Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2021; 12(21):6411-6421. doi:10.7150/jca.60102 This issue Cite
Review
Mechano-Signaling Aspects of Hepatocellular Carcinoma
Mehak Passi, Stefan Zahler ![]()
Center for Drug Research, Ludwig-Maximilians-University, Butenandtstr. 5-13, 81377 Munich, Germany.
Received 2021-3-4; Accepted 2021-8-11; Published 2021-9-3
Abstract

HCC is one of the leading causes of cancer related death worldwide and comprises about 90% of the cases of primary liver cancer. It is generally accompanied by chronic liver fibrosis characterised by deposition of collagen fibres, which, in turn, causes enhanced stiffness of the liver tissue. Changes of tissue stiffness give rise to alterations of signalling pathways that are associated to mechanical properties of the cells and the extracellular matrix, and that can be subsumed as “mechano-signaling pathways”, like, e.g., the YAP/TAZ pathway, or the SRF pathway. Stiffness of the liver tissue modulates mechanical regulation of many genes involved in HCC progression. However, mechano-signaling is still rather underrepresented in our concepts of cancer in comparison to “classical” biochemical signalling pathways. This review aims to give an overview of various stiffness induced mechano-biological aspects of HCC.
Keywords: Hepatocellular carcinoma, HCC, mechano-signaling, extracellular matrix, ECM, stiffness
Introduction
Hepatocellular carcinoma (HCC) accounts for 90% of all cases of primary liver cancer and is the third leading cause of cancer-related death worldwide [1]. Every year the incidence of occurrence of HCC in Europe and the United States increases, and currently accounts for more than 626,000 cases worldwide. Many factors such as hepatitis B virus (HBV), hepatitis C virus (HCV), alcohol and non-alcoholic steatohepatitis (NASH) contribute to the development and progression of HCC. Further, several point mutations, such as copy number aberrations (CNAs), insertions and deletions, virus integrations and gene fusions have been reported in the context of HCC [2]. Their role as drivers, however, is still unclear.
More than eighty per cent of HCC cases are associated with fibrosis induced by chronic liver injury and develop in fibrotic or cirrhotic liver. HCC is associated with the differentiation of hepatic stellate cells (HSCs) into myofibroblasts, which produce extracellular matrix (ECM). Fibrosis is generally considered as a protective response to acute liver injury and results in the replacement of injured liver parenchyma with scar tissue. Fibrosis becomes chronic and recurring if the hepatocellular injury persists. However, the role of fibrosis in promoting HCC has not been clearly established to date [3].
Generally, fibrosis is characterized by deposition of collagen fibrils of varying degree (mostly type 1 collagen) in the fibrotic liver, resulting in up to 5-fold increase of total collagen content. Fibrosis can vary from mild to bridging fibrosis and cirrhosis depending on the degree of collagen deposition. Moreover, hepatic ECM consists of many other proteins, such as non-collagenous glycoproteins (elastin, fibronectin and laminin), matricellular proteins (thrombospondins, osteopontin, tenascins, and members of the clathrin coated vesicle protein family) and connective tissue growth factor (CTGF). During liver fibrosis, many of these proteins undergo biochemical alterations [3, 4].
All these changes result in overall modulation of the hepatic extracellular matrix (ECM). They are a result of various mechanical and biochemical feedback loops that play an important role in modifying the hepatic ECM. Moreover, receptor independent mechanosensitive signal transduction pathways, such as YAP/TAZ, are involved in controlling cell survival and differentiation, and are activated by stiff ECM. The activation of the YAP/TAZ is caused via an upstream inhibition of Hippo pathway kinases on stiff substrates. Apart from the Hippo pathway, other pathways, such as Wnt/β-catenin, PTEN and the Notch signaling pathway also contribute to HCC progression [5]. Therefore, studying the role of mechanics is an important aspect of HCC when it comes to its pathogenesis.
This review focuses on discussing and highlighting various mechano-sensing processes and signaling pathways involved in the occurrence and progression of HCC. It also highlights the importance of ECM stiffness in HCC and into how it is linked to mechanosensing in HCC.
Extracellular Matrix stiffness and Hepatocellular carcinoma
During HCC there is a transition from the premalignant environment (PME) to the tumor microenvironment (TME) and the ECM undergoes a variety of changes - many of which are related to an alteration of mechanical properties. ECM not only provides mechanical strength but also regulates a variety of signaling cascades through its ability to bind to a wide variety of specific receptors such as integrins, growth factors, as well as to regulate their expression, distribution and activation [6]. The changes in the ECM components such as collagens, glycosaminoglycans, laminins, proteoglycans, and fibronectin, result in overall behavioral and phenotypic changes in the epithelial, tumor and stromal cells [6, 7]. With the help of transmembrane receptors (integrins, DDRs) surrounding cells sense these changes and regulate specific signaling pathways in response to the external stimuli [8, 9]. Increased expression of integrins and DDR2 triggers a wide variety of signaling cascades, such as phosphoinositide 3 kinase (PI3K), mitogen-activated protein kinase (MAPK) and regulates EMT of HCC [10].
Reorganization and enhanced overproduction of ECM by myofibroblasts result in the mechanically stiff microenvironment that promotes tumor cell proliferation, invasion and changes in gene expression of stromal cells by enhancing tumor progression [11, 12]. Tumor cells regulate matrix stiffness by not only influencing the degree of fibrosis but also by controlling cross-linking and expression of ECM proteins and certain enzymes such as lysyl oxidases [13, 14]. Hepatic stellate cells (HSCs) also play a major role in remodeling and maintaining ECM through secretion of major ECM proteins such as collagen, and ECM degrading enzymes called matrix metalloproteinases (MMPs) [15]. In the healthy liver, HSCs are in the quiescent state with cytoplasmic vitamin A droplets while following liver injury they lose their vitamin A droplets, and become highly activated and proliferative [16]. They cause increased production of ECM components (collagen) and other ECM proteins. They are also associated with the increased inflammation signaling and matrix degradation which all together contribute to fibrosis. In HCC, increased stiffness leads to activation of HSCs followed by the activation of MMPs and tissue inhibitor of metalloproteinases (TIMP) [17, 18].
Quiescent HSCs balance the production of ECM proteins and MMPs proteolytic activity and maintain the ECM. As a consequence of liver injury HSCs get activated causing excess collagen production and matrix degradation leading to a stiff fibrotic state [19].
In normal liver where resolution of fibrosis occurs, increased activity of MMPs leads to collagen degradation and ECM softening following the reversal of activated HSCs to their quiescent phenotype. Whereas, conversely, in the stiff state of a fibrotic liver altered characteristics of the environment such as stiffness amplify the activated HSCs phenotype further contributing to fibrosis, and later HCC development [20, 21]. Many signaling pathways in HSCs are highly mechanosensitive such as the HNF4α transcriptional network or integrin-mediated YAP activation [22, 23]. Rigidity occurring from fibrosis may affect the ECM remodeling at the protein expression and secretion level that is mediated by HSCs. Two key MMPs secreted from HSCs are MMP-2 and MMP-9 that degrade collagen [24]. Composition of ECM in fibrosis is also dependent on the sensitivity of the secretion of MMPs and TIMP (Tissue inhibitors of matrix metalloproteinase) by HSCs towards rigidity. It is known that fibrotic stiffness downregulates MMP-9 expression and secretion, and upregulate the secretion of TIMP-1 [25].
Liver stiffness to some degree correlates with increased risk of HCC development. The Young's elastic modulus E for a healthy liver ranges between 300 and 600 Pa whereas in case of fibrosis and cirrhosis development it can be 20 kPa and higher [26]. In some studies, it was found that patients with liver stiffness values of >12.5-13 kPa had a 4 to 13 fold increased risk of developing HCC [27]. Changes in matrix stiffness modulate the behavior of epithelial cells through various mechanosensitive oncogenic pathways. They also contribute to the transformation and dedifferentiation of hepatocytes, and, in addition, lead to ductular and progenitor expansion in the liver, which in turn leads to HCC. Matrix stiffness provides a niche for tumor-initiating cells (TICs) and contributes to their proliferation by a variety of mechanosensing pathways. HSCs seem to differentiate into cancer-associated fibroblasts (CAFs) under mechanical stimuli, thereby providing a permanent feed-forward loop that continues to establish a stiff tumor niche [28, 29]. This process of creation of premalignant microenvironments (PME) and the tumor microenvironment (TME) by CAFs has been reviewed very recently elsewhere [30]. Intriguingly, CAFs have recently been proposed as therapeutic target for nanocarriers [31].
Expression of LOXL (Lysyl oxidase) has been identified as an important factor controlling ECM stiffness [32]. It is upregulated by TGFβ [32], hypoxia-inducible growth factor 1 α (HIF-1α) [33], and increases stiffness by crosslinking ECM proteins. In turn, its expression increases stiffness [34], causing a positive feedback loop. In some studies, it has been shown that the inhibition of LOXL2 with a monoclonal antibody reduces stiffness, collagen deposition and tumor size by reducing fibroblast activation and fibrosis in the liver [34]. However, NASH-induced fibrosis was not reduced in large scale clinical trials [35]. Apart from the overproduction or biochemical alteration of ECM several other factors, such as high interstitial pressure as a result of hypervascularization, hyperproliferation, or cell swelling induced injury and inflammation contribute to increase stiffness of the tumors [36].
Increased stiffness promotes proliferation of cells, increases EMT (Epithelial-Mesenchymal Transition) and resistance to apoptosis, as well as the stemness of HCC cell lines [37]. At the molecular level, increasing stiffness activates multiple signaling pathways such as YAP/TAZ, β-catenin pathway, PI3K/AKT, JNK, ERK, focal adhesion kinase pathway, finally resulting in enhanced HCC proliferation and chemotherapeutic resistance [38-40]. The mechanical signaling pathways unfortunately are in part redundant and deeply interwoven, making their understanding and elucidation quite complex.
Mechanotransduction in HCC induced by ECM-stiffness
The fibrotic program in HCC is generally accomplished by fibroblast mechanosensing of the stiffened ECM. Like in other cells, mechanosensing primarily is accomplished by the activation of integrins. Integrins get activated by binding of specific ligands leading to conformational changes and the formation of focal adhesion complexes. Several adaptor proteins such as paxillin, vinculin and talin facilitate the connection of integrin to the actin cytoskeleton, translating the mechanosensitive information into changes in cell contractility and mechanoresponsive signals. Strain stiffening driven by cell contractility of ECM causes elevated levels of integrin activation, leading, e.g., to phosphorylation of the kinases Src and FAK, and thus their activation [41-43]. Increased stiffness stimulates HCC proliferation by inducing resistance towards Sorafenib through activation of β1 integrin/FAK signaling and enhancing nuclear translocation of YAP1. All these events result in the maturation of focal adhesion complexes and further activate a multitude of pathways such as MAPK, PI3K/AKT and YAP/TAZ [44, 45]. The levels of total and phosphorylated FAK are increased in HCC and are generally related to vascular invasion, tumor stage and intrahepatic metastasis [46]. Several compounds targeting FAK are under clinical and preclinical trials as specific deletion of FAK has been shown to reduce HCC proliferation and tumor-induced overexpression of cMET and β-catenin [47]. By RNA sequencing it was found that increased stiffness activates hepatic stellate cells (HSCs), leading to the release of a set of paracrine factors, including CXCL12, IL6, IL11, PDGFA and B and VEGFA [48], which all promote colorectal liver metastasis in mice by paracrine mechanisms. However, it still remains unclear, whether liver fibrosis and stiffness promote malignant transformation of hepatocytes primarily through the effect on HSCs and the hepatic tumor microenvironment [49].
Agrin a heparin sulfate proteoglycan has recently been discovered to modulate the activation of YAP, and acts as a mechanotransduction signal in HCC. It is produced by endothelial, myofibroblast, and tumor cells, and is highly expressed in human cirrhotic liver and HCC [50]. Knockdown of Agrin reduces proliferation, migration and invasion of tumor cells, and reduces the levels of EMT markers, while it induces apoptosis in vitro, as well as oncogenic signals and tumor growth in mice [51, 52]. It has been shown that Agrin activates FAK-ILK-PAK1 pathways and transduces matrix rigidity through an integrin-Lrp4/MuSK pathway, resulting in activation of YAP in HCC [53]. Agrin acts as a contributor to ECM sensing in HCC cells.
Serum Response Factor (SRF) is part of another well characterized mechanotransduction pathway. It is mediated by myocardial-like proteins (MRTF), induced by F-actin polymerization. Interestingly, SRF is not abundantly expressed by normal healthy hepatocytes and non-tumoral tissues. In contrast, in cells from high-grade human HCC and human HCC cell lines, it exhibits a strong nuclear expression [54]. SRF target gene signatures partially overlap with the mechanical stress-induced signatures from YAP/TAZ target genes. Activation of SRF in hyperproliferative nodules of hepatocytes results in HCC development [55]. On a molecular level overexpression of SRF promotes HCC cell invasion and migration by increasing expression of β-catenin and EMT genes [56, 57].
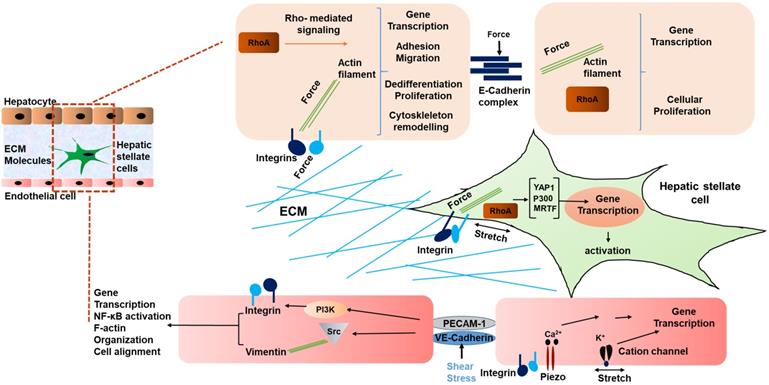
Interestingly, not only HSCs, but also liver sinusoidal endothelial cells (LSECs) have been shown to respond to stiffness and mechanosensation [58]. In a study by Liu et al, LSECs were embedded on 2D substrates underlying 3D collagen type 1 with integrated HSCs on the top. This was done to mimic the interaction of LSEC/HSC LSECs on substrates with stiffness values ranging from 140 Pa to 610 Pa. The LSECs formed capillary like structures under this condition. During the early phase of liver fibrosis angiogenesis is stimulated by the condensation of collagen fibers and generated forces by collagen remodeling that result in activation of collagen-DDR2-JAK2/P13K/AKT signaling promoting HSC activation [59]. Thus, mechanical forces in liver generated due to fibrosis indeed influence the phenotype of LSECs, and in turn contribute to the development and progression of HCC [60, 61]. Fig. 1 summarizes the various mechanical inputs related to development of HCC. All in all, the mechanical inputs described above converge at the modulation of transcriptional regulation. Table 1 summarizes genes mechanically regulated in liver disease.
Schematic representing mechanical signaling in HCC. Mechanical forces are communicated by the ECM via integrins of hepatocytes and HSCs, and between cells via E-cadherin of hepatocytes. In liver sinusoidal endothelial cells, VE-cadherin and PECAM-1 communicate shear stress between cells, causing activation of integrins. In addition, cation channels are activated by mechanical stretch on LSECs [42]. Thus, a multicellular network of force induced signaling is established.
A list of genes mechanically regulated in liver disease
| Genes mechanically regulated in liver disease | Reference |
|---|---|
| Mechanosensing receptors | |
| Integrins and Focal adhesions such as Fibronectin, vitronectin and collagens. | [42, 117, 118] |
| Adhesion receptors and cell-cell junctions such as cadherins, selectins, and CAMs | [119-121] |
| Ion channels such as TREK-1, K+ channel. | [122-124] |
| Hepatocyte nuclear factor 4 alpha HNF4α target genes | |
| Baat, F7, Gys2 | [125, 126] |
| ARP3 actin related protein 3 homolog C (ACTR3C) | [127] |
| Tubulin tyrosine ligase like family member 3 (TTLL3) | [127] |
| Actin related protein 2/3 | [42] |
| ARPC3 | [42] |
| Genes related to ECM receptor interaction | |
| ITGA1, Fibronectin1, Sorting nexin 15, Laminin, Alpha 4 | [42, 127] |
| Epithelial cell related genes | |
| CLAUDIN 12, RhoA, Src, Β1 Integrin, Phosphorylated FAK | [127] |
| Other expression of HCC genes | |
| CXCL12, IL11, IL6, PDGFA, PDGFB, VEGFA | [5, 22, 41, 128] |
| YAP1 | [90, 91] |
| Megakaryoblastic leukemia factor-1 (MKL 1) (called as myocardium related transcription factor) | [94] |
| Genes involved in mechano signalling of LSECs | |
| Β1- Integrin, Vascular endothelial growth factor (VEGFR3), CXCL1 | [53, 99, 100] |
The Hippo/YAP/TAZ pathway
The Hippo signalling pathway plays a major role in the progression and pathogenesis of HCC, linking stiffness and mechanotransduction to cancer progression. The YAP/TAZ axis seems to be a hub, where all kinds of mechanosensitive processes converge. Therefore, we will occupy ourselves with this specific pathway in greater detail. Its major components and downstream effectors are YAP and TAZ. These are involved in many autonomous functions, such as cell proliferation, differentiation, survival, development, metabolism, and cross-talk with the immune system. The role of Hippo signalling in regulation and biological functions varies from organ to organ, and between specific cell types, making it even more complex to understand. Overexpression of activated YAP results in rapid development of HCC, confirming its potential oncogenic role [62, 63].
The activity of YAP and TAZ is controlled by MST1/2 kinases. These are upstream regulators of the Hippo pathway that restrict tissue overgrowth, size and carcinogenesis by regulating YAP and TAZ activation [64]. YAP was the first protein identified with a WW domain (a motif comprising of 2 tryptophan residues), and is the key transcriptional regulator of the Hippo pathway, while TAZ is a YAP paralog (44% identity to YAP) [65, 66]. In general, MST1/MST2 kinases activate the kinases LATS1 and LATS2 by phosphorylating them at Thr1079 and Thr1041, respectively. They also phosphorylate MOB 1A and 1B kinases at Thr35 and Thr12. MOB1A/MOB1B when activated interact with LATS1 and LATS2 and lead to the autophosphorylation of LATS1 and LATS2 [67]. Both of these phosphorylation events lead to the activation of the LATS1 and LATS2 kinases. LATS kinases - when activated further - phosphorylate YAP, leading to the cytoplasmic sequestration of YAP/TAZ or ubiquitin mediated protein degradation [68, 69].
When LATS kinases are inactive, YAP/TAZ is not phosphorylated and gets translocated in the nucleus where it binds to members of the TEAD family of transcription factors, and mediates expression of target genes, which are involved in cell growth, migration, proliferation and survival, such as cysteine-rich angiogenic inducer 61 (CYR61), connective tissue growth factor (CTGF), and others [70-74] (Fig. 2). RUNX3 physically interacts with the N-terminal region of TEAD through its Runt domain. This interaction markedly reduces the DNA-binding ability of TEAD that attenuates the downstream signaling of the TEAD-YAP complex. This transcription complex also interacts with Runt-related transcription factor 3. DNA-binding ability of TEAD is reduced markedly through this interaction, which attenuates the downstream signaling of TEAD-YAP [75, 76]. In addition, YAP/TAZ also interacts with SMAD1, SMAD2/3 forming protein complexes suggesting a cross talk between the Hippo signaling pathway and the TGF-β pathway [77-79]. YAP directly interacts with the p53 promotor to enhance its expression which results in p53- dependent cycle arrest and apoptosis. It furthermore induces p21, TBox and Caspase 3 expression and inhibits the expression of anti-apoptotic factors, such as Bcl-2 and Bcl-xl. It has recently been shown that LATS1/LATS2 contributed to the tumor suppressive feature of p53 under basal conditions [80, 81]. YAP/TAZ also interact with the intracellular growth domain (ICD) of ErbB4 (one of the members of epidermal growth factor receptors in the nucleus) [82]. All these events modulate the expression of genes involved in proliferation, differentiation, development and growth. Loss of any of the core components of the Hippo pathway such as MST1/MST2, LATS1/LATS2, MOB1A/MOB1B, etc., results in upregulation in target gene transcription of YAP/TAZ-TEAD further leading to uncontrolled cellular proliferation and tissue growth [83-85].
YAP based mechanotransduction in HCC. Increased stiffness activates the YAP based Hippo signaling network. YAP/TAZ when activated, gets translocated in the nucleus and regulates the transcription of genes related to proliferation and cellular growth.
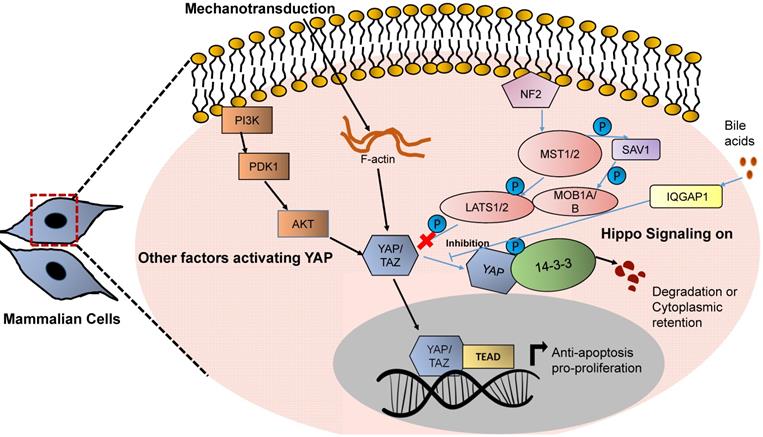
Due to fibrosis, mechanical strain occurs in the cirrhotic liver that activates YAP and TAZ, which in turn upregulate the expression of various target genes that control cellular proliferation and growth. YAP/TAZ are commonly overexpressed after deletion of Mst1/2 in hepatocytes and act as a transducer of liver tumor development and HCC [86, 87]. A major pathway of activation of YAP/TAZ, however, is a loss of phosphorylation, and, consequently, nuclear translocation of these proteins. Force transduction from the extracellular space to intracellular signaling pathways via the actin cytoskeleton is responsible for the YAP and TAZ mechanotransduction. Many mechanosensory proteins such as integrins, adherens junctions, adaptor proteins such as vinculin and talin, SRC family kinases, as well as FAK and Rho-GTPases participate in activation of YAP/TAZ. Inhibitors of actin polymerization abolish YAP/TAZ mechanotransduction [88, 89]. Overexpressing the activated form of TAZ in combination with the NRAS G12V mutation increases the formation of liver tumors, however to a lower degree than overexpressing activated YAP [90]. A list of genes regulated by YAP and upregulated in HCC can be found in Table 2.
A list of genes regulated by YAP in HCC
| YAP regulated genes in HCC | Mode of regulation | Reference |
|---|---|---|
| TEAD Transcription factor family (play key roles in normal cell growth. N-terminal region of YAP interacts with C-terminal region of TEAD protein). | ||
| RUNX2 | Downregulated | [129] |
| ITGB2 | Upregulated | [130] |
| ERBB4 | Upregulated | [131] |
| CYR61 | Downregulated | [133] |
| CTGF | Upregulated | [132] |
| AREG | Downregulated | [134] |
| MYC | Upregulated | [135] |
| Gli | Upregulated | [136] |
| Vimentin | Upregulated | [137] |
| AXL | Upregulated | [138] |
| P73 (major tumor repressor protein. YAP acts as a transcriptional co-activator of P73). | ||
| BAX | Upregulated | [139] |
| PIG3 | Downregulated | [140] |
| c-ABL | Upregulated | [141] |
| P53AIPI | Upregulated | [142] |
| ERBB-4 (EGFR family member receptor protein tyrosine kinase translocated in nucleus functioning as transcriptional regulator. It acts as a binding partner for YAP and TEAD. YAP-ERBB4 regulates organ size and tissue growth by promoting expressions of below mentioned genes). | ||
| CTGF | Upregulated | [143] |
| CYR61 | Downregulated | [133] |
| ANKRD1 | Upregulated | [137] |
| EGR1 (a nuclear protein functioning as a transcriptional regulator. It interacts with YAP and via PPXY motif of EGR-1 and WW domains induces the expression of BAX). | Downregulated | [144] |
| TBX5 (YAP, β-catenin and TBX5 forms a complex to induce the expression of transcriptional targets for cancer cell survival and transformation). | ||
| P300 | Upregulated | [144] |
| PCAF | Downregulated | [140] |
| SMADs (intracellular proteins that transduce extracellular signals from TGF-β or BMP to the nucleus, activating transcription of downstream target genes. YAP/TAZ acts as a regulator of TGF- β-SMAD signalling). | ||
| SMAD2/SMAD3 | Upregulated | [145] |
| RUNXs (members of DNA -binding transcription factor that act as regulators of development. RUNXs interact with YAP and play critical role in regulating cytoskeletal gene expression). | ||
| RUNX1/RUNX2/RUNX3 | Upregulated | [146] |
Role of YAP and TAZ in HSCs
Hepatic stellate cells (HSCs) are one of the major drivers of liver fibrosis, and are also involved in the process of liver repair after acute liver injury [91]. A fibrotic scar is formed during chronic liver injury after stellate cells are activated by excessive accumulation of ECM proteins, and trans-differentiation of quiescent HSCs into myofibroblasts. This process is generally controlled by the molecular drivers that regulate HSC activation. The Hippo signalling pathway has been recognized as one of the important pathways in stellate cell activation. During the acute liver regeneration process after hepatectomy, or ischemia- reperfusion injury and after chronic livery injury or CCl4 induced liver damage, YAP is activated in HSCs [92-94]. Sustained activation of YAP in liver fibrosis is generally due to an increase in ECM protein levels, as well as tissue stiffness [95]. In addition to HSCs, portal fibroblasts are also mechanosensitive. When seeded on polyacrylamide hydrogels both these cell types respond to increased stiffness [96-98]. Immunohistochemical analysis of murine and patient samples show that more YAP is translocated into the nucleus from cytoplasm of the activated HSCs/myofibroblasts of fibrotic livers compared to normal livers. Nuclear localization of YAP (i.e., activation) is increased when HSCs are cultured on stiff plastic surfaces for 10 h. Moreover, inhibition of YAP activation by verteprofin in vitro mitigates stiffness mediated HSC activation. All these data suggest that YAP plays an integral role in stiffness mediated HSC activation [99]. Furthermore, YAP and its transcriptional targets are upregulated in HSCs after 15 minutes of murine partial hepatectomy, as a result of elevated blood flow and shear stress, exerting mechanical forces. When stretching forces are applied to HSCs in vitro, HSCs are stimulated to produce fibronectin and also to promote fibril assembly by a β1-integrin/actin dependent mechanism [100]. YAP phosphorylation and its cytoplasmic retention has been shown to be promoted by the loss of β1 integrin in HSCs [101, 102].
Megakaryoblastic leukaemia factor-1 (MKL1), also known as myocardia related transcription factor (MRTFA), is another transcriptional regulator that responds to force changes. It is generally bound to cytoplasmic G-actin and force mediated actin assembly leads to nuclear translocation [103, 104]. Interestingly, expression of MKL1 and YAP targets is mutually dependent. This could be due to the indirect interaction of MRTF mediated and YAP mediated transcription pathways for mechanotransduction of fibroblasts [105]. It was found that disruption of transcription coactivator p300 through shRNA mediated knockdown, or a p300 inhibitor abolishes stiffness-induced HSC activation [106]. Wang et al. demonstrated that under TGFβ1 stimulation p300 was bound to TAZ and transported it to the nucleus of HSCs. In HEK cells, YAP activity was promoted by overexpression of HA-tagged p300 and its related protein CREB- binding protein (CBP) [107, 108]. Manneart and co-workers transfected HSCs with YAP siRNA in 3D aggregates and then transferred them into plastic dishes after 4 days, resulting in their inhibition [109].
LSECs and YAP
Liver sinusoidal cells (LSECs) are another non-parenchymal cells that play an essential role in liver injury by influencing regeneration and fibrosis through angiocrine signalling to stellate cells in case of acute and chronic liver damage [110]. Mechanosignaling in LSECs is induced by shear stress (caused by blood flow), stimulating them to release angiocrine factors that contribute to the development and maintenance of liver function [111]. For instance, subjecting LSECs to shear stress results in activation of β1- integrin, as well as vascular endothelial growth factor receptor 3 (VEGER3). This, in turn, causes release of hepatic growth factor and triggers proliferation and survival of hepatocytes [112, 113]. Under pathological conditions, and especially during fibrosis, mechanotransduction of LSECs thus further accelerates the progression of HCC due to angiocrine and phenotypic changes [114]. In case of chronic liver injury, a dysregulated crosstalk between hepatocytes, HSCs and LSECs contributes to liver fibrosis. Differentiated LSECs also maintain quiescence of HSCs, as revealed by co-culture experiments [115]. LSECs also maintain cell integrity in single hepatocytes after YAP activation. Angiogenesis in fibrosis is governed by YAP activation in LSECs by means of HIF-1α and VEGF-A expression [116, 117]. Moreover, apart from triggering neo-angiogenesis in the diseased liver, it is highly likely that YAP/TAZ play a major role in hepatic blood vessel formation during endothelial cell sprouting and junction maturation alike [118]. However, the role of YAP/TAZ signalling in endothelial cells during fibrosis and other liver diseases, remains to be further investigated.
Interaction of other pathways with the Hippo signalling pathway
Wnts are important proteins that play a major role in controlling many developmental and physiological processes. Their downstream effector is β-catenin, an adapter protein of cadherin type cell-cell adhesion molecules, which activates Wnt target gene expression [119]. Since exposure of cells to mechanical alterations obviously influences cell-cell contacts, Wnt-signalling has a mechano-sensitive component. Excessive activation of the Wnt/β-catenin pathway is associated with many types of cancer, including HCC and hepatoblastoma [120]. In distinct cell compartments, the Wnt and Hippo signalling pathways interact differently. In the nucleus, cooperation between YAP and β-catenin regulates gene expression e.g. in controlling the heart size, tumor transformation, and maintenance [121]. YAP/TAZ regulates phosphorylation of Dishevelled (Dvl), an important component of Wnt/β-catenin pathway, and the nuclear localization of Dvl or β-catenin, thereby inhibiting the Wnt/β-catenin activities [122]. However, it is still unknown, whether the mode of interaction between Wnt/β-catenin signalling is limited to and specific for a special cell type. The loss of activity of Mst1/2 in hepatocytes has previously been shown to lead to the activation of Wnt/β-catenin signalling in the liver and, consequently, to formation of liver tumors [123].
Notch signalling is a further pathway interacting with the Hippo pathway. Notch signalling in the liver promotes the formation of oval cells (liver stem cell) [124]. Notch signalling is highly activated in HCC patients and the expression of Notch receptor is highly regulated [125]. Direct cell-cell contact is needed for the activation of the Notch pathway, allowing the direct Notch receptor to interact with their membrane bound ligands (Jagged and Delta-like) [126]. Sequential proteolytic cleavage of notch receptors by the γ-secretase complex and a member of the ADAM family is induced by Notch ligand binding. As a result of this, NICD (Notch intracellular domain) is liberated from the membrane, and enters the nucleus. In the nucleus, it forms a ternary complex with the co-factor RBP-j and participates in the transcriptional regulation of the respective target genes [127]. Constitutively expressed hepatic NICD causes liver tumor formation in mice.
Activation of Notch signalling due to loss of activity of Mst1/2 in hepatocytes forms a positive feedback loop with YAP/TAZ, leading to severe liver enlargement and rapid HCC formation [128]. There also is a surprising inhibitory role of Wnt/β-catenin signalling to YAP/TAZ activities: in Mst1/2 null mutants, genetic removal of β-catenin in the liver significantly increases the number of tumor nodules. Mechanistically, YAP/TAZ increases the generation of Notch intracellular domain (NICD) by YAP/TAZ [129]. Notch signalling inhibits the β-TrCP-mediated degradation of TAZ and stabilizes it by forming a positive feedback loop.
A further layer of complexity is added by the fact that Wnt signalling modulates the cross-talk between Notch and YAP/TAZ: Wnt/β-catenin signalling promotes nuclear localization of DP1 (the dimerization partner of E2F transcriptional factors) through suppressing the positive feedback loop between TAZ and notch, which subsequently inhibits Notch activity [130]. Notch inhibition in vivo breaks the YAP/TAZ-Notch positive feedback loop and reduces the activity of YAP/TAZ, hepatocyte proliferation and tumor formation [131]. Therefore, there is an unexpected function of Wnt/β-catenin signaling in restricting YAP/TAZ and Notch activities that are involved in HCC initiation.
Conclusion
HCC is generally caused by intensive liver fibrosis characterised by deposition of collagen fibres and a stiffer ECM. Fibrosis leads to activation of various mechanical feedback loops and signalling pathways. One of most prominent and most studied is Hippo signalling pathway or YAP/TAZ pathway. YAP not only contributes to transcription of genes that lead to cellular proliferation but also leads to the activation of HSC and LSECs. YAP is an important protein that plays a major role in mechanotransduction. Furthermore, the Hippo signalling pathway interacts with several other pathways such as Wnt/β-catenin pathway and Notch signalling pathway that further lead to the progression of HCC. Due to the complexity and mutual entanglement of the respective signalling pathways, more focused studies are needed to further establish the relevance of receptor-independent mechanosensing signaling pathways in the development of HCC [132, 133].
From a technical perspective, the role of mechano-signaling in HCC is extremely hard to study in a clinical or in vivo setting. Recently, the awareness has grown that 2D cell cultures cannot mimic the exact physiological conditions, especially when mechanical aspects of the ECM are concerned. There is still lack of reliable and easy to handle 3D models for the study of HCC. Consequently, development of valid 3D models, which allow for access to mechanical parameters (e.g. stiffness) as well as to functional analysis with high temporal and special resolution is an important prerequisite.
Acknowledgements
The authors would like to thank Prof. Angelika Vollmar (Department of Pharmacy, Ludwig-Maximilians-Universität) for fruitful discussions and continuous support.
Funding
Mehak Passi is funded by the German Academic Exchange Service (DAAD).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Xue R, Li J, Bai F, Wang X, Ji J, Lu Y. A race to uncover a panoramic view of primary liver cancer. Cancer Biol Med. 2017;14:335-40
2. Singal AG, El-Serag HB. Hepatocellular Carcinoma From Epidemiology to Prevention: Translating Knowledge into Practice. Clin Gastroenterol Hepatol. 2015;13:2140-51
3. Affo S, Yu LX, Schwabe RF. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu Rev Pathol. 2017;12:153-86
4. Williams MJ, Clouston AD, Forbes SJ. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology. 2014;146:349-56
5. Filliol A, Schwabe RF. Contributions of Fibroblasts, Extracellular Matrix, Stiffness, and Mechanosensing to Hepatocarcinogenesis. Semin Liver Dis. 2019;39:315-33
6. Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216-9
7. Rombouts K, Carloni V. The fibrotic microenvironment as a heterogeneity facet of hepatocellular carcinoma. Fibrogenesis Tissue Repair. 2013;6:17
8. Carloni V, Luong TV, Rombouts K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: more complicated than ever. Liver Int. 2014;34:834-43
9. Cox D, Brennan M, Moran N. Integrins as therapeutic targets: lessons and opportunities. Nat Rev Drug Discov. 2010;9:804-20
10. Zhao G, Cui J, Qin Q, Zhang J, Liu L, Deng S. et al. Mechanical stiffness of liver tissues in relation to integrin beta1 expression may influence the development of hepatic cirrhosis and hepatocellular carcinoma. J Surg Oncol. 2010;102:482-9
11. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. 2012;196:395-406
12. Xie B, Lin W, Ye J, Wang X, Zhang B, Xiong S. et al. DDR2 facilitates hepatocellular carcinoma invasion and metastasis via activating ERK signaling and stabilizing SNAIL1. J Exp Clin Cancer Res. 2015;34:101
13. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT. et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891-906
14. Naba A, Clauser KR, Lamar JM, Carr SA, Hynes RO. Extracellular matrix signatures of human mammary carcinoma identify novel metastasis promoters. Elife. 2014;3:e01308
15. Lachowski D, Cortes E, Rice A, Pinato D, Rombouts K, Del Rio Hernandez A. Matrix stiffness modulates the activity of MMP-9 and TIMP-1 in hepatic stellate cells to perpetuate fibrosis. Sci Rep. 2019;9:7299
16. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14:397-411
17. Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best Pract Res Clin Gastroenterol. 2011;25:195-206
18. Moreira RK. Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med. 2007;131:1728-34
19. Caliari SR, Perepelyuk M, Cosgrove BD, Tsai SJ, Lee GY, Mauck RL. et al. Stiffening hydrogels for investigating the dynamics of hepatic stellate cell mechanotransduction during myofibroblast activation. Sci Rep. 2016;6:21387
20. Li YL, Sato M, Kojima N, Miura M, Senoo H. Regulatory role of extracellular matrix components in expression of matrix metalloproteinases in cultured hepatic stellate cells. Cell Struct Funct. 1999;24:255-61
21. Zhubanchaliyev A, Temirbekuly A, Kongrtay K, Wanshura LC, Kunz J. Targeting Mechanotransduction at the Transcriptional Level: YAP and BRD4 Are Novel Therapeutic Targets for the Reversal of Liver Fibrosis. Front Pharmacol. 2016;7:462
22. Schrader J, Gordon-Walker TT, Aucott RL, van Deemter M, Quaas A, Walsh S. et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology. 2011;53:1192-205
23. Wells RG. The role of matrix stiffness in regulating cell behavior. Hepatology. 2008;47:1394-400
24. Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI. et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15:637-46
25. Zhu J, Huang S, Wu G, Huang C, Li X, Chen Z. et al. Lysyl Oxidase Is Predictive of Unfavorable Outcomes and Essential for Regulation of Vascular Endothelial Growth Factor in Hepatocellular Carcinoma. Dig Dis Sci. 2015;60:3019-31
26. Wang M, Zhao X, Zhu D, Liu T, Liang X, Liu F. et al. HIF-1alpha promoted vasculogenic mimicry formation in hepatocellular carcinoma through LOXL2 up-regulation in hypoxic tumor microenvironment. J Exp Clin Cancer Res. 2017;36:60
27. Wu S, Zheng Q, Xing X, Dong Y, Wang Y, You Y. et al. Matrix stiffness-upregulated LOXL2 promotes fibronectin production, MMP9 and CXCL12 expression and BMDCs recruitment to assist pre-metastatic niche formation. J Exp Clin Cancer Res. 2018;37:99
28. Barry-Hamilton V, Spangler R, Marshall D, McCauley S, Rodriguez HM, Oyasu M. et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat Med. 2010;16:1009-17
29. Harrison SA, Abdelmalek MF, Caldwell S, Shiffman ML, Diehl AM, Ghalib R. et al. Simtuzumab Is Ineffective for Patients With Bridging Fibrosis or Compensated Cirrhosis Caused by Nonalcoholic Steatohepatitis. Gastroenterology. 2018;155:1140-53
30. Baglieri J, Brenner DA, Kisseleva T. The Role of Fibrosis and Liver-Associated Fibroblasts in the Pathogenesis of Hepatocellular Carcinoma. Int J Mol Sci. 2019 20
31. Kaps L, Schuppan D. Targeting Cancer Associated Fibroblasts in Liver Fibrosis and Liver Cancer Using Nanocarriers. Cells. 2020 9
32. Broders-Bondon F, Nguyen Ho-Bouldoires TH, Fernandez-Sanchez ME, Farge E. Mechanotransduction in tumor progression: The dark side of the force. J Cell Biol. 2018;217:1571-87
33. Tschumperlin DJ, Ligresti G, Hilscher MB, Shah VH. Mechanosensing and fibrosis. J Clin Invest. 2018;128:74-84
34. You Y, Zheng Q, Dong Y, Wang Y, Zhang L, Xue T. et al. Higher Matrix Stiffness Upregulates Osteopontin Expression in Hepatocellular Carcinoma Cells Mediated by Integrin beta1/GSK3beta/beta-Catenin Signaling Pathway. PLoS One. 2015;10:e0134243
35. Matthews BD, Overby DR, Mannix R, Ingber DE. Cellular adaptation to mechanical stress: role of integrins, Rho, cytoskeletal tension and mechanosensitive ion channels. J Cell Sci. 2006;119:508-18
36. Broders-Bondon F, Nguyen Ho-Bouldoires TH, Fernandez-Sanchez M-E, Farge EJJoCB. Mechanotransduction in tumor progression: The dark side of the force. 2018; 217: 1571-87.
37. Guilluy C, Swaminathan V, Garcia-Mata R, O'Brien ET, Superfine R, Burridge K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat Cell Biol. 2011;13:722-7
38. Chen JS, Huang XH, Wang Q, Chen XL, Fu XH, Tan HX. et al. FAK is involved in invasion and metastasis of hepatocellular carcinoma. Clin Exp Metastasis. 2010;27:71-82
39. Mitra SK, Schlaepfer DD. Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol. 2006;18:516-23
40. Sun Z, Guo SS, Fassler R. Integrin-mediated mechanotransduction. J Cell Biol. 2016;215:445-56
41. Dou C, Liu Z, Tu K, Zhang H, Chen C, Yaqoob U. et al. P300 Acetyltransferase Mediates Stiffness-Induced Activation of Hepatic Stellate Cells Into Tumor-Promoting Myofibroblasts. Gastroenterology. 2018;154:2209-21 e14
42. Kang N. Mechanotransduction in Liver Diseases. Semin Liver Dis. 2020;40:84-90
43. Shang N, Arteaga M, Zaidi A, Stauffer J, Cotler SJ, Zeleznik-Le NJ. et al. FAK is required for c-Met/beta-catenin-driven hepatocarcinogenesis. Hepatology. 2015;61:214-26
44. Lv X, Fang C, Yin R, Qiao B, Shang R, Wang J. et al. Agrin para-secreted by PDGF-activated human hepatic stellate cells promotes hepatocarcinogenesis in vitro and in vivo. Oncotarget. 2017;8:105340-55
45. Tatrai P, Dudas J, Batmunkh E, Mathe M, Zalatnai A, Schaff Z. et al. Agrin, a novel basement membrane component in human and rat liver, accumulates in cirrhosis and hepatocellular carcinoma. Lab Invest. 2006;86:1149-60
46. Chakraborty S, Lakshmanan M, Swa HL, Chen J, Zhang X, Ong YS. et al. An oncogenic role of Agrin in regulating focal adhesion integrity in hepatocellular carcinoma. Nat Commun. 2015;6:6184
47. Chakraborty S, Njah K, Pobbati AV, Lim YB, Raju A, Lakshmanan M. et al. Agrin as a Mechanotransduction Signal Regulating YAP through the Hippo Pathway. Cell Rep. 2017;18:2464-79
48. Park MY, Kim KR, Park HS, Park BH, Choi HN, Jang KY. et al. Expression of the serum response factor in hepatocellular carcinoma: implications for epithelial-mesenchymal transition. Int J Oncol. 2007;31:1309-15
49. Hampl V, Martin C, Aigner A, Hoebel S, Singer S, Frank N. et al. Depletion of the transcriptional coactivators megakaryoblastic leukaemia 1 and 2 abolishes hepatocellular carcinoma xenograft growth by inducing oncogene-induced senescence. EMBO Mol Med. 2013;5:1367-82
50. Kwon CY, Kim KR, Choi HN, Chung MJ, Noh SJ, Kim DG. et al. The role of serum response factor in hepatocellular carcinoma: implications for disease progression. Int J Oncol. 2010;37:837-44
51. Hilscher MB, Sehrawat T, Arab JP, Zeng Z, Gao J, Liu M. et al. Mechanical Stretch Increases Expression of CXCL1 in Liver Sinusoidal Endothelial Cells to Recruit Neutrophils, Generate Sinusoidal Microthombi, and Promote Portal Hypertension. Gastroenterology. 2019;157:193-209 e9
52. Liu L, You Z, Yu H, Zhou L, Zhao H, Yan X. et al. Mechanotransduction-modulated fibrotic microniches reveal the contribution of angiogenesis in liver fibrosis. Nat Mater. 2017;16:1252-61
53. Rafii S, Butler JM, Ding BS. Angiocrine functions of organ-specific endothelial cells. Nature. 2016;529:316-25
54. Boopathy GTK, Hong W. Role of Hippo Pathway-YAP/TAZ Signaling in Angiogenesis. Front Cell Dev Biol. 2019;7:49
55. Panciera T, Azzolin L, Cordenonsi M, Piccolo S. Mechanobiology of YAP and TAZ in physiology and disease. Nat Rev Mol Cell Biol. 2017;18:758-70
56. Sudol M. Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the Yes proto-oncogene product. Oncogene. 1994;9:2145-52
57. Varelas X. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development. 2014;141:1614-26
58. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J. et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747-61
59. Chan EH, Nousiainen M, Chalamalasetty RB, Schafer A, Nigg EA, Sillje HH. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene. 2005;24:2076-86
60. Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH. et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol Cell Biol. 2008;28:2426-36
61. Praskova M, Xia F, Avruch J. MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr Biol. 2008;18:311-21
62. Chan SW, Lim CJ, Guo K, Ng CP, Lee I, Hunziker W. et al. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 2008;68:2592-8
63. Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 2010;24:72-85
64. Chappell JC, Wiley DM, Bautch VL. Regulation of blood vessel sprouting. Semin Cell Dev Biol. 2011;22:1005-11
65. Chen CC, Chen N, Lau LF. The angiogenic factors Cyr61 and connective tissue growth factor induce adhesive signaling in primary human skin fibroblasts. J Biol Chem. 2001;276:10443-52
66. Pobbati AV, Chan SW, Lee I, Song H, Hong W. Structural and functional similarity between the Vgll1-TEAD and the YAP-TEAD complexes. Structure. 2012;20:1135-40
67. Zhao B, Ye X, Yu J, Li L, Li W, Li S. et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962-71
68. Qiao Y, Lin SJ, Chen Y, Voon DC, Zhu F, Chuang LS. et al. RUNX3 is a novel negative regulator of oncogenic TEAD-YAP complex in gastric cancer. Oncogene. 2016;35:2664-74
69. Yagi R, Chen LF, Shigesada K, Murakami Y, Ito Y. A WW domain-containing yes-associated protein (YAP) is a novel transcriptional co-activator. EMBO J. 1999;18:2551-62
70. Alarcon C, Zaromytidou AI, Xi Q, Gao S, Yu J, Fujisawa S. et al. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell. 2009;139:757-69
71. Grannas K, Arngarden L, Lonn P, Mazurkiewicz M, Blokzijl A, Zieba A. et al. Crosstalk between Hippo and TGFbeta: Subcellular Localization of YAP/TAZ/Smad Complexes. J Mol Biol. 2015;427:3407-15
72. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ. et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012;151:1457-73
73. Strano S, Munarriz E, Rossi M, Castagnoli L, Shaul Y, Sacchi A. et al. Physical interaction with Yes-associated protein enhances p73 transcriptional activity. J Biol Chem. 2001;276:15164-73
74. Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J. et al. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat Cell Biol. 2008;10:837-48
75. Haskins JW, Nguyen DX, Stern DF. Neuregulin 1-activated ERBB4 interacts with YAP to induce Hippo pathway target genes and promote cell migration. Sci Signal. 2014;7:ra116
76. Nishio M, Hamada K, Kawahara K, Sasaki M, Noguchi F, Chiba S. et al. Cancer susceptibility and embryonic lethality in Mob1a/1b double-mutant mice. J Clin Invest. 2012;122:4505-18
77. Lee KP, Lee JH, Kim TS, Kim TH, Park HD, Byun JS. et al. The Hippo-Salvador pathway restrains hepatic oval cell proliferation, liver size, and liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:8248-53
78. Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J. et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19:27-38
79. Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y. et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009;16:425-38
80. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M. et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179-83
81. Kim W, Khan SK, Liu Y, Xu R, Park O, He Y. et al. Hepatic Hippo signaling inhibits protumoural microenvironment to suppress hepatocellular carcinoma. Gut. 2018;67:1692-703
82. Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development. 2011;138:3907-14
83. Hagenbeek TJ, Webster JD, Kljavin NM, Chang MT, Pham T, Lee HJ. et al. The Hippo pathway effector TAZ induces TEAD-dependent liver inflammation and tumors. Sci Signal. 2018 11
84. Kordes C, Sawitza I, Gotze S, Herebian D, Haussinger D. Hepatic stellate cells contribute to progenitor cells and liver regeneration. J Clin Invest. 2014;124:5503-15
85. Swiderska-Syn M, Xie G, Michelotti GA, Jewell ML, Premont RT, Syn WK. et al. Hedgehog regulates yes-associated protein 1 in regenerating mouse liver. Hepatology. 2016;64:232-44
86. Dechene A, Sowa JP, Gieseler RK, Jochum C, Bechmann LP, El Fouly A. et al. Acute liver failure is associated with elevated liver stiffness and hepatic stellate cell activation. Hepatology. 2010;52:1008-16
87. Konishi T, Schuster RM, Lentsch AB. Proliferation of hepatic stellate cells, mediated by YAP and TAZ, contributes to liver repair and regeneration after liver ischemia-reperfusion injury. Am J Physiol Gastrointest Liver Physiol. 2018;314:G471-G82
88. Li Z, Dranoff JA, Chan EP, Uemura M, Sevigny J, Wells RG. Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology. 2007;46:1246-56
89. Olsen AL, Bloomer SA, Chan EP, Gaca MD, Georges PC, Sackey B. et al. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am J Physiol Gastrointest Liver Physiol. 2011;301:G110-8
90. Mannaerts I, Leite SB, Verhulst S, Claerhout S, Eysackers N, Thoen LF. et al. The Hippo pathway effector YAP controls mouse hepatic stellate cell activation. J Hepatol. 2015;63:679-88
91. Martin K, Pritchett J, Llewellyn J, Mullan AF, Athwal VS, Dobie R. et al. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat Commun. 2016;7:12502
92. Huang X, Yang N, Fiore VF, Barker TH, Sun Y, Morris SW. et al. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. 2012;47:340-8
93. Simonetto DA, Yang HY, Yin M, de Assuncao TM, Kwon JH, Hilscher M. et al. Chronic passive venous congestion drives hepatic fibrogenesis via sinusoidal thrombosis and mechanical forces. Hepatology. 2015;61:648-59
94. Zhao XH, Laschinger C, Arora P, Szaszi K, Kapus A, McCulloch CA. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J Cell Sci. 2007;120:1801-9
95. Foster CT, Gualdrini F, Treisman R. Mutual dependence of the MRTF-SRF and YAP-TEAD pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. 2017;31:2361-75
96. Ding BS, Cao Z, Lis R, Nolan DJ, Guo P, Simons M. et al. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature. 2014;505:97-102
97. Hata S, Hirayama J, Kajiho H, Nakagawa K, Hata Y, Katada T. et al. A novel acetylation cycle of transcription co-activator Yes-associated protein that is downstream of Hippo pathway is triggered in response to SN2 alkylating agents. J Biol Chem. 2012;287:22089-98
98. Wang Y, Tu K, Liu D, Guo L, Chen Y, Li Q. et al. p300 Acetyltransferase Is a Cytoplasm-to-Nucleus Shuttle for SMAD2/3 and TAZ Nuclear Transport in Transforming Growth Factor beta-Stimulated Hepatic Stellate Cells. Hepatology. 2019;70:1409-23
99. Lorenz L, Axnick J, Buschmann T, Henning C, Urner S, Fang S. et al. Mechanosensing by beta1 integrin induces angiocrine signals for liver growth and survival. Nature. 2018;562:128-32
100. Kostallari E, Shah VH. Angiocrine signaling in the hepatic sinusoids in health and disease. Am J Physiol Gastrointest Liver Physiol. 2016;311:G246-51
101. Elpek GO. Angiogenesis and liver fibrosis. World J Hepatol. 2015;7:377-91
102. Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D. et al. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol. 2017;66:212-27
103. Park JA, Kwon YG. Hippo-YAP/TAZ signaling in angiogenesis. BMB Rep. 2018;51:157-62
104. Zhang C, Bian M, Chen X, Jin H, Zhao S, Yang X. et al. Oroxylin A prevents angiogenesis of LSECs in liver fibrosis via inhibition of YAP/HIF-1alpha signaling. J Cell Biochem. 2018;119:2258-68
105. Kim W, Khan SK, Yang Y. Interacting network of Hippo, Wnt/beta-catenin and Notch signaling represses liver tumor formation. BMB Rep. 2017;50:1-2
106. de La Coste A, Romagnolo B, Billuart P, Renard CA, Buendia MA, Soubrane O. et al. Somatic mutations of the beta-catenin gene are frequent in mouse and human hepatocellular carcinomas. Proc Natl Acad Sci U S A. 1998;95:8847-51
107. Hansen CG, Moroishi T, Guan KL. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 2015;25:499-513
108. Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL. et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458-61
109. Tanimizu N, Mitaka T. Re-evaluation of liver stem/progenitor cells. Organogenesis. 2014;10:208-15
110. Geisler F, Strazzabosco M. Emerging roles of Notch signaling in liver disease. Hepatology. 2015;61:382-92
111. Chen H, Liu D, Yang Z, Sun L, Deng Q, Yang S. et al. Adrenergic signaling promotes angiogenesis through endothelial cell-tumor cell crosstalk. Endocr Relat Cancer. 2014;21:783-95
112. Luo K. Signaling Cross Talk between TGF-beta/Smad and Other Signaling Pathways. Cold Spring Harb Perspect Biol. 2017 9
113. Totaro A, Castellan M, Di Biagio D, Piccolo S. Crosstalk between YAP/TAZ and Notch Signaling. Trends Cell Biol. 2018;28:560-73
114. Moya IM, Halder G. Hippo-YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat Rev Mol Cell Biol. 2019;20:211-26
115. Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q. et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A. 2010;107:1437-42
116. Masuzaki R, Tateishi R, Yoshida H, Goto E, Sato T, Ohki T. et al. Prospective risk assessment for hepatocellular carcinoma development in patients with chronic hepatitis C by transient elastography. Hepatology. 2009;49:1954-61
117. Tucker GC. Inhibitors of integrins. Curr Opin Pharmacol. 2002;2:394-402
118. Mathieu S, Manneville JB. Intracellular mechanics: connecting rheology and mechanotransduction. Curr Opin Cell Biol. 2019;56:34-44
119. Ingber DE. Tensegrity: the architectural basis of cellular mechanotransduction. Annu Rev Physiol. 1997;59:575-99
120. Klezovitch O, Vasioukhin V. Cadherin signaling: keeping cells in touch. F1000Res. 2015;4:550
121. St Croix B, Sheehan C, Rak JW, Florenes VA, Slingerland JM, Kerbel RS. E-Cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1). J Cell Biol. 1998;142:557-71
122. Sukharev SI, Blount P, Martinac B, Blattner FR, Kung C. A large-conductance mechanosensitive channel in E. coli encoded by mscL alone. Nature. 1994;368:265-8
123. Patel AJ, Honore E, Maingret F, Lesage F, Fink M, Duprat F. et al. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. 1998;17:4283-90
124. Lesage F, Maingret F, Lazdunski M. Cloning and expression of human TRAAK, a polyunsaturated fatty acids-activated and mechano-sensitive K(+) channel. FEBS Lett. 2000;471:137-40
125. Desai SS, Tung JC, Zhou VX, Grenert JP, Malato Y, Rezvani M. et al. Physiological ranges of matrix rigidity modulate primary mouse hepatocyte function in part through hepatocyte nuclear factor 4 alpha. Hepatology. 2016;64:261-75
126. Xia T, Zhao R, Feng F, Song Y, Zhang Y, Dong L. et al. Gene expression profiling of human hepatocytes grown on differing substrate stiffness. Biotechnol Lett. 2018;40:809-18
127. Gortzen J, Schierwagen R, Bierwolf J, Klein S, Uschner FE, van der Ven PF. et al. Interplay of Matrix Stiffness and c-SRC in Hepatic Fibrosis. Front Physiol. 2015;6:359
128. Nakagomi R, Tateishi R, Masuzaki R, Soroida Y, Iwai T, Kondo M. et al. Liver stiffness measurements in chronic hepatitis C: Treatment evaluation and risk assessment. J Gastroenterol Hepatol. 2019;34:921-8
129. Guo L, Teng L. YAP/TAZ for cancer therapy: opportunities and challenges (review). Int J Oncol. 2015;46:1444-52
130. Zhi X, Zhao D, Zhou Z, Liu R, Chen C. YAP promotes breast cell proliferation and survival partially through stabilizing the KLF5 transcription factor. Am J Pathol. 2012;180:2452-61
131. Wang EY, Cheng JC, Thakur A, Yi Y, Tsai SH, Hoodless PA. YAP transcriptionally regulates ErbB2 to promote liver cell proliferation. Biochim Biophys Acta Gene Regul Mech. 2018
132. Cheng JC, Wang EY, Yi Y, Thakur A, Tsai SH, Hoodless PA. S1P Stimulates Proliferation by Upregulating CTGF Expression through S1PR2-Mediated YAP Activation. Mol Cancer Res. 2018;16:1543-55
133. LaQuaglia MJ, Grijalva JL, Mueller KA, Perez-Atayde AR, Kim HB, Sadri-Vakili G. et al. YAP Subcellular Localization and Hippo Pathway Transcriptome Analysis in Pediatric Hepatocellular Carcinoma. Sci Rep. 2016;6:30238
134. Ahn EY, Kim JS, Kim GJ, Park YN. RASSF1A-mediated regulation of AREG via the Hippo pathway in hepatocellular carcinoma. Mol Cancer Res. 2013;11:748-58
135. Cho Y, Park MJ, Kim K, Kim SW, Kim W, Oh S. et al. Reactive oxygen species-induced activation of Yes-associated protein-1 through the c-Myc pathway is a therapeutic target in hepatocellular carcinoma. World J Gastroenterol. 2020;26:6599-613
136. Liu S, Miao R, Zhai M, Pang Q, Deng Y, Liu S. et al. Effects and related mechanisms of serotonin on malignant biological behavior of hepatocellular carcinoma via regulation of Yap. Oncotarget. 2017;8:47412-24
137. Yang N, Chen T, Wang L, Liu R, Niu Y, Sun L. et al. CXCR4 mediates matrix stiffness-induced downregulation of UBTD1 driving hepatocellular carcinoma progression via YAP signaling pathway. Theranostics. 2020;10:5790-801
138. Xu MZ, Chan SW, Liu AM, Wong KF, Fan ST, Chen J. et al. AXL receptor kinase is a mediator of YAP-dependent oncogenic functions in hepatocellular carcinoma. Oncogene. 2011;30:1229-40
139. Huo X, Zhang Q, Liu AM, Tang C, Gong Y, Bian J. et al. Overexpression of Yes-associated protein confers doxorubicin resistance in hepatocellullar carcinoma. Oncol Rep. 2013;29:840-6
140. Kim MK, Jang JW, Bae SC. DNA binding partners of YAP/TAZ. BMB Rep. 2018;51:126-33
141. Avruch J, Zhou D, Bardeesy N. YAP oncogene overexpression supercharges colon cancer proliferation. Cell Cycle. 2012;11:1090-6
142. Liu AM, Xu MZ, Chen J, Poon RT, Luk JM. Targeting YAP and Hippo signaling pathway in liver cancer. Expert Opin Ther Targets. 2010;14:855-68
143. Urtasun R, Latasa MU, Demartis MI, Balzani S, Goni S, Garcia-Irigoyen O. et al. Connective tissue growth factor autocriny in human hepatocellular carcinoma: oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology. 2011;54:2149-58
144. Wang J, Tang X, Weng W, Qiao Y, Lin J, Liu W. et al. The membrane protein melanoma cell adhesion molecule (MCAM) is a novel tumor marker that stimulates tumorigenesis in hepatocellular carcinoma. Oncogene. 2015;34:5781-95
145. Miao HL, Pan ZJ, Lei CJ, Wen JY, Li MY, Liu ZK. et al. Knockdown of GPC3 inhibits the proliferation of Huh7 hepatocellular carcinoma cells through down-regulation of YAP. J Cell Biochem. 2013;114:625-31
146. Wang H, Du YC, Zhou XJ, Liu H, Tang SC. The dual functions of YAP-1 to promote and inhibit cell growth in human malignancy. Cancer Metastasis Rev. 2014;33:173-81
Author contact
![]() Corresponding author: Prof. Stefan Zahler, E-mail: stefan.zahleruni-muenchen.de; Phone: +49-89-218077196.
Corresponding author: Prof. Stefan Zahler, E-mail: stefan.zahleruni-muenchen.de; Phone: +49-89-218077196.