patients. Mononuclear cells were isolated by density gradient centrifugation using Lymphoprep™ (Solarbio, China) and cultured in RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg/ml streptomycin (1 × P/S). The clinical information of clinical samples was described in Supplementary Table S1.
Cell viability assay
The indicated cells were planted in the 96-well plates (5 × 104 cells / well) and treated with different concentrations of CHI or combined with BTZ for 48 hours. Cell viability was assessed using the CCK-8 cell proliferation kit according to the manufacturer's instructions (Dojindo Laboratories, Kumamoto, Japan).
Quantification of the synergism of CHI with BTZ
XG1 and ARP-1 cells were treated with different doses of CHI and BTZ in monotherapy and in combinations for 48 hours. The following doses of CHI (in μmol/L) and BTZ (in nmol/L) were used: 0:0, 0.5:0.5, 1:1, 2:2, 4:4, 8:8, 16:16, 32:32. The combination index (CI) was calculated by CompuSyn for Drug Combinations and General Dose-Effect Analysis (ComboSyn, Inc. 599 MillRun, Paramus, NJ, 07653, USA), which was based on the Chou Talalay method [22] with the following interpretation: CI > 1: antagonistic effect, CI = 1: additive effect and CI < 1: synergistic effect.
Soft agar colony forming assay
The effect of CHI and BTZ on ARP-1 cells was determined using a soft agar colony forming assay [23]. Briefly, 1 × 106 ARP-1 cells were treated with 1.0 μmol/L of CHI, 5.0 nmol/L of BTZ or the combination for 24 hours. Then the cells were counted and 1.5 × 103 cells were plated in duplicate containing 0.5 mL of RPMI 1640 medium with 20% FBS, 0.28% agarose (low gelling temperature, Dalian Meilun Biotechnlogy Co., China) into 12-well tissue-culture plates, where pre-placed with 1.0 mL of RPMI 1640 medium with 20% FBS, 0.58% agarose at the bottom. After 8 days of incubation at 37°C in a 95% humidified atmosphere containing 5% CO2, colonies were counted using an inverted microscope and a graph was plotted.
Cell cycle and cell apoptosis detection
For the cell cycle assay, indicated cells were treated with different doses of CHI or combined with BTZ for 24 hours, and 2 × 105 cells were collected and fixed with 75%-80% ethanol at -20 °C for 24 hours. After centrifugation, cells were washed with phosphate buffer solution (PBS), and then incubated with propidium iodide (PI) and RNase-A (US Everbright Inc, USA) for 30 minutes at room temperature. DNA contents were analyzed by flow cytometry. For the cell apoptosis assay, cells were treated with different doses of CHI or combined with BTZ for 48 hours. Cells (2 × 105) were harvested at various intervals after treatment, washed with ice-cold PBS and resuspended in 400 μL binding buffer. 20 μg/mL FITC-Annexin V and 5 μg/mL PI were added and cells were incubated for 15 minutes in a dark environment, according to the manufacturer's instructions of FITC-Annexin V and propidium iodide (PI) apoptosis kit (US Everbright Inc, USA).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated using Trizol reagent (Invitrogen, USA). Gene expression (mRNA) was analyzed using the Mastercycler ep realplex2 (Eppendorf, GER) in a two-step real-time PCR (95 °C for 5 minutes, followed by 40 cycles of 95 °C for 10 seconds and 60 °C for 30 seconds). All PCR reactions were run in triplicate, and mRNA levels of target genes relative to β-actin were calculated using the 2-ΔΔCT method. The primer sequences used for PCR are listed in Supplementary Table S2.
Western blot
The following antibodies were used: antibodies against β-actin, cleaved caspase 3, cleaved caspase 8, cleaved PARP-1, γ-H2AX, HDAC1, HDAC2 and HDAC8 (Affinity, USA). Samples were prepared and resolved on 10% SDS-PAGE, and blotted onto 0.22 μm PVDF membranes (EMD Millipore Corporation, Billerica, MA, USA). Blots were visualized using the enhanced chemiluminescence (ECL) reagents (Thermo Scientific, USA).
Transfection of HDAC1 cDNA
ARP-1 cells treated with 1.0 μmol/L CHI or not were transfected with HDAC1 cDNA (Origene Technologies Inc., Rockville, MD, USA) using FuGENE HD Transfection Reagent according to the manufacturers' protocol. To determine the efficiency of cDNA, HDAC1 expression was assessed by Western blot assays.
Immunofluorescence assay
ARP-1 cells were cultured with 1.0 μmol/L CHI alone or combined with 5.0 nmol/L BTZ for 24 hours. Cells were harvested and dropped in the glass coverslips, then were fixed with 4% paraformaldehyde for 20 minutes, followed by three PBS rinses, permeabilized with 0.1% Triton X-100 (Sigma) for 15 minutes and blocked with 5% BSA in PBS for 1 hour at room temperature. The samples were then stained overnight at 4 °C with primary antibody against phospho-H2AX (1: 200, Affinity, USA), followed by incubation with Alexa Fluor 594 goat anti-rabbit IgG (Affinity, USA) for 1 hour at room temperature in the dark, and then were counterstained using DAPI (Sigma, USA). Subsequently, the coverslips were mounted on the glass slides. The cells were scanned and images were captured by confocal fluorescence microscope (Olympus, Japan).
Measurement of ROS Generation
Cells were pretreated with or without 15.0 mmol/L N-Acetyl-L-cysteine (NAC) for 2 hours at 37 °C and then incubated with various drugs for indicated times. Then the cells were washed with PBS, resuspended in RPMI 1640 medium containing 10.0 μmol/L of 2,7-dichlorodihydro-fluorescein diacetate (DCFH-DA) (Beyotime, China), and incubated at 37 °C for 20 minutes. Fluorescence intensity was assessed using a flow cytometer (BD, USA).
Myeloma xenograft mouse model
Mice experiment was performed under the protocol approved by the Institutional Animal Care and Use Committee of Central South University (NO. 2019sydw0105) in accordance with the Guidelines for the Care and Use of Laboratory Animals. NOD-Prkdcscid Il2rgtm1/Bcgen mice (deficient in mature T lymphocytes, B lymphocytes and NK cells) were purchased from Jiangsu Biocytogen Co., Ltd. Mice were raised in a super pathogen-free condition. MM xenograft mouse model was established by subcutaneous injection of 1 × 106 ARP-1 cells (in 100 μL of Matrigel-cell suspension mixture) into the left abdomen of mouse. Ten days later, when tumors became palpable, mice were randomized into four groups: Vehicle (control group), CHI (15.0 mg/kg), BTZ (1.0 mg/kg), and CHI (15.0 mg/kg) combined with BTZ (1.0 mg/kg). Tumor-bearing mice were treated with vehicle control, CHI (15.0 mg/kg by intraperitoneal injection (i.p), every other day for 14 days), BTZ (1.0 mg/kg i.p. 3 days weekly, interval administration). Caliper measurements of the tumor diameters were performed 3 days weekly, and the tumor volume was estimated as the volume of an ellipse using the following formula: 4/3π × (a/2) × (b/2)2. Tumor volume were monitored every 3 days. The animals were sacrificed at the end of the study when tumor volume reached 2000 mm3, at which time the tumors were excised, weighed, subjected to histological analysis and extraction of protein for Western blot analysis.
Statistical analysis
All data are expressed as means ± standard deviation (SD) and are representative of at least three separate experiments. The two-tailed Student t-test and one-way ANOVA were used for comparisons of two or more than two groups, respectively. p < 0.05 was considered statistically significant. GraphPad Prism 7.0 software (CA, USA) was used for the analysis.
Results
High expression of HDAC1 is sensitive to Chidamide in MM cells
As CHI was a subtype selective HDACi against HDAC1, 2, 3 and 10 [11], we first investigated the mRNA levels of Class I HDACs (HDAC1, 2, 3 and 8) and HDAC10 in MM ARP-1 and XG1 cells treated with different concentrations of CHI (0.5 μmol/L, 1 μmol/L, 2 μmol/L). Among these HDACs, qRT-PCR results showed that the expression of HDAC1 was the most significant inhibition by CHI in both ARP-1 and XG1 cells (Figure 1A). Then we focused the basal mRNA level of HDAC1 in different MM cell lines. As shown in Figure 1B, among these eight MM cell lines (XG1, ARP-1, OCI-my5, KMS-11, KMS-28, RPMI-8226, ANBL6-WT, ANBL6-BR), ARP-1 showed the highest HDAC1 expression at mRNA level, while XG1 showed the lowest. ANBL6-BR was BTZ-resistant cells with a higher HDAC1 expression than ANBL-6 wild-type (ANBL6-WT) cells (Figure 1B). Yet it was noteworthy that the protein levels of HDAC1 were not completely consistent with the mRNA levels in these myeloma cell lines (Supplementary Figure S1). Subsequently, these eight MM cell lines were treated with different doses of CHI (0.5 - 32.0 μmol/L) for 48 hours. CCK-8 results showed a dose-dependent pattern of CHI cytotoxicity and the IC50 values of CHI were ranged from 1.1 to 12.9 μmol/L (Figure 1C). ARP-1 was the cell line that most sensitive to CHI among these eight MM cell lines, and the IC50 values of CHI in those MM cell lines were negatively correlated with the mRNA expression of HDAC1 (Figure 1D), suggesting that the cytotoxicity of CHI on MM cells is partly dependent on the HDAC1 expression. To confirm the effect of CHI on the expression of HDAC1 in MM cells, ARP-1 and XG1 were treated with CHI for 48 hours, Western blot showed that CHI was effective in decreasing HDAC1 in both ARP-1 and XG1 cells (Figure 1E), and moderately or faintly decreased the expression of HDAC2 or HDAC8 protein in ARP-1 and XG1 cells, respectively (Supplementary Figure S2). Furthermore, ARP-1 with the higher HDAC1 expression was more sensitive to CHI than XG1, because the mRNA and protein expression of HDAC1 in ARP-1 cells was decreased by CHI at lower dose (Figure 1F). Thus, CHI could be a promising drug for MM patients with high expression of HDAC1 in tumor cells.
High expression of HDAC1 is sensitive to Chidamide in MM cells. (A) Relative mRNA levels of HDACs in ARP-1 and XG1 cells treated with different concentrations of CHI by qRT-PCR. (B) Relative mRNA levels of HDAC1 in MM cell lines by qRT-PCR. (C) CCK8 analysis of viability of MM cell lines treated with different concentrations of CHI. (D) The growth inhibition IC50 values in MM cell lines shows a negative correlation with relative mRNA levels of HDAC1. (E) The protein levels of HDAC1 were decreased with the increasing concentrations of CHI treatment for 48 hours in ARP-1 and XG1 cells. (F) The quantitation of mRNA and protein levels of HDAC1 in ARP-1 and XG1 cells treated with CHI. Error bars indicate mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; NS, not significant.
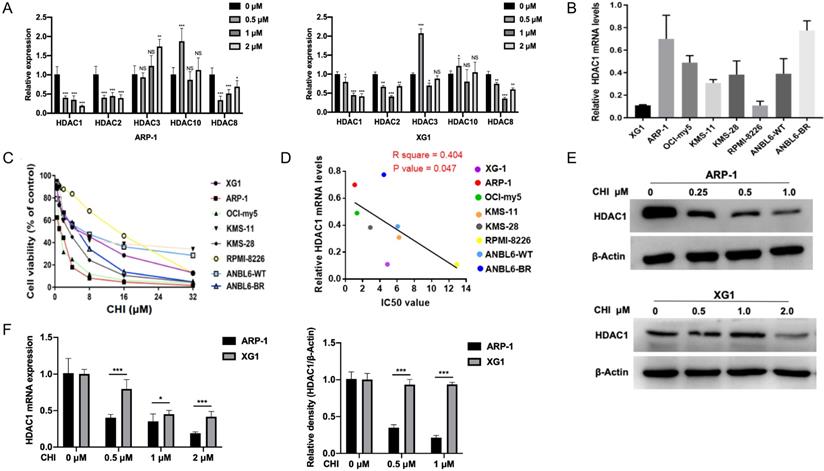
Chidamide inhibits proliferation and induces cell apoptosis and G0/G1 arrest in MM cells
To further evaluate the effect of CHI on MM, ARP-1 cells were treated with CHI (0.5-32.0 μmol/L) for 48 hours, and then cell viabilities were examined at different time points (24, 48, or 72 hours) using CCK-8. The result demonstrated a dose-dependent and time-dependent pattern of CHI cytotoxicity in ARP-1 cells (Figure 2A). Meanwhile, the clonogenic soft agar assay showed that the colonies were dramatically decreased in ARP-1 after treatment with CHI (Figure 2B). To further validate the role of HDAC1 in the cytotoxicity of CHI on MM cells, ARP-1 cells treated with 1.0 μmol/L CHI or not were transfected with HDAC1 cDNA for 48 hours. Western blot revealed that the expression of HDAC1 protein was successfully upregulated by transfection of HDAC1 cDNA (Figure 2C), and overexpression of HDAC1 could inhibit the anti-proliferation effect caused by CHI (Figure 2D).
Chidamide inhibits proliferation and induces cell apoptosis and G0/G1 arrest in MM cells. (A) CCK8 analysis of viability of ARP-1 cells treated with different concentrations of CHI (0, 2.0 µmol/L, 8.0 µmol/L, 32.0 µmol/L) for 24, 48, 72 hours, respectively. (B) Colony forming assay showed the colony formation rate of cells treated with different concentrations of CHI (0, 1.0 μmol/L, 4.0 µmol/L) for 48 hours in ARP-1 cells. (C) The expression of HDAC1 in ARP-1 cells treated with 1.0 µmol/L CHI or HDAC1 cDNA or not by Western blot. (D) CCK-8 analysis of viability of ARP-1 cells treated with 1.0 µmol/L CHI or HDAC1 cDNA or not. (E) Annexin V-FITC/PI double staining analysis of XG1 and ARP-1 cells treated with different concentrations of CHI (0, 0.5 µmol/L, 1.0 µmol/L, 2.0 µmol/L) for 48 hours. Percentages of MM cell apoptosis based on three independent experiments. (F) Cell cycle analysis of XG1 and ARP-1 cells treated with different concentrations of CHI (0, 0.5 µmol/L, 1.0 µmol/L, 2.0 µmol/L) for 24 hours. Percentages of the subpopulation of MM cells at different cell phases based on three independent experiments. Error bars indicate mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001.
Subsequently, we tried to determine the effect of CHI on apoptosis and cell cycles. ARP-1 and XG1 were treated with CHI (0, 0.5, 1, 2 μmol/L), and then apoptotic cells were examined by Annexin-V staining. Dose-dependent increases of apoptotic cells were observed in both ARP-1 and XG1, moreover, more apoptotic cells were detected in ARP-1 cells compared with XG1 cells when they were treated with the same dose of CHI (Figure 2E). Cell cycles results from flow cytometry (PI staining) analysis showed that the percentage of G1 phase was significantly increased in both ARP-1 and XG1 treating with CHI, while the percentages of S phase and G2 phase were decreased (Figure 2F), indicating that CHI induced G1 arrest in MM cells. Collectively, our results showed that inhibited proliferation and induced cell apoptosis and G0/G1 arrest in MM cells.
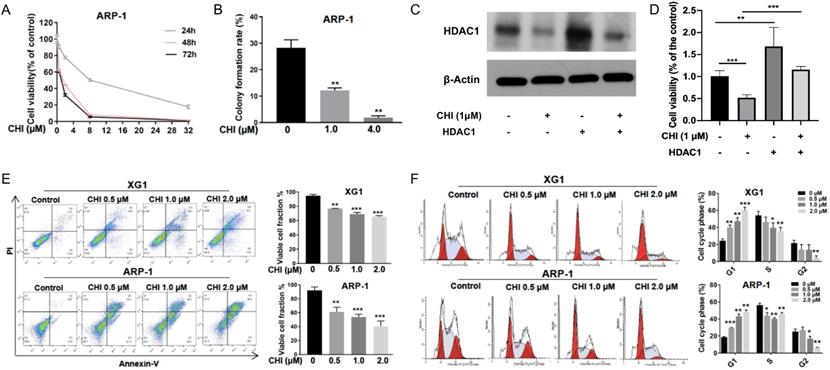
Chidamide increases Bortezomib effects synergistically in antagonizing MM cell growth
Previous studies had determined that over-expression of HDAC1 promoted BTZ-resistance in MM cells [20]. Our above data indicated CHI decreased HDAC1 in MM cells and its effect on MM cells was positively correlated with HDAC1 expression. We thus speculated that CHI might be synergistic with BTZ in MM cells. The synergistic effect of CHI and BTZ was investigated by using the Chou-Talalay method [22]. XG1 and ARP-1 were treated with different concentrations of CHI and BTZ in monotherapy, and in combination for 48 hours, then CCK-8 assay was performed to examine cell viability and the combination index (CI) was calculated by Chou-Talalay method. As shown in Figure 3A-C, the combination of CHI and BTZ significantly accelerated cell death in both XG1 and ARP-1 cells compared with CHI or BTZ alone, moreover, both two MM cell lines displayed significant synergistic effects with CI in the synergistic range (0.3-0.7). Meanwhile, soft agar colony forming assay showed that a significant decrease in clonogenic ability was observed in ARP-1 after treatment with combination of CHI and BTZ compared with single-agent alone (Figure 3D). Above data showed that CHI induced apoptosis and G1 arrest in MM cells, we wondered whether combination of CHI and BTZ increased apoptosis and G1 arrest in MM cells. As a result, increased apoptotic cells were observed in both ARP-1 and XG1 treated with combination of CHI and BTZ than that observed in CHI or BTZ alone (Figure 3E) and increased G1 arrest was also observed in ARP-1 with the same treatment (Figure 3F). Subsequently, the synergistic effect of CHI and BTZ was explored in bone marrow monocytes derived from two newly diagnosed and one relapsed MM patients. As shown in Figure 3G and 3H, decreased cell viability and increased apoptotic cells were obtained in those cells treated with combination of CHI and BTZ compared with CHI or BTZ alone. These findings indicated that CHI was synergistic with BTZ in antagonizing MM cell growth.
Chidamide increases Bortezomib effects synergistically in antagonizing MM cell growth. (A) The dose-effect curve of CHI and BTZ as single agent and in combinations in XG1 and ARP-1 cells using the Chou-Talalay method. (B) The observed CI of combination in the experiments performed and the line gives an estimation of the CI for the combination in XG1 and ARP-1 cells. (C) CCK8 analysis of the combination of CHI (0.5 µmol/L) and BTZ (2.0 nmol/L) in XG1 and ARP-1 cells after 48 hours of treatment. (D) Colony forming assay showed the colony formation rate of cells treated with CHI (1.0 µmol/L) combined with BTZ (5.0 nmol/L) for 48 hours in ARP-1 cells. (E) Flow cytometry (Annexin V/PI) analysis of the apoptosis of the combination of CHI (0.5 µmol/L) and BTZ (2.0 nmol/L) in the MM cell lines XG1 and ARP-1 after 48 hours of treatment. (F) Cell cycle analysis of ARP-1 cells treated with CHI (0.5 µmol/L) and BTZ (2.0 nmol/L) for 24 hours. (G, H) MM cells derived from two patients with newly diagnosed MM and a patient with relapsed/refractory MM were treated in vitro for 36 hours with CHI (1.0 μmol/L) and BTZ (5.0 nmol/L) in monotherapy and in combination. CCK8 and Flow cytometry (Annexin V/PI) were used to analyze the proliferation and apoptosis. Error bars indicate mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001.
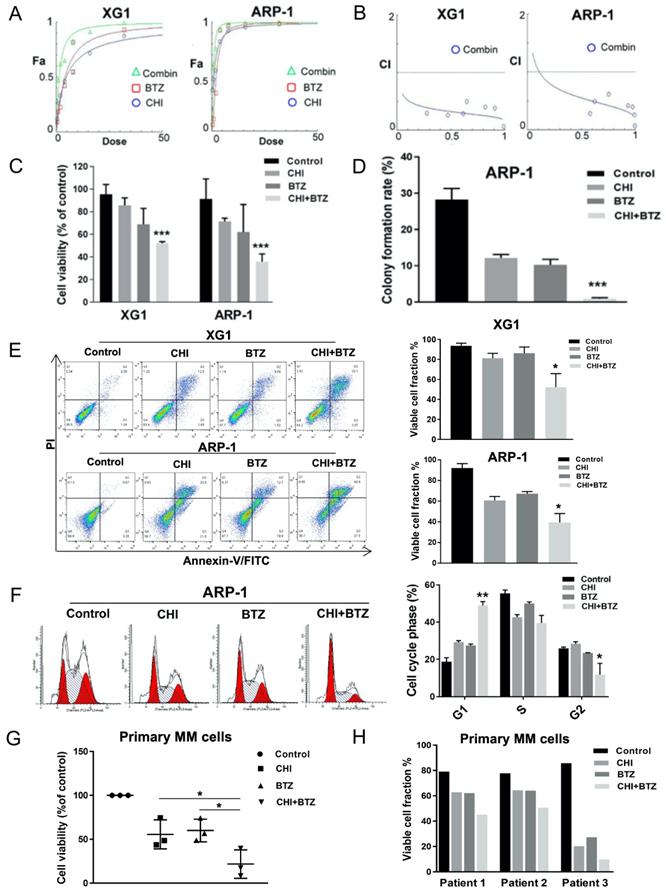
A combination of Chidamide and Bortezomib increases production of ROS dependent DNA damage and the changes of cell apoptosis and cycle pathway in MM cells
Previous studies had reported that proteasome inhibitor was synergistic with HDAC inhibitor through increasing ROS [24, 25]. CHI was found to induce ROS-dependent cell death in leukemia cells [11]. Thus, we hypothesized that increase of ROS might be the main mechanism by which CHI synergistic with BTZ in MM cells. To confirm this hypothesis, ARP-1 was pre-treated with or without NAC, followed by treatment with CHI or in combination with BTZ. ROS assay showed that the combination of CHI and BTZ significantly increased ROS level in ARP-1 as compared with CHI or BTZ alone, moreover, combination of CHI and BTZ-induced ROS was substantially abrogated by NAC (Figure 4A). Meanwhile, the proliferative toxicities of CHI or BTZ alone and in combination on MM cells were evidently reversed by NAC (Figure 4B), suggesting that the cell death at least in part, was ROS-dependent. It was widely accepted that ROS leads to cell death mainly through inducing DNA damage. We subsequently examined the expression of γ-H2AX, a DNA damage marker, in XG1 and ARP-1 cells after treatment with CHI and BTZ alone or in combination. Western blot demonstrated that increased γ-H2AX was observed in MM cells in response to combination of CHI and BTZ compared with those cells treated with CHI or BTZ alone (Figure 4C), which was also validated by immunofluorescence staining of γ-H2AX in ARP-1 cells (Figure 4D). In addition, the expression of cleaved caspase-3, cleaved caspase-8 and cleaved PARP increased, while the expression of HDAC1 decreased in XG1 and ARP-1 cells treated with the combination of CHI and BTZ (Figure 4E, F). Thus, we thought that the synergistic anti-tumor effect of CHI and BTZ may be related with the increased production of ROS dependent DNA damage and the changes of cell apoptosis and cycle pathway in MM cells.
A combination of Chidamide and Bortezomib increases production of ROS dependent DNA damage and the changes of cell apoptosis and cycle pathway in MM cells. (A) ARP-1 cells were pretreated with or without NAC (15.0 mmol/L) for 2 hours at 37°C and then incubated with CHI (1.0 µmol/L) and/or BTZ (5.0 nmol/L) for 24 hours, then ROS generation was detected. (B) ARP-1 cells were pretreated with or without 15 mmol/L NAC and then treated with Chidamide or Bortezomib alone or in combination, and cell viabilities were evaluated using CCK-8 assays. (C) The expression of γ-H2AX in ARP-1 cells treated with CHI (1.0 µmol/L) and/or BTZ (5.0 nmol/L) were determined by Western blot. (D) Representative images of γ-H2AX (Red) and nuclei (Blue) in ARP-1 cells treated with single agent or combination for 24 hours by immunofluorescence assay. Scale bars represent 20 µm. (E, F) Western blot analysis of the expressions of cleaved caspase3, cleaved caspase8, cleaved PARP-1 and HDAC1 in XG1 (E) and ARP-1 (F) cells after 48 hours treatment with single agent or in combination. Error bars indicate mean ± SD. **p < 0.01.
Chidamide and Bortezomib synergistically inhibits MM cell growth in vivo
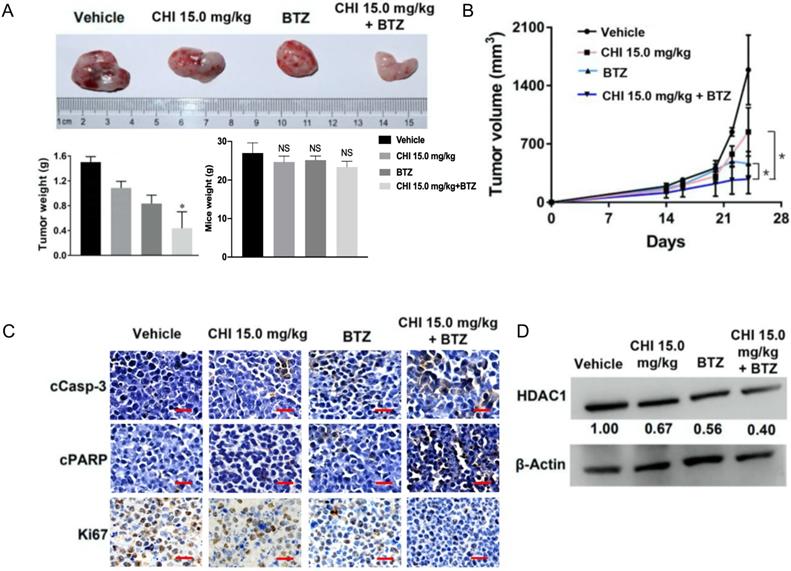
Above data determined the synergistic effect of CHI and BTZ in vitro, we next explored whether the synergistic effect was applied to in vivo MM model. Then, 1 × 106 ARP-1 was injected subcutaneously into the left abdomen of B-NDG® mice (NOD-Prkdcscid Il2rgtm1/Bcgen). After 10 days, all mice were divided into four groups randomly (n=3), and then treated with CHI (15.0 mg/kg), BTZ (1.0 mg/kg), combination of CHI and BTZ, and PBS, respectively. Tumor progression was monitored by tumor volume. As shown in Figure 5A and 5B, compared to the vehicle group or single drug group, a significant decrease in tumor volume and tumor weight was observed in the combination of CHI and BTZ group and there were no significant differences in mice weight. Next, we examined the expression of proliferation markers Ki67 and apoptotic markers cleaved Caspase-3 and cleaved PARP in tumors using immuno-histochemical. Tumors from mice treated with the combination of CHI and BTZ showed a decrease in the expression of cell Ki-67 and increase of cleaved Caspase-3 and cleaved PARP compared with vehicle, CHI or BTZ alone (Figure 5C). In addition, Western blot revealed that the expression of HDAC1 was downregulated in tumors treated with the combination of CHI and BTZ (Figure 5D). These data suggested that CHI and BTZ synergistically inhibited MM cell growth in vivo.
Chidamide and Bortezomib synergistically inhibits MM cell growth in vivo. (A, B) Efficacy of single-agent and combination treatment of CHI with BTZ in a human myeloma model in B-NDG mice. Tumor volumes, tumor weight and mice weight in ARP-1 cells xenografted mice model following treatment with single agent CHI and combinations with BTZ were shown (n=3). (C) Immunohistochemical analyses with anti-cleaved caspase-3, anti-cleaved PARP and anti-Ki-67 induced by the treatment with single agent and in combinations in vivo. (D) Western blot analysis of the expression of HDAC1 in the xenografts treated with single agent and in combinations. Error bars indicate mean ± SD. **p < 0.01; NS, not significant.
Discussion
MM is an incurable monoclonal plasma cell disorder [26]. Drug-resistance induced relapse is the most important cause for the death of MM patient. Thus, it is urgent demand to develop novel drugs against MM. CHI, a novel orally HDACi has been approved by the CFDA for the treatment of PTCL [14], was found to be potential therapeutic agent in MM in vitro and in vivo [15, 17, 27]. In the present study, we showed for the first time that combination of CHI and BTZ significantly increased cell death compared with CHI or BTZ alone. In addition, the underlying mechanisms by which CHI synergizes with BTZ was explored.
Multiple HDACs were over-expressed in MM cells, and played important roles in MM progression and drug-resistance [5, 20]. Several HDACis especially class I HDACis have achieved excellent treatment result in MM [28-30]. In this study, the effect of CHI, one of class I HDACis targeting HDAC1, 2, 3 and 10, on MM cells were investigated. Our data showed that CHI was effective in inducing cell death in MM cell lines, and the cytotoxicity of CHI on MM cell lines was positively correlated with the expression of HDAC1 in those MM cell lines. Meanwhile, overexpression of HDAC1 could reverse the anti-proliferation effect caused by CHI, which suggested that CHI affected MM cells partly dependent on HDAC1. It had been widely shown that HDAC inhibitors could induce apoptosis and cell cycle arrest, all of which eventually resulted in an inhibition of cell proliferation [11, 31]. Here, our data also confirmed that CHI could induce apoptosis and G0/G1 arrest in MM cells.
BTZ, as the first proteasome inhibitor, has achieved good treatment effect in MM therapy, however, the development of BTZ-resistance attenuated its long-term utility [2, 19, 32]. HDAC1 was found to be positively associated with BTZ-resistance, moreover, over-expression of HDAC1 promoted resistance to BTZ in MM cell, implicating that targeting HDACs might sensitize MM cells to BTZ [20, 33, 34]. In fact, the synergistic anti-MM activities between HDACi and BTZ have been observed in several preclinical studies [24, 28]. Pan-HDACi had shown clear benefit in patients with relapsed or refractory MM when combined with BTZ [35, 36]. It was also showed that normal hematopoietic cells were largely unaffected when exposed to combination of BTZ and HDACi [37]. These findings implicated that combination of HDACis and BTZ could be a promising therapeutic strategy for MM. In this study, we found that combination of CHI and BTZ significantly inhibited cell growth and increased apoptotic cells as compared with CHI or BTZ alone in MM cell lines as well as primary MM cells derived from 3 MM patients. Moreover, the synergistic effect of CHI and BTZ on MM was further evidenced by decreased tumor growth in MM xenograft mice treated with combination of CHI and BTZ compared with those mice treated with single drug. Taken together, our findings confirmed the synergistic effect of CHI and BTZ on MM, and provided a promising therapeutic strategy for MM.
CHI was able to induce ROS-dependent cell death in leukemia cells [11, 38] and previous studies have indicated that BTZ synergized with HDACis via increasing ROS level in MM cells [39, 40]. In this study, MM cells treated with combination of CHI and BTZ showed higher level of ROS than those cells treated with single reagent. In addition, increased γ-H2AX, cleaved Caspase-3, cleaved PARP were also observed in MM cells treated with combination of CHI and BTZ compared with CHI or BTZ alone. Therefore, we concluded that CHI synergized with BTZ through increasing ROS dependent DNA damage and the changes of cell apoptosis and cycle pathway in MM cells.
In summary, our current study found that the anti-tumor effect of CHI was partly depended on the expression of HDAC1 in MM cells and CHI combined with BTZ exhibited synergistic effect in antagonizing MM cell growth in vitro and in vivo. Moreover, our data showed increase of ROS dependent DNA damage and the changes of cell apoptosis and cycle pathway were the potential mechanisms by which CHI synergized with BTZ in MM cells. In general, our findings provide a novel promising therapeutic strategy for MM patients, especially for those MM patients resistant to BTZ.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81570117, 81700125), the Natural Science Foundation of Hunan Province (2018JJ2651), and the Medical Science Research Special Fund of Medical and Health Public Welfare Foundation of Beijing (YWJKJJH-KYJJ-B17464).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Rollig C, Knop S, Bornhauser M. Multiple myeloma. Lancet. 2015;385:2197-208
2. Chim CS, Kumar SK, Orlowski RZ, Cook G, Richardson PG, Gertz MA. et al. Management of relapsed and refractory multiple myeloma: novel agents, antibodies, immunotherapies and beyond. Leukemia. 2018;32:252-62
3. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693-705
4. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834-40
5. Mithraprabhu S, Kalff A, Chow A, Khong T, Spencer A. Dysregulated Class I histone deacetylases are indicators of poor prognosis in multiple myeloma. Epigenetics. 2014;9:1511-20
6. Mithraprabhu S, Khong T, Jones SS, Spencer A. Histone deacetylase (HDAC) inhibitors as single agents induce multiple myeloma cell death principally through the inhibition of class I HDAC. Br J Haematol. 2013;162:559-62
7. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769-84
8. Hideshima T, Richardson PG, Anderson KC. Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther. 2011;10:2034-42
9. Richardson PG, Hungria VT, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A. et al. Panobinostat plus bortezomib and dexamethasone in previously treated multiple myeloma: outcomes by prior treatment. Blood. 2016;127:713-21
10. Laubach JP, Schjesvold F, Mariz M, Dimopoulos MA, Lech-Maranda E, Spicka I. et al. Efficacy and safety of oral panobinostat plus subcutaneous bortezomib and oral dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma (PANORAMA 3): an open-label, randomised, phase 2 study. Lancet Oncol. 2021;22:142-54
11. Gong K, Xie J, Yi H, Li W. CS055 (Chidamide/HBI-8000), a novel histone deacetylase inhibitor, induces G1 arrest, ROS-dependent apoptosis and differentiation in human leukaemia cells. Biochem J. 2012;443:735-46
12. Gu R, Liu T, Zhu X, Gan H, Wu Z, Li J. et al. Development and validation of a sensitive HPLC-MS/MS method for determination of chidamide (epidaza), a new benzamide class of selective histone deacetylase inhibitor, in human plasma and its clinical application. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;1000:181-6
13. Lu X, Ning Z, Li Z, Cao H, Wang X. Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis Res. 2016;5:185-91
14. Shi Y, Jia B, Xu W, Li W, Liu T, Liu P. et al. Chidamide in relapsed or refractory peripheral T cell lymphoma: a multicenter real-world study in China. J Hematol Oncol. 2017;10:69
15. Liu Z, Jing Q, Wang Y, Li Y, Mi F, Xiang C. et al. The short-term effect of histone deacetylase inhibitors, chidamide and valproic acid, on the NFkappaB pathway in multiple myeloma cells. Int J Mol Med. 2019;43:285-93
16. He J, Chen Q, Gu H, Chen J, Zhang E, Guo X. et al. Therapeutic effects of the novel subtype-selective histone deacetylase inhibitor chidamide on myeloma-associated bone disease. Haematologica. 2018;103:1369-79
17. Yuan XG, Huang YR, Yu T, Jiang HW, Xu Y, Zhao XY. Chidamide, a histone deacetylase inhibitor, induces growth arrest and apoptosis in multiple myeloma cells in a caspase-dependent manner. Oncol Lett. 2019;18:411-9
18. Adams J. The proteasome: a suitable antineoplastic target. Nat Rev Cancer. 2004;4:349-60
19. Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006;107:4907-16
20. Kikuchi J, Wada T, Shimizu R, Izumi T, Akutsu M, Mitsunaga K. et al. Histone deacetylases are critical targets of bortezomib-induced cytotoxicity in multiple myeloma. Blood. 2010;116:406-17
21. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV. et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15:e538-48
22. Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440-6
23. Pandey MK, Gowda K, Sung SS, Abraham T, Budak-Alpdogan T, Talamo G. et al. A novel dual inhibitor of microtubule and Bruton's tyrosine kinase inhibits survival of multiple myeloma and osteoclastogenesis. Exp Hematol. 2017;53:31-42
24. Feng R, Oton A, Mapara MY, Anderson G, Belani C, Lentzsch S. The histone deacetylase inhibitor, PXD101, potentiates bortezomib-induced anti-multiple myeloma effect by induction of oxidative stress and DNA damage. Br J Haematol. 2007;139:385-97
25. Gao L, Gao M, Yang G, Tao Y, Kong Y, Yang R. et al. Synergistic Activity of Carfilzomib and Panobinostat in Multiple Myeloma Cells via Modulation of ROS Generation and ERK1/2. Biomed Res Int. 2015;2015:459052
26. Mimura N, Hideshima T, Anderson KC. Novel therapeutic strategies for multiple myeloma. Exp Hematol. 2015;43:732-41
27. Sun Y, Li J, Xu Z, Xu J, Shi M, Liu P. Chidamide, a novel histone deacetylase inhibitor, inhibits multiple myeloma cells proliferation through succinate dehydrogenase subunit A. Am J Cancer Res. 2019;9:574-84
28. Ocio EM, Vilanova D, Atadja P, Maiso P, Crusoe E, Fernandez-Lazaro D. et al. In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica. 2010;95:794-803
29. Laubach JP, San-Miguel JF, Hungria V, Hou J, Moreau P, Lonial S. et al. Deacetylase inhibitors: an advance in myeloma therapy? Expert Rev Hematol. 2017;10:229-37
30. Richardson PG, Mitsiades CS, Laubach JP, Hajek R, Spicka I, Dimopoulos MA. et al. Preclinical data and early clinical experience supporting the use of histone deacetylase inhibitors in multiple myeloma. Leuk Res. 2013;37:829-37
31. Singh AK, Bishayee A, Pandey AK. Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients. 2018;10:731
32. Moreau P, Richardson PG, Cavo M, Orlowski RZ, San Miguel JF, Palumbo A. et al. Proteasome inhibitors in multiple myeloma: 10 years later. Blood. 2012;120:947-59
33. Senese S, Zaragoza K, Minardi S, Muradore I, Ronzoni S, Passafaro A. et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol Cell Biol. 2007;27:4784-95
34. Keshelava N, Davicioni E, Wan Z, Ji L, Sposto R, Triche TJ. et al. Histone deacetylase 1 gene expression and sensitization of multidrug-resistant neuroblastoma cell lines to cytotoxic agents by depsipeptide. J Natl Cancer Inst. 2007;99:1107-19
35. Greig SL. Panobinostat: A Review in Relapsed or Refractory Multiple Myeloma. Target Oncol. 2016;11:107-14
36. Kaufman JL, Fabre C, Lonial S, Richardson PG. Histone deacetylase inhibitors in multiple myeloma: rationale and evidence for their use in combination therapy. Clin Lymphoma Myeloma Leuk. 2013;13:370-6
37. Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004;10:3839-52
38. Li Y, Wang Y, Zhou Y, Li J, Chen K, Zhang L. et al. Cooperative effect of chidamide and chemotherapeutic drugs induce apoptosis by DNA damage accumulation and repair defects in acute myeloid leukemia stem and progenitor cells. Clin Epigenetics. 2017;9:83
39. Jagannath S, Dimopoulos MA, Lonial S. Combined proteasome and histone deacetylase inhibition: A promising synergy for patients with relapsed/refractory multiple myeloma. Leuk Res. 2010;34:1111-8
40. Lipchick BC, Fink EE, Nikiforov MA. Oxidative stress and proteasome inhibitors in multiple myeloma. Pharmacol Res. 2016;105:210-5
Author contact
![]() Corresponding author: Fangping Chen, Department of Hematology, Xiangya Hospital, Central South University, Changsha, Hunan, China. Tel.: +8613707485350; E-mail: xychenfpedu.cn.
Corresponding author: Fangping Chen, Department of Hematology, Xiangya Hospital, Central South University, Changsha, Hunan, China. Tel.: +8613707485350; E-mail: xychenfpedu.cn.