Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2022; 13(2):527-540. doi:10.7150/jca.61385 This issue Cite
Research Paper
SIX1 Predicts Poor Prognosis and Facilitates the Progression of Non-small Lung Cancer via Activating the Notch Signaling Pathway
Shanshan Huang, Wanling Lin, Lei Wang, Yuan Gao, Xun Yuan, Peng Zhang, Yuan Chen ![]() , Qian Chu
, Qian Chu ![]()
Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Received 2021-4-8; Accepted 2021-11-30; Published 2022-1-1
Abstract

Background: Many transcription factors involved in embryonic development and reactivated in tumors are considered potential prognostic biomarkers and novel therapeutic targets in various cancers. Sine oculis homeobox homolog 1 (SIX1), a developmentally restricted transcriptional regulator, plays a critical role during tumor initiation and development. However, the prognostic value and biological function of SIX1 in non-small cell lung cancer (NSCLC) remain unclear.
Methods: Bioinformatic analyses were conducted to investigate the expression of SIX1 in cancer and adjacent normal tissues of NSCLC and further explore the correlations between SIX1 expression and clinical outcomes. Western blotting and RT-PCR analysis were performed to detect of SIX1 expression level in NSCLC cell lines and normal bronchial epithelial cell. EdU, CCK-8, clonal formation assay, wound healing and transwell assay were performed to explore the effects of gain- or loss-of-function of SIX1 on cellular proliferation, migration and invasion in vitro. Gene set enrichment analysis (GSEA) was used to identify the potential signaling pathways involved in SIX1 mediated biological function and the correlation was confirmed by western blotting and RT-PCR analysis. In vivo experiment was conducted to further validate the tumor-promoting effects of SIX1.
Results: Bioinformatic analysis indicated that SIX1 was markedly upregulated in NSCLC tissues of and positively correlated with poor prognosis of patients with NSCLC. Ectopic expression of SIX1 facilitated proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT) of NSCLC cells. On the contrary, knocking down SIX1 exhibited the opposite effects. Mechanistic studies suggested that SIX1 activated the Notch pathway to promote the malignant biological behaviors of NSCLC, which could be reversed by inhibiting the Notch signaling with γ-secretase inhibitor.
Conclusions: SIX1 could facilitate multiple malignant biological behaviors by activating the Notch signaling pathway and function as a promising prognostic biomarker.
Keywords: SIX1, non-small cell lung cancer, Notch signaling, epithelial-mesenchymal transition.
Introduction
Lung cancer is the leading cause of cancer-related death worldwide. According to the latest data from Global Cancer Statistics, there were approximately 2.2 million new cases and 1.8 million deaths of lung cancer in 2020, accounting for approximately 1 in 10 (11.4%) cancers diagnosed and 1 in 5 (18.0%) deaths [1]. Approximately 85% of lung cancers are diagnosed as non-small cell lung cancer (NSCLC), including lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and large cell lung cancer (LCLC), etc [2]. Despite the rapid development of advanced diagnostic methods and precise individual therapeutic regimens, uncontrolled proliferation and metastasis still lead to poor prognosis of patients with NSCLC. Therefore, an in-depth understanding of the molecular mechanisms involved in tumor progression is crucial for developing novel therapies for patients with NSCLC.
Aberrant activation of developmental genes in mature tissues is one of the important causes of tumorigenesis. Certainly, many homeobox genes, which were known to be critical for specific cells development, displayed deregulated expression in cancer [3-5]. Evidences have suggested that their inappropriate activation played a complex causal role in carcinogenesis and tumor progression [6]. Homeobox transcription factor SIX1, which belongs to the SIX families, is essential for the development of many organs, including the eye, ear, muscle and kidney [7, 8]. In recent decades, SIX1 has been reported to be overexpressed in numerous human cancers, such as breast cancer [9, 10], ovarian cancer [11], colorectal cancer [12], thyroid carcinoma [13], and prostate cancer [14], leading to a more aggressive cell phenotype and poorer prognosis. Mimae T et al. showed that SIX1 was 136-fold upregulated in microinvasion cancer cells compared to lepidic growth cancer cells in minimally invasive adenocarcinomas using a laser capture microdissection system [15], suggesting that SIX1 may be a reliable progressive biomarker in NSCLC. However, the prognostic value and biological function of SIX1 in NSCLC required further investigation.
In this study, we revealed that SIX1 level was significantly elevated in NSCLC, and the high level of SIX1 was correlated with shortened time to relapse and decreased overall survival (OS). Overexpression of SIX1 could promote proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT) of NSCLC cells, whereas knockdown of SIX1 exhibited the opposite effects. In addition, we found that SIX1 could activate the Notch signaling pathway in NSCLC, and inhibition of the Notch signaling pathway with γ-secretase inhibitor could reverse a series of SIX1-mediated malignant phenotypes. In summary, our results indicated that SIX1 might serve as a potential prognostic biomarker for NSCLC, and suggested that the SIX1/Notch axis might be a promising target for combating NSCLC progression.
Materials and Methods
Data mining and analysis
The TIMER database (http://timer.cistrome.org/) was used to analyze the mRNA expression of SIX1 in pan-cancer. The Oncomine database (http://www.oncomine.org) was used to further analyze the mRNA levels of SIX1 in cancer tissues and adjacent normal tissues of NSCLC. The UALCAN database (http://ualcan.path.uab.edu/) exhibited comprehensive cancer transcriptome and clinical patient data (pulled from TCGA). GSE31210 was downloaded from the GEO database to analyze the correlation between the level of SIX1 and prognosis. The potential related signaling pathway was detected through gene set enrichment analysis (GSEA).
Cell Cultures and reagents
The human NSCLC cell lines A549, H1299, PC-9, H460 and the normal bronchial epithelial cell (HBE) were obtained from the oncology laboratory of Tongji Hospital, Wuhan, China. All cells were cultured in RPMI 1640 medium (HyClone, Logan, UT) supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Grand Island, NY), and then stored in a 37 °C humidified incubator with 5% CO2. The γ-secretase inhibitor DAPT was purchased from MedChemExpress (Monmouth Junction, NJ).
Western blotting analysis
Cells were lysed with RIPA lysis buffer (Beyotime, Shanghai, China) on ice for half an hour. The supernatant was collected after centrifugation at 12 000 g at 4℃ for 20 min. Protein concentration was measured by the BCA protein assay kit (Beyotime, Shanghai, China). The denatured proteins were separated by SDS‐PAGE gels and transferred to the 0.45 μm PVDF membranes (Millipore, Billerica, USA). The membranes were blocked with 5% BSA for 1 h at room temperature. Next, the membranes were incubated with primary antibodies including SIX1 (#16960), E-cadherin (#3195), Vimentin (#5741), Notch1 (#4380), Hes1 (#11988), Notch2 (#5732), (1:1000, Cell Signaling Technology, Beverly, MA, USA), Tubulin mouse monoclonal antibody (#66031‐1‐Ig), (1:5000, Proteintech, Wuhan, China) at 4°C overnight, and then incubated with corresponding secondary antibodies (1:5000, Boster, Wuhan, China) for 1 hour at room temperature. Finally, West Pico Plus Chemiluminescent Substrate (Thermo Scientific, Marietta, OH, USA) was used to detect the indicated proteins. Image J (National Institutes of Health, Bethesda, MA, USA) was used to analyze the intensity of the blotting bands.
Real-time PCR (RT-PCR) analysis
Total RNA was extracted from cells with TRIzol reagent (Takara, Shiga, Japan) and then reverse transcribed into cDNA using a PrimeScript RT-PCR kit (Takara, Japan). Real-time PCR analysis was conducted using SYBR Premix Ex Taq (Takara, Japan) with a 7900 Real-time PCR system (Applied Biosystems, USA). The △△CT method was adopted to calculate the relative mRNA expression of indicated genes, which was further normalized to the β-actin mRNA levels. The sequences of the primers were shown in Table 1.
The sequences of primers used for experiments in this study.
| Primer | Sequence 5'-3' |
|---|---|
| SIX1 Forward Primer | CTGCCGTCGTTTGGCTTTAC |
| SIX1 Reverse Primer | GCTCTCGTTCTTGTGCAGGT |
| β-actin Forward Primer | CATGTACGTTGCTATCCAGGC |
| β-actin Reverse Primer | CTCCTTAATGTCACGCACGAT |
| Notch1 Forward Primer | GAGGCGTGGCAGACTATGC |
| Notch1 Reverse Primer | CTTGTACTCCGTCAGCGTGA |
| Notch2 Forward Primer | CCTTCCACTGTGAGTGTCTGA |
| Notch2 Reverse Primer | AGGTAGCATCATTCTGGCAGG |
| Hes1 Forward Primer | TCAACACGACACCGGATAAAC |
| Hes1 Reverse Primer | GCCGCGAGCTATCTTTCTTCA |
| E-cadherin Forward Primer | CGAGAGCTACACGTTCACGG |
| E-cadherin Reverse Primer | GGGTGTCGAGGGAAAAATAGG |
| Vimentin Forward Primer | GACGCCATCAACACCGAGTT |
| Vimentin Reverse Primer | CTTTGTCGTTGGTTAGCTGGT |
Cell transfection with lentivirus
Cells were cultured overnight to 60% confluence in RPMI-1640 containing 10% FBS before transfection. Subsequently, the cell culture medium was replaced with a serum-free medium containing the lentiviruses (GeneChem, Shanghai, China) at a multiplicity of infection of 20 and polybrene (GeneChem) at a final concentration of 5 μg/ml. Cells as negative control were transduced with corresponding empty vector lentiviruses in the same manner. According to the cell status, the cells were transferred to a complete medium at 12-24h after transfection and further cultured for another 48 h. Then cells were screened with 2 μg/ml puromycin for one week.
Cell transfection with siRNA
Transfection with siRNA was performed with Lipofectamine 2000 (Invitrogen) according to the instructions. The small interfering RNAs used to knockdown SIX1 were:
siSIX1-1:
Sense, GCUACUGCUUCAAGGAGAATT,
Antisense, UUCUCCUUGAAGCAGUAGCTT,
siSIX1-2:
Sense, GCGACCACCUGCACAAGAATT,
Antisense, UUGUUGUGCAGGUGGUCGCTT,
siSIX1-3:
Sense, GUGCGAGGUUCUGCAGCAATT,
Antisense, UUGCUGCAGAACCUCGCACTT.
Immunofluorescence staining
A total of 3 × 104 cells were seeded in 24-well plates and incubated at 37 °C overnight. Cells were washed with PBS and then fixed with 4% polyformaldehyde for 15 min. Next, cells were permeabilized with 0.5% Triton X-100 for 20 min and blocked with 5% BSA for 30 min at room temperature. After blocking, cells were incubated with diluted primary antibodies (SIX1, 1:400, #16960, Vimentin, 1:400, #5741) at 4 °C overnight. The next day, cells were incubated with Alexa Fluor Cy3-conjugated secondary antibody (1:200, Promoter, Wuhan, China) at room temperature for 1 h and then incubated with DAPI for 5 min. Finally, the cells were covered with anti-fluorescence quenching agents and photographed under a fluorescence microscope. Quantification of fluorescence intensities was conducted using Image J software. The exposure times and microscope settings were consistent for each captured images. Three random areas were selected to take three images in each independent experiment, and three independent experiments were repeated.
Cell counting kit‐8 (CCK8) assay
The transfected cells were plated into 96-well plates with 2 × 103 cells/100 μl per well. After the cells attached to the well, all cells incubated for another 0, 24, 48, or 72 hours. Then, 100ul serum-free medium containing 10% CCK8 was added to each well and incubated for another 2 h. The absorbance was measured at 450 nm via a Power Wave XS microplate reader (BioTek, Winooski, VT, USA). Each experiment was conducted at least three times.
EdU assay
The transfected cells were plated into 96-well plates with 1 × 104 cells/100 μl per well. After the cells were attached to the well, various reagents were added sequentially according to the EdU kit's instructions (RiboBio, Guangzhou, China). Edu positive cells were observed under the fluorescence microscope.
Transwell assays
Transwell 24‐well plates with an 8‐μm diameter filter (Corning, Corning, NY, USA) were used for the experiments. For the transwell invasion assay, the filters were precoated with 50 μl of diluted extracellular matrix (ECM) gel overnight. 5 × 104 cells (migration) and 1 × 105 cells (invasion) in 200 μl serum‐free RPMI‐1640 medium were placed in the upper chamber and 700 μl RPMI‐1640 medium with 20% FBS was placed in the lower chamber. The plates were then cultured for another 24 hours or 48 hours. Next, the upper cells were fixed with 4% paraformaldehyde and stained with 0.3% crystal violet for 15 min. Finally, the cells passing through the aperture were examined and counted under the microscope.
Immunohistochemistry (IHC)
At the endpoint of the observation, the tumor tissues were removed and then fixed in 4% paraformaldehyde. The fixed tissues were then embedded in paraffin and sectioned at 4μm thickness. After that, the sections were deparaffinized, rehydrated, and stained using the primary antibodies (SIX1, 1:100, CST; Hes1, 1:2000, CST; Vimentin, 1:200, CST; Ki-67, 1:400, Abcam) at 4 °C overnight. The next day, sections were incubated with the corresponding secondary antibody at 37 °C for 30 min. Finally, images were captured under the microscope. The protein expression was evaluated according to the staining intensity and percentage of positive cells by using an H-score method [16].
Animal experiments
Five‐week‐old male BALB/c nude mice were used for the construction of the xenograft model in vivo. Cells (5×10^6/125 μL PBS) were injected subcutaneously into the right flanks of each mouse (five mice per group). Tumor volumes were measured in two dimensions every three days using digital calipers and calculated using the following formula: tumor volume (mm3) = 1/2 (length (mm) × (width (mm))2). After 24 days of injection, the mice were sacrificed, and the tumor tissues were fixed and embedded for further analyses.
Statistical analysis
All quantitative data are presented as the mean values ± SD from at least three independent experiments and statistical analyses were undertaken by GraphPad 8.0. The survival analysis was performed using GraphPad 8.0. Significant differences were determined by a two-tailed Student's t-test or one-way ANOVA analysis. Pearson's correlation was used to measure the strength of association between two variables. p < 0.05 was considered significant. *p< 0.05, **p < 0.01, ***p < 0.001.
Results
Elevated mRNA expression of SIX1 in many types of cancer
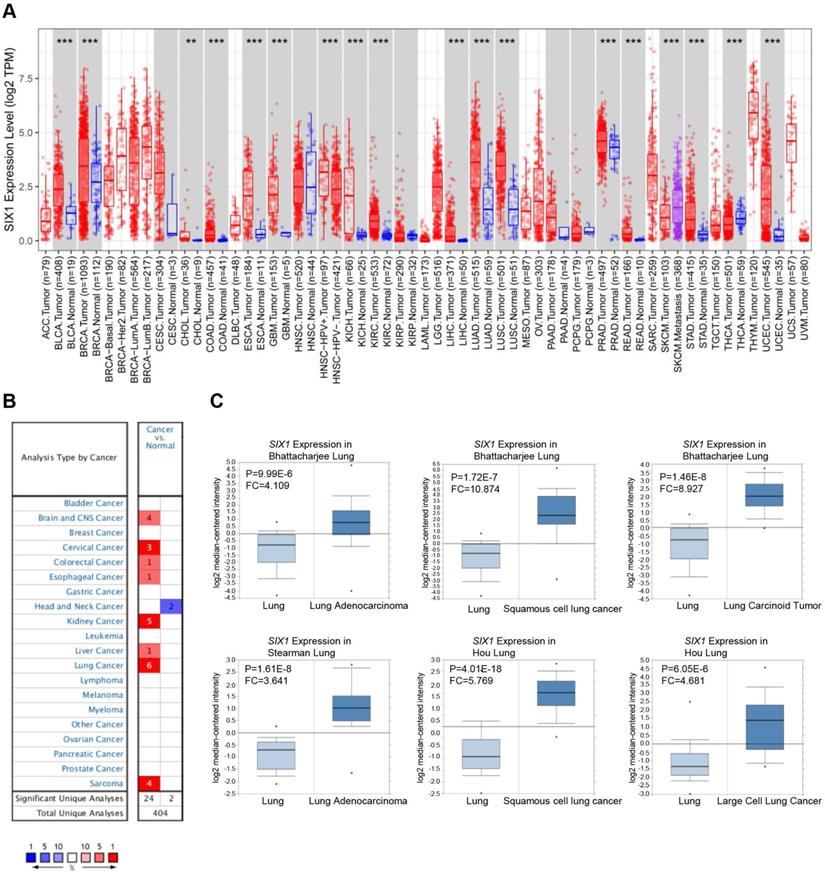
First, the expression of SIX1 in cancers and matched adjacent normal tissues were compared using the TIMER database. The results suggested that SIX1 mRNA levels were remarkably upregulated in many cancer tissues (BLCA, BRCA, CHOL, COAD, ESCA, GBM, KICH, KIRC, LIHC, LUAD, LUSC, PRAD, READ, STAD, and LUEC), compared with the corresponding adjacent normal tissues. Moreover, SKCM metastatic tissues showed higher SIX1 mRNA level than SKCM primary tumor tissues (Fig. 1A). We also examined the transcriptional level of SIX1 in a variety of malignancies using the Oncomine database. The database contains 26 significant, unique analyses. In 24 of the 26 unique analyses, SIX1 expression was elevated in cancer tissues than in normal tissues, including 6 analyses related to lung cancer (Fig. 1B). In these 6 analyses of lung cancer from Bhattacharjee al., Stesrman et al. and Hou et al., SIX1 expression levels in NSCLC, including LUAD, LUSC, lung carcinoid tumor, and LCLC, were significantly upregulated in cancer tissues (Fig. 1C).
SIX1 mRNA expression in pan-cancer. (A) SIX1 expression in multiple cancers were analyzed using the TIMER database. (B) SIX1 expression in multiple cancers were analyzed using the Oncomine database. (C) SIX1 expression was increased in LUAD, LUSC, lung carcinoid tumor and LCLC tumor tissues. *p< 0.05, **p < 0.01, ***p < 0.001.
SIX1 predicted poor clinical outcomes in NSCLC
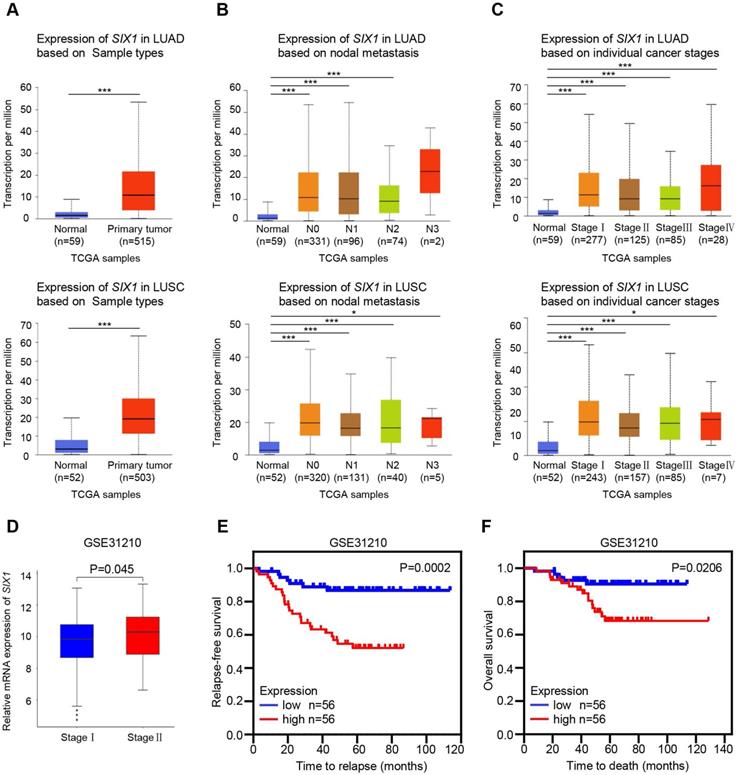
The UALCAN and GEO databases were used to further analyze the SIX1 expression in NSCLC. Consistently, TCGA-LUAD and TCGA-LUSC samples in the UALCAN database also showed elevated mRNA level of SIX1 in lung cancer tissues (Fig. 2A). Although there was no significant correlation between the SIX1 expression and lymph node metastasis or tumor-node-metastasis (TNM) stage when N0, N1, N2, and N3, or stage I, II, III, and IV were compared, the expression of SIX1 was markedly higher in patients with TCGA-LUAD and TCGA-LUSC than normal controls in a subgroup analysis based on nodal metastasis and disease stages, (Fig. 2B and 2C). In addition, a data mining based on the GSE31210, a database that included 226 primary LUAD of pathological stage I-II, showed that the SIX1 expression was higher in patients in stage II than in stage I (Fig. 2D). Moreover, survival analysis revealed that high mRNA expression of SIX1 was associated with the short time to relapse and poor OS in NSCLC (Fig. 2E and 2F). These data suggested that SIX1 might be a potential prognostic indicator in NSCLC.
Correlation of SIX1 expression and clinicopathological features in NSCLC patients. (A) SIX1 was significantly upregulated in LUAD and LUSC in the UALCAN database. (B) SIX1 expression in LUAD and LUSC with different lymph node metastatic states. (C) SIX1 expression in LUAD and LUSC with stage Ⅰ to stage Ⅳ diseases. (D) SIX1 expression in patients with different stages in the GSE31210 database. (E) The correlation between SIX1 expression and RFS in NSCLC patients. (F) The correlation between SIX1 expression and OS in NSCLC patients. *p< 0.05, **p < 0.01, ***p < 0.001.
SIX1 promoted proliferation of NSCLC cells
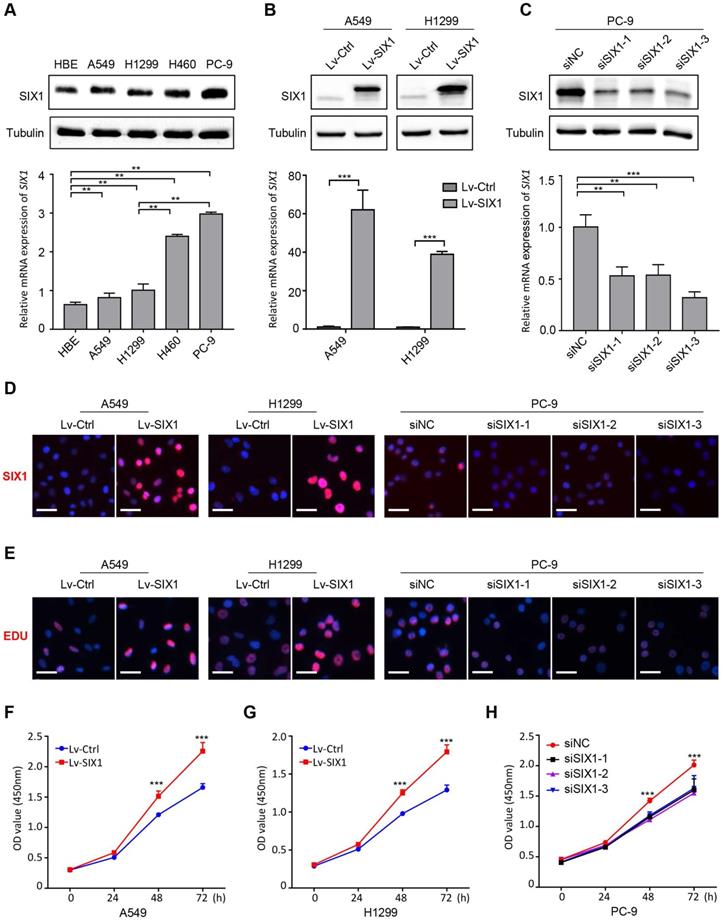
Based on the above results, we then conducted a series of gain-of-function and loss-of-function experiments in NSCLC cells to explore the potential role of SIX1 in NSCLC. The endogenous protein and mRNA expression of SIX1 in four NSCLC cell lines (A549, H1299, H460, and PC-9) and a normal human bronchial epithelial cell HBE were measured by western blotting and RT-PCR. Consistent with the analysis of clinical samples, SIX1 expression was generally upregulated in NSCLC cells compared with HBE. The endogenous expression of SIX1 was the highest in PC-9 cells, whereas A549 and H1299 cells harbored lower SIX1 protein and mRNA level (Fig. 3A). Therefore, the lentivirus overexpressing SIX1 was transfected into A549 and H1299 cells, while the siRNA knocking down SIX1 was applied to PC-9 cells. The structure of the lentiviral vector overexpressing SIX1 and its transfection efficiency in NSCLC cell lines were shown in Fig. S1. We found that SIX1 was efficiently overexpressed in A549 and H1299 cells and inhibited in PC-9 cells, and validated by western blotting, RT-PCR and immunofluorescence staining (Fig. 3B-D, Fig. S2). We proceeded to evaluate the effect of SIX1 on NSCLC cells viability by EdU assays. The data showed that overexpression of SIX1 contributed to more EdU positive cells, while knockdown of SIX1 exhibited an opposite effect (Fig. 3E). Additionally, CCK-8 assays also showed an increased cell growth in SIX1-overexpressing NSCLC cells, while knockdown of SIX1 diminished cell proliferation in vitro (Fig. 3F-H). Collectively, the above results demonstrated that SIX1 could positively regulate the proliferation of NSCLC cells.
SIX1 facilitates NSCLC cells growth in vitro. (A) The protein and mRNA level of SIX1 on baseline in four NSCLC cells and one normal bronchial epithelial cell. (B) SIX1 expression in A549 and H1299 cells transfected with SIX1-overexpressing lentivirus. (C) SIX1 expression in PC-9 cells transfected with SIX1 siRNA. (D) Immunofluorescence staining of SIX1 in indicated cells. (E) DNA synthesis in indicated cells by EdU assays. (F) (G) (H) The growth curves of indicated cells by CCK-8 assays. *p< 0.05, **p < 0.01, ***p < 0.001. Graphs show mean ± SD.
SIX1 contributed to migration, invasion and EMT of NSCLC cells
The above database analysis showed that SIX1 was elevated in metastatic SKCM tissues than in primary SKCM tumor tissues. In addition, upregulated SIX1 might be associated with late stage in NSCLC. Therefore, we next investigated the effect of SIX1 on metastasis in NSCLC cells. GSEA was performed based on RNA-sequencing data from the TCGA lung cancer database. The samples were divided into the SIX1-low group and SIX1-high group according to the quartile of SIX1 expression (Fig. 4A, SIX1-low: Q1, SIX1-high: Q4). GSEA revealed that the gene sets relevant to metastasis were enriched in samples with high SIX1 expression (Fig. 4B and 4C). Next, we conducted transwell and wound healing experiments to investigate the effects of SIX1 on cell migration and invasion in NSCLC in vitro. Evidently, A549 cells with SIX1 overexpression exhibited stronger migration and invasive abilities compared with control cells, while knockdown of SIX1 dramatically inhibited migration and invasion in PC-9 cells (Fig. 4D, Fig. S3).
SIX1 induced EMT and promoted migration and invasion. (A) SIX1 expression in the high-SIX1 sample compared with the low-SIX1 counterparts of the TCGA lung cancer database. (B, C) SIX1 expression was positively correlated with metastasis-related genes in NSCLC by GSEA. (D) Cell migration and invasion assays of indicated cells by transwell chamber. (E) The expression of E-cadherin and Vimentin was changed after up- or down- regulation of SIX1 in NSCLC cells. *p< 0.05, **p < 0.01, ***p < 0.001. Graphs show mean ± SD.
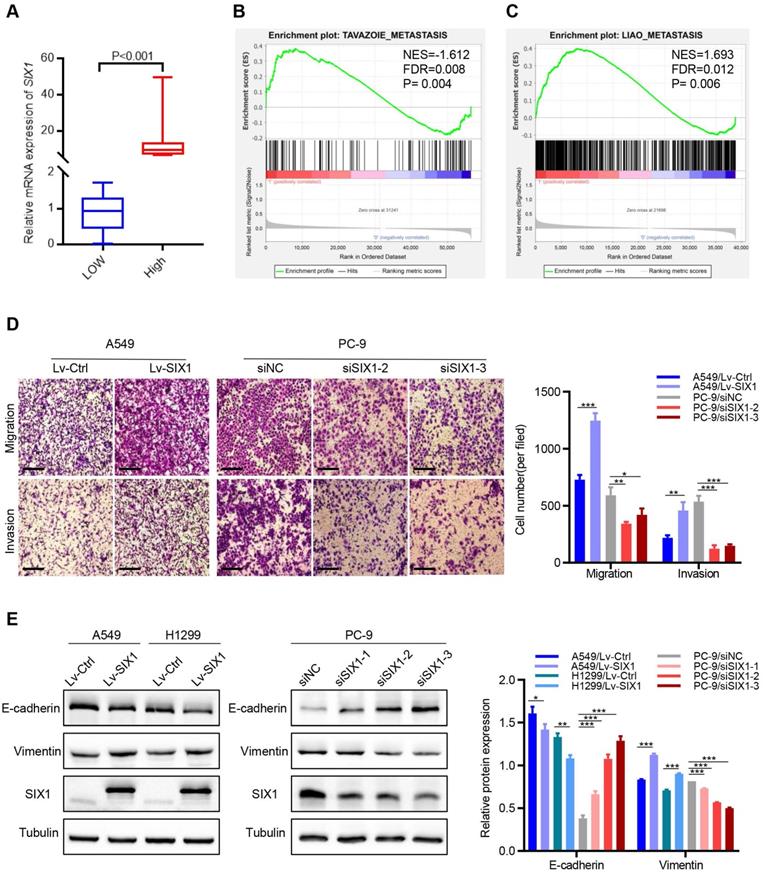
EMT has been reported to be critical for the increased cellular motility and invasiveness. Therefore, we further investigated the potential effect of SIX1 on EMT processes in NSCLC. We found that the protein levels of epithelial marker (E-cadherin) were notably decreased, and the level of a mesenchymal marker (Vimentin) was significantly increased in A549 and H1299 cells with SIX1 overexpression. Conversely, knockdown of SIX1 induced the up-regulation of E-cadherin and down-regulation of Vimentin in PC-9 cells (Fig. 4E). Similar results were found in the immunofluorescence assay of Vimentin (Fig. S4). In conclusion, the results confirmed that SIX1 could promote the migration, invasion, and EMT process of NSCLC cells.
SIX1 activated the Notch signaling pathway in NSCLC cells
Among the various pathways revealed by GSEA enrichment analysis, we observed that the Notch signaling pathway was positively correlated with the expression of SIX1 in NSCLC (Fig. 5A). Previous studies have demonstrated that the activated Notch signaling pathway played critical roles in regulating multiple malignant biological behaviors in NSCLC. Therefore, we tested whether SIX1 might exert its biological functions by activating the Notch signaling pathway. We first detected the correlation between SIX1 and the Notch pathway target gene, Hes1, in the TCGA database. We found that SIX1 and HES1 mRNA expressions were positively correlated in both LUAD and LUSC (Fig. 5B and 5C). Furthermore, overexpression of SIX1 robustly increased the protein levels of Notch1, Notch2 and Hes1 in A549 and H1299 cells (Fig. 5D), but without significant change of Notch1 and Notch2 mRNA levels, despite an increase in the expression of Hes1 mRNA (Fig. S5). In contrast, knockdown of SIX1 in PC-9 cells remarkably decreased the protein levels of Notch1, Notch2, and Hes1 (Fig. 5E). However, when Notch1 was knocked down, EMT was notably reversed in A549 cells, while the expression of SIX1 did not change (Fig. S6). These results suggested that SIX1 might act as an upstream to activate the Notch signaling pathway in NSCLC cells.
SIX1 activates the Notch signaling pathway in NSCLC cells. (A) The SIX1 expression was positively correlated with the Notch signaling pathway in NSCLC by GSEA. (B) The SIX1 and HES1 mRNA expression was positively correlated in TCGA LUAD samples. (C) The SIX1 and HES1 mRNA expression was positively correlated in TCGA LUSC samples. (D) (E) The protein level of Notch1, Notch2 and Hes1 in indicated cells. (F) The protein level of Notch1, Notch2, Hes1 E-cadherin, Vimentin and SIX1 upon treated with Notch pathway inhibitor (DAPT, 10uM). (G) Cell migration and invasion assays of indicated cells with or without the DAPT treatment by transwell chamber. *p< 0.05, **p < 0.01, ***p < 0.001. Graphs show mean ± SD.
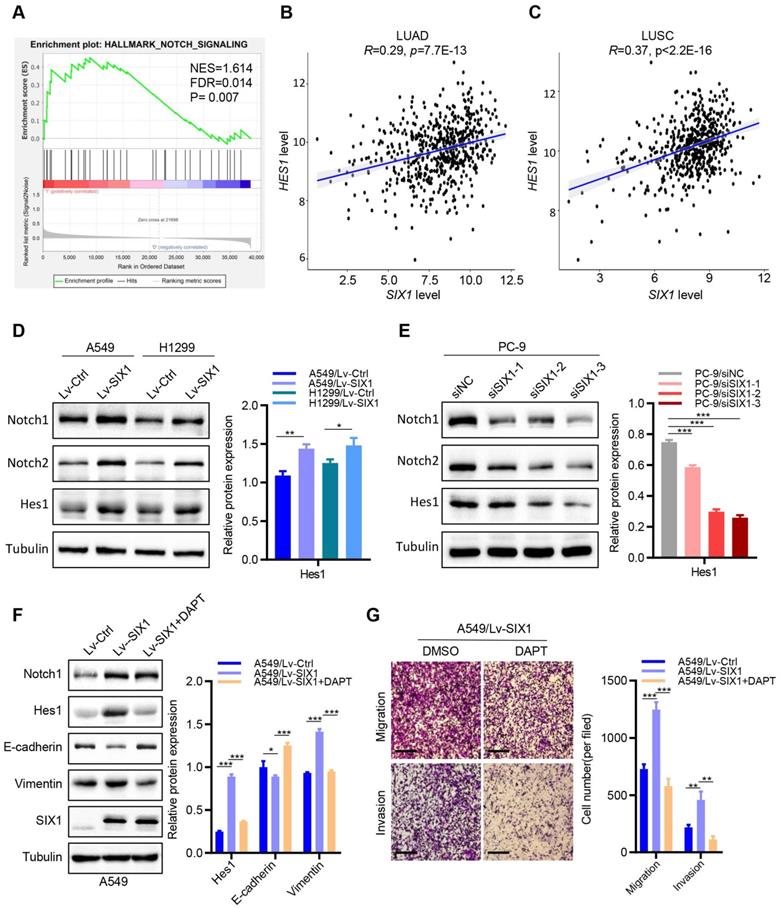
Based on the above results that overexpression of SIX1 could promote proliferation, migration, invasion, and EMT processes of NSCLC cells in vitro, we next investigated the effects of the Notch signaling inhibitor (a γ-secretase inhibitor, DAPT) [17] on SIX1-induced malignant biological behaviors. Interestingly, clonal formation analysis showed that DAPT significantly reduced SIX1-induced proliferation in A549 cells (Fig. S7). In addition, DAPT drastically attenuated the increased expression of Hes1 and Vimentin caused by the upregulation of SIX1, while the E-cadherin expression was elevated. Similarly, DAPT had no significant effect on the expression of SIX1. Furthermore, the abilities of cell migration and invasion were measured by transwell assays. We found that cell migration and invasion were weakened in the LV-SIX1 +DAPT group compared with the Lv-SIX1 group (Fig. 5F and 5G). Collectively, overexpression of SIX1 could promote the activation of the Notch signaling pathway, while inhibition of the Notch signaling could reduce the carcinogenic effects caused by SIX1 in NSCLC.
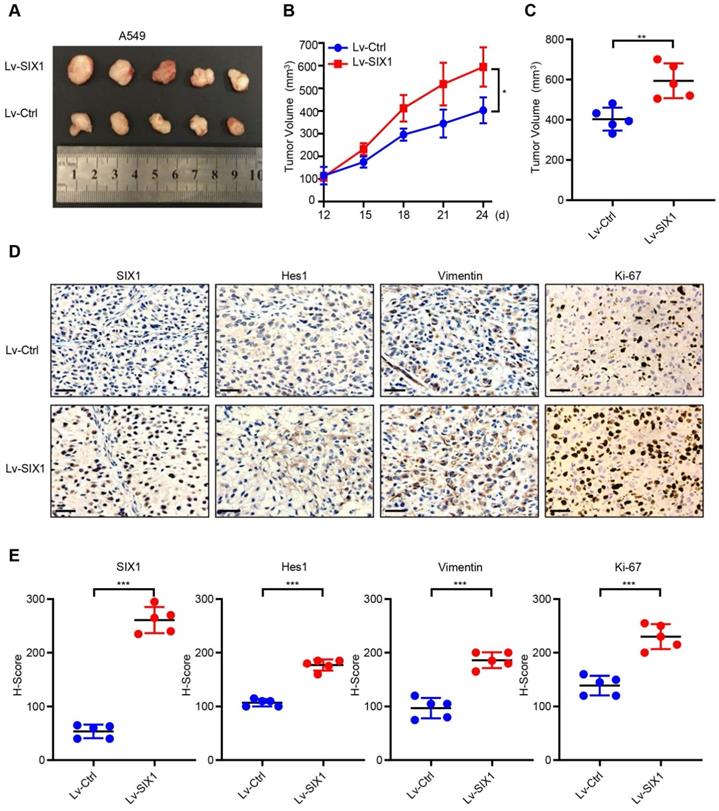
SIX1 facilitated tumor growth of NSCLC cells in vivo
To further investigate the potential oncogenic effects of SIX1, we generated the subcutaneous tumor by injecting A549/Lv-Ctrl cells and A549/Lv-SIX1 cells (5×106 cells/mouse) in BALB/c nude mice. The tumor size was measured from day 12 and recorded every three days, and a growth curve was plotted. Tumor tissue was removed from both groups on day 24, the endpoint of observation, and the mean tumor size was quantified. The results showed that overexpression of SIX1 significantly facilitated tumor growth in vivo (p< 0.05, Fig. 6A-C). We further performed IHC staining of tumor tissues on both groups. Data confirmed that SIX1 was overexpressed in the Lv-SIX1 tumor tissues compared with the control group, mainly located at the nucleus. Importantly, Hes1, Vimentin, and Ki-67 levels were also higher than those in the control groups (Fig. 6D and 6E). Therefore, these findings provide evidence that SIX1 could also act as an oncogene that induced Notch signaling activation and promote tumor growth and EMT process in vivo.
SIX1 facilitates the growth of A549 cells in vivo. (A) Tumor xenografts of each group. n=5. (B) Tumor growth curve in each group. (C) Tumor volumes on day 24 were shown for groups containing 5 mice each. (D) (E) IHC staining was performed to determine the expression of SIX1, Hes1, Vimentin and Ki-67 in the tumor. *p< 0.05, **p < 0.01, ***p < 0.001. Graphs show mean ± SD.
Discussion
Aberrant activation of embryonic developmental pathways is a pervasive tumor-promoting mechanism. Previous studies have indicated that SIX1, a transcription factor involved in embryonic development, is frequently dysregulated in numerous human cancers, and its increased expression is associated with poor clinical outcomes [18]. Usually, SIX1 is highly expressed during embryogenesis and rarely expressed in normal adult tissues [19]. SIX1 is expressed in the distal epithelium and stroma of lung branching airways, and its absence can lead to differentiation of lung stroma and fibroblasts, leading to defective development of lung septum [20]. In studies on NSCLC, although the upregulation of SIX1 has been observed to promote preinvasive-to-invasive LUAD progression [15], the expression status, prognostic value, and biological function of SIX1 in NSCLC remain to be further studied.
Consistent with previous studies, we also confirmed that the expression of SIX1 in NSCLC tissues was higher than that in normal tissues through the analysis of multiple databases [21, 22]. However, previous studies yielded inconsistent results regarding the prognostic value of SIX1 in NSCLC. Liu Q et al. reported that the higher expression of SIX1 was associated with the greater possibility of the tumorigenesis, but it had no correlation with node metastasis, TNM stage and OS [21]. A previous study conducted by Xia et al. showed that the expression of SIX1 was associated with large tumor size, advanced tumor stage, and distant metastasis of NSCLC [22]. Moreover, Mimae T et al. showed that the upregulation of SIX1 could contribute to preinvasive-to-invasive transition in ADC [15]. In our study, we did not find a significant correlation between the SIX1 expression and lymph node metastasis or TNM stage in TCGA database. However, our data mining based on the GSE31210 showed that the high SIX1 expression was associated with advanced tumor stage and a high level of SIX1 was correlated with shortened time to relapse and decreased OS in patients with NSCLC. The possible explanation was the difference in populations of patients and the difference in the cut-off values in different studies. The samples of the study of Xia et al., Mimae T et al, and GSE31210 were from Asian populations, while most samples included in the TCGA database came from American population. Furthermore, we divided sample into the SIX1-low group and SIX1-high group according to the quartile of SIX1 expression rather than the median expression. Recently, several studies have confirmed DACH1 functions as tumor suppressor in NSCLC and many other cancers [23-25]. In addition, a recent study in breast cancer showed that a combination of SIX1 with DACH1 could effectively distinct patients with high relapse risk within 3, 5, and 8 years [26]. Although SIX1 might serve as a promising biomarker for NSCLC prognosis to some extent, a more comprehensive predictive model is helpful to predict the prognosis of patients with NSCLC.
Accumulating evidence has suggested that SIX1 was involved in tumor initiation and progression by regulating multiple activities of cancer cells. SIX1 is known to be essential for cell growth and proliferation by enhancing the transcription of a range of target genes. For example, SIX1 was overexpressed in breast cancer and led to increased cell proliferation by upregulating cyclin A1 [9, 27]. H. Ford et al. proposed that SIX1 was involved in the control of the G2/M transition of the cell cycle in the breast cancer cell, thereby enhancing the tumorigenic potential [28]. SIX1 activated the cyclin D1 and c-Myc to control cell proliferation, survival, and motility in rhabdomyosarcoma [29]. SIX1 could promote proliferation via upregulating the connective tissue growth factor (CTGF) in glioblastoma cells [30]. Additionally, recent studies have shown that SIX1 directly increases the transcription of glycolysis genes and enhances the glycolysis of cancer cells to promote tumor growth [31]. Similarly, we found that overexpression of SIX1 could promote the proliferation and tumor growth of NSCLC, while knockdown of SIX1 exhibited the opposite effect. Therefore, SIX1 might be a potential target for controlling the growth of NSCLC.
Distant metastasis was a significant challenge in the battle against cancers. Previous studies have indicated that EMT, a reversible dynamic process in which epithelial cells lose the intercellular adhesion and transit into aggressive mesenchymal cells, could enhance the motility and invasiveness of cancer cells, thus promoting the metastasis and progression of tumors [32, 33]. EMT was usually characterized by the downregulation of epithelial cell markers (E-cadherin, Desmoplakin, and Occludin) and upregulation of mesenchymal cell markers (Vimentin, N-cadherin, and Fibronectin) [34]. SIX1-induced EMT has been observed in many human cancers. SIX1 induced human mammary carcinoma cells to undergo EMT and metastasis through TGF-β signaling [35]. SIX1 promotes EMT in colorectal cancer through ZEB1 activation [36]. Zhu et al. showed that SIX1 could transcriptionally activate the Vimentin, thereby promoting gastric cancer cell migration and invasion [37]. Here, we found that SIX1 promoted the migration and invasion of NSCLC cells. Upregulated SIX1 expression showed an increased expression of Vimentin and a decreased expression of E-cadherin, while downregulated SIX1 resulted in the opposite result. These data demonstrated that SIX1 might promote NSCLC progression by inducing EMT.
Notch signaling is an evolutionarily conserved pathway that is critical for the development and homeostasis in various tissues [38]. Previous studies have also indicated that the Notch signaling pathway was involved in carcinogenesis and tumor progression in numerous malignant cancers by regulating cell proliferation, differentiation, and apoptosis [39]. In addition, the Notch signaling played an essential role in the occurrence of lung cancer and cross-talked with multiple transcriptional factors to trigger EMT, thereby promoting the progression of NSCLC [40]. In this study, we found that the Notch signaling pathway was significantly enriched in the group with high SIX1 expression, and the level of HES1, the downstream target gene of Notch signaling, was positively correlated with SIX1 in NSCLC. Further studies showed that SIX1 could activate the Notch signaling and its downstream oncogenes. Importantly, SIX1-driven malignant biological behaviors could be blocked by the Notch pathway inhibitor DAPT in NSCLC cells. In summary, we demonstrated that SIX1 might promote NSCLC progression by activating the Notch signaling pathway. Given that transcription factors are notoriously difficult to target directly, our results suggested that inhibition of Notch signaling by a γ-secretase inhibitor (DAPT) might be a promising therapeutic strategy for SIX1-driven NSCLC patients in the future.
However, there were still some limitations in our research. First, it has been reported that the gain-of-function mutations and amplification of SIX1 enhanced its ability to promote tumor growth [41, 42]. Therefore, more studies are needed to investigate the mechanisms leading to the SIX1 re-expression in NSCLC. Second, we found that overexpression of SIX1 induced an increase in Notch1, Notch2, and Hes1 protein levels without significant change of Notch1 and Notch2 mRNA levels, despite an increase in the expression of Hes1 mRNA. These results indicated that SIX1 might be involved in the post-transcriptional regulation of the Notch receptor, and further studies are needed to explore the underlying mechanism. Third, additional oncogenic signaling pathways regulated by SIX1 need to be investigated in NSCLC, which is essential for a comprehensive understanding of SIX1's role in NSCLC and the development of appropriately targeted drugs in NSCLC.
In conclusion, we demonstrated that SIX1 was markedly upregulated in tumor tissues than the corresponding adjacent normal tissues in NSCLC. Elevated SIX1 expression was correlated with poor prognosis. Overexpression of SIX1 in NSCLC contributed to cell proliferation, migration, invasion, and the dynamic properties of EMT. Moreover, we found that SIX1 could activate the Notch signaling pathway in NSCLC, and inhibition of the Notch signaling pathway with γ-secretase inhibitor could reverse a series of SIX1-mediated malignant phenotypes. Overall, our findings suggested that SIX1 might serve as a prognostic biomarker for NSCLC and highlighted the potential value of the SIX1/Notch axis as a promising target for combating NSCLC progression.
Abbreviations
SIX1: Sine oculis homeobox homolog 1; NSCLC: non-small cell lung cancer; GSEA: Gene set enrichment analysis; EMT: epithelial-mesenchymal transition; LUAD: lung adenocarcinoma; LUSC: lung squamous cell carcinoma; LCLC: large cell lung cancer; FBS: fetal bovine serum; IHC: immunohistochemistry.
Supplementary Material
Supplementary figures.
Acknowledgements
We are grateful to Yu Jin, Piao Li, and Yongbiao Huang for their help in animal experiments and suggestions for this manuscript. This work was supported by National Natural Science Foundation of China (Numbers 82072597, 81974483, 82072589). We gratefully acknowledge all the participants in this study.
Ethical approval
All animal experiments in this study were overseen and approved by the Ethical committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology (IRB ID: TJH-201812003).
Author Contributions
Shanshan Huang, Qian Chu conceived and designed the study. Shanshan Huang and Wanling Lin performed the experiments in vitro. Shanshan Huang and Lei Wang performed the animal experiments. Shanshan Huang drafted the manuscript. All authors read and approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209-49
2. Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C. et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29:iv192-iv237
3. Samuel S, Naora H. Homeobox gene expression in cancer: insights from developmental regulation and deregulation. Eur J Cancer. 2005;41:2428-37
4. Liu Y, Han N, Zhou S, Zhou R, Yuan X, Xu H. et al. The DACH/EYA/SIX gene network and its role in tumor initiation and progression. Int J Cancer. 2016;138:1067-75
5. Kong D, Liu Y, Liu Q, Han N, Zhang C, Pestell RG. et al. The retinal determination gene network: from developmental regulator to cancer therapeutic target. Oncotarget. 2016;7:50755-65
6. Abate-Shen C. Deregulated homeobox gene expression in cancer: cause or consequence? Nat Rev Cancer. 2002;2:777-85
7. Zheng W, Huang L, Wei ZB, Silvius D, Tang B, Xu PX. The role of Six1 in mammalian auditory system development. Development. 2003;130:3989-4000
8. Chai L, Yang J, Di C, Cui W, Kawakami K, Lai R. et al. Transcriptional activation of the SALL1 by the human SIX1 homeodomain during kidney development. J Biol Chem. 2006;281:18918-26
9. Coletta RD, Christensen K, Reichenberger KJ, Lamb J, Micomonaco D, Huang L. et al. The Six1 homeoprotein stimulates tumorigenesis by reactivation of cyclin A1. Proc Natl Acad Sci U S A. 2004;101:6478-83
10. Xu M, Zhang X, Zhang S, Piao J, Yang Y, Wang X. et al. SPOCK1/SIX1axis promotes breast cancer progression by activating AKT/mTOR signaling. Aging (Albany NY). 2020;13:1032-50
11. Behbakht K, Qamar L, Aldridge CS, Coletta RD, Davidson SA, Thorburn A. et al. Six1 overexpression in ovarian carcinoma causes resistance to TRAIL-mediated apoptosis and is associated with poor survival. Cancer Res. 2007;67:3036-42
12. Li Z, Tian T, Hu X, Zhang X, Li L, Nan F. et al. Targeting Six1 by lentivirus-mediated RNA interference inhibits colorectal cancer cell growth and invasion. Int J Clin Exp Pathol. 2014;7:631-9
13. Kong D, Li A, Liu Y, Cui Q, Wang K, Zhang D. et al. SIX1 Activates STAT3 Signaling to Promote the Proliferation of Thyroid Carcinoma via EYA1. Front Oncol. 2019;9:1450
14. Zeng J, Shi R, Cai CX, Liu XR, Song YB, Wei M. et al. Increased expression of Six1 correlates with progression and prognosis of prostate cancer. Cancer Cell Int. 2015;15:63
15. Mimae T, Okada M, Hagiyama M, Miyata Y, Tsutani Y, Inoue T. et al. Upregulation of notch2 and six1 is associated with progression of early-stage lung adenocarcinoma and a more aggressive phenotype at advanced stages. Clin Cancer Res. 2012;18:945-55
16. Cho YA, Kim EK, Heo SJ, Cho BC, Kim HR, Chung JM. et al. Alteration status and prognostic value of MET in head and neck squamous cell carcinoma. J Cancer. 2016;7:2197-206
17. Cui D, Dai J, Keller JM, Mizokami A, Xia S, Keller ET. Notch Pathway Inhibition Using PF-03084014, a gamma-Secretase Inhibitor (GSI), Enhances the Antitumor Effect of Docetaxel in Prostate Cancer. Clin Cancer Res. 2015;21:4619-29
18. Wu W, Ren Z, Li P, Yu D, Chen J, Huang R. et al. Six1: a critical transcription factor in tumorigenesis. Int J Cancer. 2015;136:1245-53
19. Blevins MA, Towers CG, Patrick AN, Zhao R, Ford HL. The SIX1-EYA transcriptional complex as a therapeutic target in cancer. Expert Opin Ther Targets. 2015;19:213-25
20. Lu K, Reddy R, Berika M, Warburton D, El-Hashash AH. Abrogation of Eya1/Six1 disrupts the saccular phase of lung morphogenesis and causes remodeling. Dev Biol. 2013;382:110-23
21. Liu Q, Li A, Tian Y, Liu Y, Li T, Zhang C. et al. The expression profile and clinic significance of the SIX family in non-small cell lung cancer. J Hematol Oncol. 2016;9:119
22. Xia Y, Zhu Y, Ma T, Pan C, Wang J, He Z. et al. miR-204 functions as a tumor suppressor by regulating SIX1 in NSCLC. FEBS Lett. 2014;588:3703-12
23. Liu Q, Li A, Yu S, Qin S, Han N, Pestell RG. et al. DACH1 antagonizes CXCL8 to repress tumorigenesis of lung adenocarcinoma and improve prognosis. J Hematol Oncol. 2018;11:53
24. Wu K, Chen K, Wang C, Jiao X, Wang L, Zhou J. et al. Cell fate factor DACH1 represses YB-1-mediated oncogenic transcription and translation. Cancer Res. 2014;74:829-39
25. Chu Q, Han N, Yuan X, Nie X, Wu H, Chen Y. et al. DACH1 inhibits cyclin D1 expression, cellular proliferation and tumor growth of renal cancer cells. J Hematol Oncol. 2014;7:73
26. Dong B, Yi M, Luo S, Li A, Wu K. RDGN-based predictive model for the prognosis of breast cancer. Exp Hematol Oncol. 2020;9:13
27. Coletta RD, Christensen KL, Micalizzi DS, Jedlicka P, Varella-Garcia M, Ford HL. Six1 overexpression in mammary cells induces genomic instability and is sufficient for malignant transformation. Cancer Res. 2008;68:2204-13
28. Ford HL, Landesman-Bollag E, Dacwag CS, Stukenberg PT, Pardee AB, Seldin DC. Cell cycle-regulated phosphorylation of the human SIX1 homeodomain protein. J Biol Chem. 2000;275:22245-54
29. Yu Y, Davicioni E, Triche TJ, Merlino G. The homeoprotein six1 transcriptionally activates multiple protumorigenic genes but requires ezrin to promote metastasis. Cancer Res. 2006;66:1982-9
30. Tian T, Li A, Lu H, Luo R, Zhang M, Li Z. Six1 promotes glioblastoma cell proliferation and invasion by upregulation of connective tissue growth factor. Am J Cancer Res. 2015;5:1823-30
31. Li L, Liang Y, Kang L, Liu Y, Gao S, Chen S. et al. Transcriptional Regulation of the Warburg Effect in Cancer by SIX1. Cancer Cell. 2018;33:368-85 e7
32. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442-54
33. Lu W, Kang Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev Cell. 2019;49:361-74
34. Yang MH, Baek SH, Chinnathambi A, Alharbi SA, Ahn KS. Identification of protocatechuic acid as a novel blocker of epithelial-to-mesenchymal transition in lung tumor cells. Phytother Res. 2021;35:1953-66
35. Farabaugh SM, Micalizzi DS, Jedlicka P, Zhao R, Ford HL. Eya2 is required to mediate the pro-metastatic functions of Six1 via the induction of TGF-beta signaling, epithelial-mesenchymal transition, and cancer stem cell properties. Oncogene. 2012;31:552-62
36. Ono H, Imoto I, Kozaki K, Tsuda H, Matsui T, Kurasawa Y. et al. SIX1 promotes epithelial-mesenchymal transition in colorectal cancer through ZEB1 activation. Oncogene. 2012;31:4923-34
37. Zhu Z, Rong Z, Luo Z, Yu Z, Zhang J, Qiu Z. et al. Circular RNA circNHSL1 promotes gastric cancer progression through the miR-1306-3p/SIX1/vimentin axis. Mol Cancer. 2019;18:126
38. Siebel C, Lendahl U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol Rev. 2017;97:1235-94
39. Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S. et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer Lett. 2015;369:20-7
40. Yuan X, Wu H, Han N, Xu H, Chu Q, Yu S. et al. Notch signaling and EMT in non-small cell lung cancer: biological significance and therapeutic application. J Hematol Oncol. 2014;7:87
41. Walz AL, Ooms A, Gadd S, Gerhard DS, Smith MA, Guidry Auvil JM. et al. Recurrent DGCR8, DROSHA, and SIX homeodomain mutations in favorable histology Wilms tumors. Cancer Cell. 2015;27:286-97
42. Reichenberger KJ, Coletta RD, Schulte AP, Varella-Garcia M, Ford HL. Gene amplification is a mechanism of Six1 overexpression in breast cancer. Cancer Res. 2005;65:2668-75
Author contact
![]() Corresponding authors: Qian Chu (E-mail: qianchutjmu.edu.cn) and Yuan Chen (E-mail: chenyuan008com)
Corresponding authors: Qian Chu (E-mail: qianchutjmu.edu.cn) and Yuan Chen (E-mail: chenyuan008com)