Impact Factor ISSN: 1837-9664

 Global reach, higher impact
Global reach, higher impactJ Cancer 2022; 13(4):1272-1281. doi:10.7150/jca.64996 This issue Cite
Research Paper
Novel Insights into Pediatric Acute Lymphoblastic Leukemia Ophthalmic Relapses from a Nationwide Cohort Study
Solenne Le Louet1 ![]() , Véronique Icart2, Marion Strullu3, Arnaud Petit4, Claire Freycon5, Pascale Blouin6, Jill Serre6, Nicolas Rama7, Yves Reguerre8, Christophe Piguet9, Marlène Pasquet10, Audrey David11, Pauline Simon12, Marilyne Poiree13, Liana Carausu14, Fanny Rialland15, Wadih Abouchahla16, Paul Saultier17, Stéphane Ducassou18, Julie Valduga19, André Baruchel3, Yves Bertrand1, Carine Domenech1,7
, Véronique Icart2, Marion Strullu3, Arnaud Petit4, Claire Freycon5, Pascale Blouin6, Jill Serre6, Nicolas Rama7, Yves Reguerre8, Christophe Piguet9, Marlène Pasquet10, Audrey David11, Pauline Simon12, Marilyne Poiree13, Liana Carausu14, Fanny Rialland15, Wadih Abouchahla16, Paul Saultier17, Stéphane Ducassou18, Julie Valduga19, André Baruchel3, Yves Bertrand1, Carine Domenech1,7
1. Institute of Pediatric Hematology and Oncology, Hospices Civils de Lyon, University-Lyon1, Lyon, France
2. Department of Pediatric Ophtalmology, Hopital Nord Ouest de Villefranche sur Saône, Gleize, France
3. Department of Pediatric Hematology, Hôpital Robert-Debré, Paris, France
4. Department of Pediatric Hematology, Hôpital Armand-Trousseau, Paris, France
5. Department of Pediatric Hematology, CHU Grenoble Alpes, Grenoble, France
6. Department of Pediatric Hematology, CHRU de Tours, Tours, France
7. Apoptosis, Cancer and Development Laboratory - Center of Research of Cancerology of Lyon, INSERM U1052-CNRS UMR5286, Lyon, France
8. Department of Pediatric Hematology, Centre hospitalier Félix-Guyon, Saint Denis de La Réunion, France
9. Department of Pediatric Hematology, CHU de Limoges, Limoges, France
10. Department of Pediatric Hematology, CHU de Toulouse, Toulouse, France
11. Department of Pediatric Hematology, CHU de Saint Etienne, Saint Etienne, France
12. Department of Pediatric Hematology, CHU de Besançon, Besançon, France
13. Department of Pediatric Hematology, CHU de Nice, Nice, France
14. Department of Pediatric Hematology, CHU de Brest, Brest, France
15. Department of Pediatric Hematology, CHU de Nantes, Nantes, France
16. Department of Pediatric Hematology, CHU de Lille, Lille, France
17. Department of Pediatric Hematology, Hôpital de la Timone, Marseille, France
18. Department of Pediatric Hematology, CHU de Bordeaux, Bordeaux, France
19. Department of Pediatric Hematology, CHRU de Nancy, Nancy, France
Received 2021-7-15; Accepted 2021-11-30; Published 2022-1-24
Abstract

Ten to fifteen percent of children with acute lymphoblastic leukemia (ALL) relapse following treatment. Of these, less than 2% display ophthalmic relapses, which owing to their scarcity, are largely undocumented, leaving clinicians with few diagnostic and therapeutic recommendations, despite serious functional sequelae. We conducted a French multicenter retrospective study to collect all clinical, radiological, biological, and therapeutic data, and outcomes for children with ALL ophthalmic relapses. From 2000 to 2020, 20 ophthalmic relapses occurring after first-line therapy performed before January 1st, 2017 were included in our study: 14 B-ALL and 6 T-ALL. Fifteen patients (75%) had concomitant involvement of the central nervous system, and 11 (55%) a combined bone marrow relapse. Only 1 had an isolated ophthalmic relapse. Eight children (40%) died, 7 from a refractory disease and 1 from toxic death, and 4 patients relapsed. With a median follow-up of 63.1 months, 8 patients are currently alive in continuous complete remission with only 2 displaying severe ophthalmic sequelae. Although rare, ophthalmic relapse could have a significant impact on the functional prognosis of survivors. Their management must be multidisciplinary, with a central role given to ophthalmologists.
Keywords: Childhood, ophthalmic relapse, ALL
Introduction
Acute lymphoblastic leukemia (ALL) is the most common pediatric cancer (75-80% of childhood leukemia). Although overall survival at 5 years reaches 85-90%,[1] 10-15% of children relapse and display a poor prognosis that has not significantly improved over the last two decades.[2-6] The main sites of relapse are the bone marrow (BM) and the central nervous system (CNS). Combined or isolated extramedullary involvements may also be observed in the testis, ovary, eye and kidney.[7] Ophthalmic relapses, the incidence rate of which is estimated at 2%[8,9], have essentially been reported in case studies on children.[10-13] The clinical signs are in no way specific and may vary according to the part of the eye involved at relapse, as well as the mechanism of blastic invasion (meningeal tropism by the optic nerve or vascular involvement). Leukemic involvement of the retina is frequent[14] and could be responsible for a progressive or sudden decrease in visual acuity, and in some cases complete loss. Other sites of involvement may include the orbit, cornea, anterior chamber, vitreous, choroid, sclera and may result in painful red eyes and hypopyon.[15] This non-specific pattern of symptoms may be falsely attributed to a viral infection or a refractive disorder by ophthalmologists, resulting in a delay in diagnostic and therapeutic management, with potential serious functional sequelae.
It is well established that the prognosis of relapses depends on their location and the duration of the first complete remission (CR1).[16,17] Studies suggest that children with ocular relapse, either alone or in combination with a medullary involvement, should be treated with an intensive chemotherapy regimen that includes intrathecal chemotherapy with local ocular radiotherapy of at least 20 Gy.[10-12,18] However, data are limited to clinical case descriptions with a short follow-up and no information on the risk of secondary relapse and functional sequelae. Despite the rarity of these leukemic events, their prognosis is severe, and the specific treatment margins (including radiotherapy) are not well defined in relapse protocols. Moreover, little is known about their biological characteristics, hampering the adjustment of preventive treatment. Based on a national pediatric cohort, this study describes the early symptoms indicative of ALL ophthalmic relapse, the treatments received as first-line management and during relapse, and the outcomes of these children. This study thus sets the basis for improving the development of future therapeutic strategies.
Materials and Methods
Design and inclusion criteria
We performed a national multicenter retrospective study in the 28 pediatric hematology centers of the “Société Française de lutte contre les Cancers et les Leucémies de l'Enfant et de l'Adolescent” (SFCE). All patients (younger than 20 years) who suffered from an isolated or combined ALL ophthalmic relapse between 2000 and 2020 were included in our study, except patients who received first-line therapy after January the 1st, 2017. Patients were treated according to national and European ALL protocols[19,20] in frontline and at relapse. All demographic, clinical, biological and radiological data, therapies and outcomes were collected from the time of ALL diagnosis onwards.
Definitions
After achieving CR1, ophthalmic relapse was defined as the appearance of clinical symptoms of active ocular disease, associated with an abnormal ophthalmic examination (fundus retinography, optical coherence tomography (OCT) or B-ultrasonography). Clinical symptoms with or without optic nerve infiltration, included red or painful eye, decreased visual acuity, visual field amputation, exophthalmia and diplopia. The eye segment involvement was defined in the Supplemental file 1. Cytological confirmation, performed by an anterior/posterior segment puncture was not necessary for diagnosis of ophthalmic relapse if documented by CNS involvement in lumbar puncture. CNS relapse was defined by at least five leukocytes per microliter in cerebrospinal fluid and cytological evaluation, demonstrating the presence of lymphoblasts or clinical evidence of cranial nerve involvement, regardless of the number of cells (status CNS3). Combined relapse was defined by an ophthalmic relapse associated with BM involvement and /or CNS involvement. BM involvement was diagnosed according to the morphological detection of lymphoblasts, and also at the level of the minimal residual disease (MRD) through flow cytometric profiles or using Ig-TCR clone-specific probes defined at diagnosis.
Overall survival was defined as the time from initial ALL diagnosis to death or last follow-up.
Ethics statement
The patient's legal guardian signed an informed consent before treatment, which included authorization for the use of their data. Data collection was performed in accordance with patient confidentiality. Additionally, parents were informed of this retrospective study by an information note sent by mail or given in person, and did not object. This procedure complied with the Declaration of Helsinki and with the MR004 (“Commission Nationale de l'informatique et des libertés” n°2211365). The SFCE ethics committee approved the study (CL-2020-4).
Statistical analysis
Descriptive statistics were reported in terms of absolute frequencies and percentages. The distribution of data was described in terms of a median value. Sample odds ratios (OR) were calculated with the R package Epi. P-values < 0.05 were considered to be significant.
Survival analyses included the interval between initial ALL diagnosis and the occurrence of death or the date of last examination, with a five-year follow-up. The cut-off date for these analyses was June the 15th, 2021. Survival rates were estimated using the Kaplan-Meier method.
Results
Demographic and clinical data of ALL at initial diagnosis
Between 2000 and 2020, a total of 20 patients were included (10 boys and 10 girls) (Table 1). Among them, 14 had a B common precursor (BCP)-ALL and 6 had a T-ALL, including one patient with mixed phenotype acute leukemia at diagnosis that evolved into lymphoid at relapse. The overall median age was 6.7 years (range, 0.36 to 15.7 years) at initial diagnosis, with 5.1 years for patients with BCP-ALL and 14.3 years for T-ALL. Five (25%) children had good prognostic features at 1st diagnosis (age, leukocytosis, hyperdiploidy (> 50 chromosomes), ETV6-RUNX1, CNS1 status). Among the 20 patients, 6 displayed a CNS3 status, and 12 had hyperleukocytosis (>50G/l). In the case of (BCP)-ALL (n = 14), 2 patients had a t(1;19) translocation, 2 had an IKZF1 deletion, 1 a KMT2A rearrangement and 1 patient had a Philadelphia chromosome. A summary of the risk factors according to immunophenotype is presented in Table 1.
ALL initial characteristics of the 20 children with ophthalmic relapse
| Patient characteristics | BCP-ALL (n=14) (% of cases) | T-ALL (n=6) (% of cases) | |
|---|---|---|---|
| Male | 7 (50) | 3 | |
| Female | 7 (50) | 3 | |
| Age median [min, max] | 5.1 y [0.4,12.6] | 14.3 y [7.3;15.7] | |
| < 1 years | 1 (7) | 0 | |
| 1-10 years | 10 (71) | 2 (33) | |
| ≥ 10 years | 3 (21) | 4 (67) | |
| WBC at diagnosis | 7 (41) | ||
| < 50 G/L | 8 (57) | 0 | |
| > 50 G/L | 6 (43) | 6 (100) | |
| NCI risk group | |||
| Standard risk | 6 (43) | - | |
| High risk | 8 (57) | - | |
| Status of CNS | |||
| CNS 1 | 11 (79) | 2 (33) | |
| CNS 2 | 0 | 1 (17) | |
| CNS 3 | 3 (21) | 3 (50) | |
| Karyotype | |||
| Hyperdiploid | 3 (21) | 0 | |
| Complex | 0 | 1 (17) | |
| Molecular rearrangement | 1 | ||
| t(12;21)/ETV6-RUNX1 | 2 (14) | 0 | |
| t(1;19)/E2A-HLF | 2 (14) | 0 | |
| t(9.22)/BCR-ABL | 1 (7) | 0 | |
| KMT2A rearrangement | 1 (7) | 0 | |
| IKZF deletion | 2 /14 (14) evaluated | 0 | |
| Treatment group of 1rst line | |||
| Low risk | 4 (28) | 0 | |
| Average risk | 5 (36) | 0 | |
| High or very high risk | 5 (36) | 6 (100) | |
| Treatment before ophthalmic relapse | |||
| HSCT | 3 (21) | 1 (17) | |
| Radiotherapy | 3* (21) | 1# (17) | |
| Response (/evaluated cases) | |||
| PPR | 1/10 (10) | 4 | |
| MRD TP1 > 10-3 | 6/11 (55) | 1/5 | |
| MRD TP2 > 10-3 | 2/10 (20) | 0/5 |
Values are the number of patients (%), unless indicated otherwise; Abbreviations: WBC: white blood cell; CNS: central nervous system; HSCT: hematopoietic stem cell transplantation; MRD: minimal residual disease measured after induction therapy (TP1) or after consolidation (TP2); NCI: National Cancer Institute risk group classification. Standard risk: WBC count less than 50,000/μL and age 1 to younger than 10 years. High risk: WBC count 50,000/μL or greater and/or age 10 years or older. PPR: D8 poor prednisone response. *: 2 patients benefited from a 12 Grays total body irradiation (TBI) as a component of HSCT conditioning regimen, and 1 received a craniospinal irradiation due to his CNS3 status (EsPhALL protocol); #: this patient received a 12 Gy TBI before HSCT in 1rst complete remission.
Four patients (20%) were included in the standard risk group (BCP-ALL n = 4; T-ALL = 0), 5 in the average risk group and 11 in the high-risk group according to different French frontline protocols. Ten patients (50%) displayed criteria of poor response to treatment (poor prednisone response at day 8 n = 5; minimal residual disease (MRD) at the end of induction >10-3 n = 7, MRD at the end of consolidation >10-3 n = 2), including 2 with good prognostic factors at diagnosis. Two patients received immunotherapy with blinatumomab due to a chemo refractory disease in first-line therapy.
Four children underwent allogenic hematopoietic stem cell transplantation (HSCT) before ophthalmic relapse: 3 in CR1, due to their severe and refractory disease, and 1 after initial CNS leukemia relapse. Four children had cranial radiotherapy before ophthalmic relapse: 3 had undergone a 12 Gy total body irradiation (TBI) in the conditioning regimen of HSCT and 1 had received prophylactic cranial irradiation of 12 Gy according to his CNS status (EsPhALL protocol). The 4th patient who underwent HSCT before ophthalmic relapse did not receive TBI as he was an infant.
Clinical presentation and treatment of ophthalmic relapses (Table 2)
Ophthalmic relapse was the 1st relapse for 18 children. In the 2 other patients, ophthalmic relapse occurred after a BM relapse and a CNS relapse. The median age at ophthalmic relapse was 9.9 years (range 1.8 to 18.6 years). Relapse was diagnosed with a median delay of 24.7 months (range, 5.5 to 129.4 months) after the initial ALL diagnosis, occurring earlier in patients with T-ALL (9.9 months vs 33.1 months for patients with BCP-ALL; p value = 0.06). Eleven children (55%) relapsed early (< 30 months after initial diagnosis). Characteristics of ophthalmic relapses are described in Table 2. The eye segment involvement was anterior for 6 children. Ophthalmic symptoms appeared progressively (more than 15 days) for 8 children, whereas the remaining 12 (60%) had acute symptoms. Patients displayed variable clinical manifestations: the most frequent symptoms being reduced visual acuity (n = 12) and ocular pain (n = 5). Six patients had a bilateral ophthalmic involvement (30%). Nine of the 16 patients with available fundus images (56%) had papilledema related to blastic tumor invasion (Fig. 1), 1 (6%) had exudative retinal detachment (sign of choroidal damage). None of our patients displayed signs of leukemic retinopathy, specifically no extensive ocular hemorrhage which is a sign of bone marrow failure. At ophthalmic relapse, 15 children (75%) had a concomitant involvement of the CNS. Among our 20 patients, 18 had undergone brain imaging. The images of 8 of them showed signs of retro orbital invasion: contrast of the optic nerves n = 5; contrast of meningeal n = 1; intracerebral process n = 2 (Fig. 2). Among the remaining 10 patients, 5 had an abnormal ocular contrast on cerebral magnetic resonance imaging indicative of leukemia cell invasion, without sign of retro orbital invasion (lens n = 1, retinal n = 1, conjunctival n= 1, orbital wall n = 2) and 5 patients had a normal imaging. Eleven patients (55%) had a combined BM involvement (cytological n = 7; MRD > 10-2 n = 2; MRD < 10-2 n = 2). Only 1 patient had an isolated ophthalmic relapse. This patient relapsed with an isolated peri-macular lesion 7 months after HSCT, yet displayed a normal cerebral MRI, cerebrospinal fluid (CSF) analysis and negative BM MRD. Despite the lack of documentation on posterior chamber puncture, he was treated with HD-MTX combined with protontherapy before CNS relapse 10 months later. Only 6 patients had cytological confirmation of ocular involvement (4 anterior chamber paracentesis and 2 palpebral biopsy).
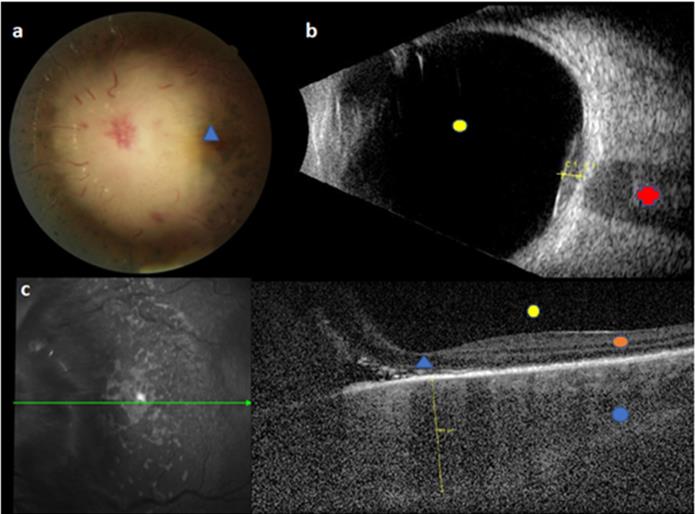
Initial images of BCP-ALL ophthalmic relapse for a child with normal brain imaging and no blast at CSF. This figure reveals a sub-macular choroidal hypertrophy and choroidal infiltration above the optic nerve. a. Fundus retinography: choroidal infiltration above optic nerve; all big papillary vessels and papilla are hidden by the choroidal infiltration, blue triangle = macula. b. B-ultrasonography: C1 = infiltration above optic nerve; red cross = optic nerve; yellow circle = vitreous cavity. c. left picture: fundus retinography; green arrow = horizontal line of OCT. Right picture: horizontal OCT: yellow line = sub-macular choroidal hypertrophy; blue circle = choroid, orange circle = retina; yellow circle = vitreous cavity, blue triangle = macula. Legend: ALL: acute lymphoblastic leukemia; CSF: cerebrospinal fluid; OCT: optical coherence tomography.
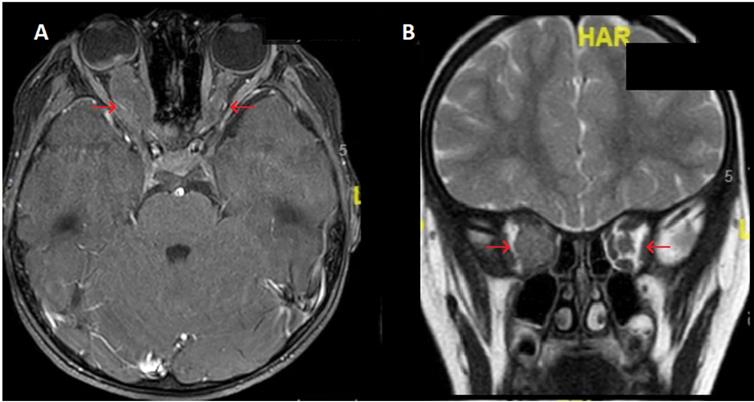
Brain magnetic resonance imaging (MRI) of BCP-ALL ophthalmic relapse for a child with massive infiltration of the right optic nerve. Images show a major circumferential thickening of the homogeneous right optic nerve with little contrast with lesser involvement of the left optic nerve. A. Axial THRIVE sequences after gadolinium injection B. Coronal T2. Legend: ALL: acute lymphoblastic leukemia
Characteristics of ALL ophthalmic relapses of 20 children
| Characteristics | Patients, n = 20 (%) |
|---|---|
| Median time from ALL diagnosis [min, max] | 24.7 m [5.5, 129.4] |
| Median Age [min, max] | 9.9 y [1.8, 18,6] |
| Localization of ophthalmological symptoms | |
| Unilateral | 14 (70) |
| Bilateral | 6 (30) |
| Time to onset of symptoms | |
| Gradual (>15 days) | 8 (40) |
| Acute (<15 days) | 12 (60) |
| The most frequent initial symptoms | |
| Decreased visual acuity | 12 (60) |
| Ocular pain | 5 (25) |
| Exophthalmos | 2 (10) |
| Sign of associated meningeal involvement | |
| Headache | 1 (5) |
| Photophobia | 3 (15) |
| The most frequent ophthalmological examination | |
| Papilloedema | 9/17 (53) |
| Hypopyon | 4 (20) |
| Combined medullary involvement | 11 (55) |
| Cytological | 7 (35) |
| MRD ≥ 10-2 | 2 (10) |
| MRD < 10-2 | 2 (10) |
| Combined CNS involvement | 15 (75) |
| CSF involvement | 12 (60) |
| Retroorbital damage at brain imaging | 8/18 (44) |
| Current status | |
| Complete remission | 8 (40) |
| New relapse or palliative state | 4 (20) |
| Deceased | 8 (40) |
Values are the number of patients (%), unless indicated otherwise. Abbreviations: MRD: minimal residual disease; CNS: central nervous system; CSF: cerebrospinal fluid
Complete treatment of the 20 patients with an ophthalmic relapse is described in Table 3. All patients received repeated triple intrathecal therapy (ITT) (hydrocortisone, methotrexate, aracytine) and combined induction therapy including dexamethasone and high dose methotrexate (HD-MTX). However, 2 patients did not receive HD-MTX due to a septic cause or macrophage activation syndrome and myelitis. Half of the cohort underwent allogenic HSCT after remission, including one patient with a second HSCT. Four patients (20%) received autologous chimeric antigen receptor T (CAR-T) cell therapy. A total of 13 children (65%) underwent radiotherapy: 8 underwent TBI of 12 Gy in the conditioning regimen of HSCT, among which 6 patients received an additional cranial boost. Five patients received cranial irradiation without TBI including 1 patient with an 18 Gy protontherapy. Seven children did not receive cranial irradiation including one infant < 2 years of age at ophthalmic relapse and 2 for refractory diseases. Finally, one patient underwent enucleation.
Characteristics, treatments and final outcome of patients with ophthalmic relapse (n = 20)
| 1st line | Previous Relapse | Ophthalmic relapse | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IP | Blina | RT | HSCT | Relapse delay (m) | HD- MTX (g/m²) | DEX | Triple IT | Blina | CAR-Tcells | TBI/ cranial boost | Isolated CRT (Gy) | HSCT | Status | ||
| (Gy) | |||||||||||||||
| P1 | B | - | - | - | - | 64.3 | 1 | Yes | Yes | - | - | 12/6 | - | Yes | CR2 |
| P2 | B | - | - | - | - | 55.2 | 1 | Yes | Yes | - | - | - | 18* | - | CR2 |
| P3 | B | - | - | - | - | 19.7 | 1 | Yes | Yes | - | - | - | - | - | Died |
| P4 | B | - | - | - | CHT + Bortezomib | 74.9 | 5 | Yes | Yes | Yes | - | - | - | Relapse | |
| P5 | B | - | - | - | - | 13.8 | 5 | Yes | Yes | - | - | 12/24 | - | Yes | CR2 |
| P6 | B | - | - | - | - | 34.0 | 1 | Yes | Yes | - | - | - | 24** | - | Died |
| P7 | B | Yes | TBI 12Gy | Yes | - | 16.1 | 8 then 5 | Yes | Yes | - | Yes | - | Proton 18Gy | - | Relapse |
| P8 | B | - | - | - | - | 29.6 | 5 | Yes | Yes | - | - | - | 18 | - | CR2 |
| P9 | B | - | - | - | CHT, TBI, HSCT | 58.3 | 5 | Yes | Yes | - | Yes | - | - | - | Relapse |
| P10 | B | - | - | - | - | 33.9 | 5 | Yes | Yes | Yes | - | 12/6 | - | Yes | Died |
| P11 | B | - | - | - | - | 53.4 | 1 | Yes | Yes | - | 12/12 | - | Yes | Died | |
| P12 | MPAL | - | - | Yes | - | 17.1 | 3 | Yes | Yes | - | Yes | - | - | - | Relapse |
| P13 | B | Yes | - | - | - | 5.5 | 8 | Yes | Yes | - | - | 12 | - | Yes | CR2 |
| P14 | B | - | 12Gy CS | - | - | 32.3 | 5 | Yes | Yes | - | 12 | - | Yes | Died | |
| P15 | T | - | - | - | - | 6.9 | 0# | Yes | Yes | - | - | - | - | Yes | Died |
| P16 | T | - | - | - | - | 11.3 | 0# | Yes | Yes | - | - | - | - | - | Died |
| P17 | T | - | TBI 12Gy | Yes | - | 129.4 | 5 | Yes | Yes | - | - | - | - | Yes | CR2 |
| P18 | T | - | - | - | - | 6.1 | 5 | Yes | Yes | - | - | 12/6 | Yes | CR2 | |
| P19 | T | - | - | - | - | 12.6 | 8 then 5 | Yes | Yes | - | - | - | 24 | - | Died |
| P20 | T | - | - | - | - | 8.4 | 1 | Yes | Yes | - | - | 12/6 | - | Yes | CR2 |
Abbreviations: Blina: Blinatumomab; CAR: chimeric antigen receptor; CHT: Chemotherapy; CR2: second complete remission; ; CRT: cranial irradiation; CS: Craniospinal; DEX: dexamethasone (10mg/m2/d); Gy: Gray; HSCT: hematopoietic stem cell transplantation; HD-MTX: High dose Methotrexate; IP: immunophenotype; IT: intrathecal; m: months; MPAL: mixed phenotype acute leukemia; RT: radiotherapy; TBI: total body irradiation including in HSCT conditioning regimen; - : not applicable
Relapse delay is delay between the diagnosis and the ophthalmic relapse;
# P15 and P16 did not received HD-MTX due to septic cause (P15) or macrophagic activation syndrome and myelitis (P16)
*P2 received craniospinal irradiation 18Gy
**P6 received cranial irradiation (24 Gy), right ocular irradiation (18 Gy), spinal axis (18Gy) and suffered a 2nd relapse treated with chemotherapy and enucleation, died before HSCT
Outcome
Eight children from our cohort (40%) died: one from toxic death related to HSCT and 7 from refractory diseases including 4 patients who reached medullary remission, albeit with the persistence of an ocular invaded sanctuary site. Among them, 4 patients did not undergo HSCT: 1 because of a late isolated extramedullary relapse and 3 because a new remission could not be achieved. Median time to death after ophthalmic relapse was 13.0 months (range 3.6 to 39.4 months).
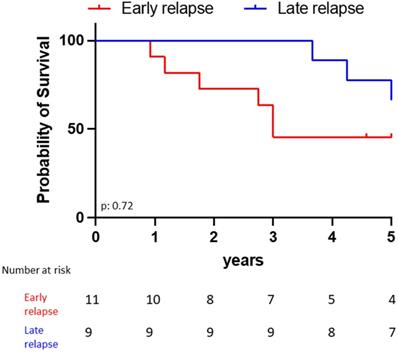
The estimated 5-year survival rate after first ALL diagnosis was 63.6% for the late ophthalmic relapses vs 55.6% for the early ones (p = 0.72, Fig. 3). All patients who received CAR-T cell infusion achieved initial ophthalmic remission. However, 4 patients relapsed when they lost their CAR-T cells between 4 to 6 months after the injection (extra medullary isolated relapse n = 2 (including one periorbital), medullary relapse n = 2).
Five years overall survival from initial ALL diagnosis according to the occurrence of early (n=11) versus late (n=9) ophthalmic relapse.
The 8 surviving children in 2nd complete remission (CR2) (40%) had a median follow-up of 63.1 months after ophthalmic relapse (range 12.3 months to 13.7 years). Two patients had severe ophthalmic sequelae (collapsed unilateral visual acuity with scotoma), and both presented a decrease in visual acuity at relapse. This was unilateral and brutal for one, bilateral and progressive for the other. Importantly, the loss of visual acuity was accompanied by a pale scarred papilla with retinal nerve fiber atrophy.
Characteristics of ALL at initial diagnosis were similar between the patients who died (n = 8), or who are currently experiencing a new relapse or in palliative care (n = 4), and the 8 patients in CR2 (Table 4). T-ALL ophthalmic relapses did not have a worse prognosis than BCP-ALL (p = 0.64). Combined medullary (cytological and molecular) relapse was not a significant worse prognostic factor, though it was associated with an odds ratio (OR) of 3.12 (p = 0.36). Furthermore, the use of radiotherapy including TBI (with or without cranial boost) was not associated (p = 0.16) with a better prognosis. Only 2 patients currently in continuous complete remission did not undergo a HSCT associated with TBI after their ophthalmic relapse, but benefited from a cranial irradiation (18 Gy).
Risk (OR) to decease or to have a refractory disease when an ophthalmic relapse occurs (n = 20)
| Characteristics | Patients with new relapse or death n = 12* (%) | Patients alive in CR n = 8** (%) | OR | P value |
|---|---|---|---|---|
| Characteristics of 1st ALL | ||||
| Age at ALL diagnosis, median | 5.7 y | 8.5 y | ||
| [min, max] | [0.36; 14.8] | [2.4;15.7] | ||
| Immunophenotype | ||||
| B | 9(75) | 5(62.5) | 1.74 | 0.64 |
| T | 3(25) | 3(37.5) | ||
| CNS Status | ||||
| CNS 1 | 7 (58) | 6 (75) | ||
| CNS 2 | 1 (8) | 0 | 2.14 | 0.64 |
| CNS 3 | 4 (33) | 2 (25) | ||
| HSCT | 3 (25) | 1 (12.5) | ||
| Radiotherapy | 3 (25) | 1 (12.5) | ||
| Characteristics of ophthalmic relapses | ||||
| Time to ophthalmic relapse after ALL, median [min, max] | 26.0m [6.9;74.9] | 21.7 m [5.5;129.4] | ||
| Unilateral | 9 (75) | 5 (62.5) | 1.74 | 0.64 |
| Acute appearance symptoms (<15 days) | 9 (75) | 3 (37.5) | 4.56 | 0.16 |
| Hypopyon | 4 (33) | 0 | - | 0.11 |
| CNS involvement | 9 (75) | 6 (75) | 1 | 1 |
| Medullary involvement | 8 (67) | 3 (37.5) | 3.12 | 0.36 |
| cytological | 6 (50) | 1 (12.5) | ||
| molecular | 2(17) | 2 (25) | ||
| Treatment after ophthalmic relapse | ||||
| HSCT | 4 (33) | 6 (75) | 0.18 | 0.17 |
| TBI + HSCT | 3 (25) | 5 (63) | 0.22 | 0.17 |
| TBI + Cranial Boost + HSCT | 2 (17) | 4 (50) | 0.22 | 0.16 |
*Among the 12 patients: 8 deceased, 4 patients recently relapsed after a RC2
**8 patients = patients among the 12 live patients, excluding the 4 patients in palliative treatment or who recently relapsed. Abbreviations: ALL: acute lymphoblastic leukemia; CAR: chimeric antigen receptor; CNS: central nervous system; HSCT: hematopoietic stem cell transplantation; m: months
Discussion
Due to therapeutic improvement in pediatric ALL, only 10 to 15% of children will relapse, and of these, only a few will suffer from an ophthalmic relapse. We report here results of an original study on 20 patients, based on the nationwide multicentric inclusion of patients treated in SFCE centers between 2000 and 2020. Most of our knowledge about ophthalmic relapses has so far mainly arisen from case studies in which T-ALL were rarely described, contrasting with our present study in which 30% of ophthalmic relapse occurred with the T phenotype, though this did not constitute a worse prognostic factor than BCP-ALL. This rate was also higher than that reported by Pui et al.[21]
Likewise, clinical manifestations of ophthalmic involvement in children with ALL relapse were described in case reports, but the prognosis of visual acuity was under-investigated.[22] Even if the pathophysiology is not yet well understood, different mechanisms (more or less related) could explain the variety of ocular symptoms, without specific lesions, observed. Indeed, relapses may have arisen from the CNS, but could also have occurred through the blood, following the rupture of the ocular blood barriers. In the case of “vascular” ophthalmic relapses, clinical manifestations are generally blastic hypopyon, “masquerade syndrome” or choroidal located tumor (diffused hypertrophy and retinal perivascular leukemic infiltrates) that may be associated with a red eye, a painful eye and/or a decrease in visual acuity. In contrast, involvement of the CNS is evidenced by leopard spot retinopathy and/or optic nerve infiltration.[23] Even though papilledema is well documented in the case of intracranial hypertension, it can also be secondary to a direct optic disc infiltration or due to an optic neuropathy induced by opportunistic infection. Lastly, we also observed ophthalmic relapses in our cohort occurring after immunotherapy, including blinatumomab or HSCT, indicative of a possible mechanism of immune resistance from leukemic cells. We distinguished two different patterns involved in leukemic ophthalmic relapses: leukemic retinopathy versus leukemic cell infiltration. Leukemic retinopathy is due to manifestations following BM insufficiency such as retinal hemorrhage or hyperviscosity (roth spots, cotton-wool spots, retinal venous dilatation). Furthermore, ophthalmic relapse is rare and leukemic infiltration is often difficult to diagnose in this context. Hence, the role of ophthalmologists is fundamental, to perform a quick and complete examination, including advanced ocular imaging such as optical coherence tomography (OCT) and fluorescein angiography of retinal vasculature, which may improve early recognition of ocular infiltration.
In view of the seriousness of ophthalmic relapses, it is essential to try to prevent them during first-line treatment. To date, first-line treatment is stratified according to clinical and biological data and on MRD results.[24-26] Based on strategies aimed at minimizing toxicity, the therapeutic program implemented by various groups, such as the EORTC-CLG and the St Jude, includes stopping CNS prophylactic radiation, and has led to some success.[6,20,27-31] This should be compensated by intensifying systemic chemotherapy with agents that exhibit good CNS penetration and intrathecal therapy. Indeed, prophylaxis of extramedullary leukemia relapses is an important part of first-line treatment. The efficacy of dexamethasone,[20,32] HD-MTX, IT, and Asparaginase for some groups, were demonstrated to prevent CNS relapse, including patients in the low/standard risk groups, and these treatments are part of ongoing European trials.[33-35] Relapse protocols are less well delineated than first-line treatment, and will probably be guided by whole genome sequencing of leukemia cells in future years, with the development of targeted therapies and immunotherapies. Regardless of the site of relapse, patients most often require systemic and intrathecal reinduction chemotherapy. Upon achieving CR2, patients should continue intensive chemotherapy. In addition, some children with poor prognostic factors such as early relapse or high MRD results at the disease evaluation points, may benefit from HSCT after achieving a new CR. The most effective practice is a myeloablative conditioning regimen with TBI, as demonstrated recently.[36,37]
In our study, the survival rate of patients is consistent with the overall post-relapse survival rate described in previous studies, including patients with combined medullary relapses.[38,39] The survivors displayed severe functional consequences, including extensive decreased visual acuity to blindness. Visual sequelae are mainly due to retinal, choroidal or optic nerve lesions that can evolve into serious optic atrophy, as well as to toxic therapy (radiation).
Our study proposes new insights into pediatric ALL ophthalmic relapses. Despite their rarity, ophthalmic involvement may be the only early manifestation of leukemic relapse. The diagnosis of any ophthalmic symptomatology concerning a child with a history of ALL should be considered as an ophthalmic relapse until proven otherwise. An urgent ophthalmic consultation (visual acuity, occulomotricity, pupillary defect, slit lamp) is necessary. All oculo-orbital structures should be fully checked by conducting a bilateral fundus examination after pupil dilation, an OCT capable of detecting early choroidal lesions, a B-ultrasonography and a visual field.[40] Lastly, an angiography (fluorescein or indocyanine green) should be performed to visualize the vascularisation. However, this latter examination is limited due to allergic risks, its invasive nature and the required participation of the child. The ophthalmic consultation must be followed immediately by brain imaging in order to check for retro-orbital damage, especially the contrast of the optic nerves. No corticosteroids (including topical) should be administered prior to diagnostic sampling, including CSF (with cytospin), BM evaluation and anterior chamber paracentesis if needed. The diagnosis of BM relapse currently relies on cytological analysis (blast cells ≥ 5%). However, we recommend that it should be considered as soon as the disease is detectable by flow cytometry and molecular biology (Ig TCR) if at least 1% of lymphoblasts are detected by cytological analysis, as recently proposed by the international consensus of the Pont-di-Legnio Consortium.[41] Once the diagnosis is confirmed, treatment is a priority. To our knowledge, formal trials have not been performed to evaluate the best treatment options for a rapid clearance of CSF blasts. However, we recommend to use systemic chemotherapies based on good drug penetration into the CNS: DXM (10 mg/m2/day) and HD-MTX associated with repeated triple intra-thecal (steroids, aracytine, MTX) every 4 days until reaching CSF remission. When a HSCT is necessary after obtaining CR, a 12 Gy TBI in conditioning regimen is advised. Due to the small number of patients included in this study, it was difficult to draw conclusions about the therapeutic interest of cranial boost and local treatment by ocular radiotherapy or enucleation. In our study, only one patient underwent enucleation, which did not prevent the progression of the disease. This therapy should thus not be considered as a routine, but may be utilized when the damage is localized in spite of aesthetic sequelae. Indeed, the sequelae would be added to adverse effects of HSCT and TBI, such as radiation retinopathy, cataract or glaucoma. Alternative options that could be envisaged to limit side effects would be protontherapy and contactotherapy. According to the literature, cranial boost does not seem to influence the risk of relapse in children[43], and concerns exist over potential severe adverse events.[44] In addition, our study included 3 patients who were treated with Blinatumomab. It would be interesting to complete our findings with further studies to evaluate the efficacy of immunotherapy in these cases of extramedullary relapse. Indeed, even if our 4 patients infused with CAR-T cells experienced a new relapse, Pillai et al reported the regression of a soft-tissue mass involving the eyelid and periorbital region without any therapeutic intervention, consistent with delayed but active tumor cell killing by CAR-T cells.[45]
In conclusion, the management of ophthalmic relapse should be multidisciplinary, with a central role given to ophthalmologists, supported by the use of ocular imaging such as OCT, in the detection of relapse. Future studies should define optimal management approaches for ophthalmic relapses.
Abbreviations
ALL: acute lymphoblastic leukemia; BCP-ALL: B-cell precursor acute lymphoblastic leukemia; BM: bone marrow; CAR: chimeric antigen receptor; CNS: central nervous system; CSF: cerebrospinal fluid; CR: complete remission; CR1: 1st complete remission; CR2: 2nd complete remission; D: day; DFS: disease-free survival; HD-MTX: high dose methotrexate; HSCT: hematopoietic stem cell transplantation; IT: intrathecal; ITT: triple intrathecal therapy; MRD: minimal residual disease; MRI: magnetic resonance imaging; MTX: methotrexate; OR: odds ratios; M: months; OCT: optical coherence tomography; TBI: total body irradiation; Y: years.
Supplementary Material
Supplementary file.
Acknowledgements
We gratefully thank Dr RAY-LUNVEN for her help, her expert advice, and encouragement for this project.
We also warmly thank Dr MANSHIP for her careful proofreading of our manuscript.
Funding source: The project was supported by “Enfants Cancers Santé” which financially supports the Société Française de lutte contre les Cancers de l'Enfant et de l'Adolescent.
Author Contributions
Dr Domenech and Dr Solenne Le Louët conceptualized and designed the study, conducted data analyses, drafted the initial manuscript, reviewed, and revised the manuscript.
Dr Rama carried out the data statistical analyses and reviewed and revised the manuscript.
Dr Icart, Dr Strullu, Pr Petit, Dr Freycon, Dr Blouin, Dr Serre, Dr Rama, Dr Reguerre, Dr Piguet, Dr Pasquet, Dr David, Dr Simon, Dr Poiree, Dr Carausu, Dr Rialland, Dr Abouchahla, Dr Saultier, Dr Ducassou, Dr Valduga provided study materials and patients, contributed to the analysis and interpretation of data and reviewed the manuscript.
Dr Icart, Pr Bertrand and Pr Baruchel critically reviewed and revised the manuscript for important intellectual content.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. 2020;105:2524-39
2. Hunger SP, Lu X, Devidas M. et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children's oncology group. J Clin Oncol. 2012;30:1663-9
3. Pui C-H, Yang JJ, Hunger SP. et al. Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J Clin Oncol. 2015;33:2938-48
4. Schrappe M, Bleckmann K, Zimmermann M. et al. Reduced-Intensity Delayed Intensification in Standard-Risk Pediatric Acute Lymphoblastic Leukemia Defined by Undetectable Minimal Residual Disease: Results of an International Randomized Trial (AIEOP-BFM ALL 2000). J Clin Oncol. 2018;36:244-53
5. Maloney KW, Devidas M, Wang C. et al. Outcome in Children With Standard-Risk B-Cell Acute Lymphoblastic Leukemia: Results of Children's Oncology Group Trial AALL0331. J Clin Oncol. 2020;38:602-12
6. Jeha S, Pei D, Choi J. et al. Improved CNS Control of Childhood Acute Lymphoblastic Leukemia Without Cranial Irradiation: St Jude Total Therapy Study 16. J Clin Oncol. 2019;37:3377-91
7. Gaynon PS. Childhood acute lymphoblastic leukaemia and relapse. Br J Haematol. 2005;131:579-87
8. Bunin N, Rivera G, Goode F, Hustu HO. Ocular relapse in the anterior chamber in childhood acute lymphoblastic leukemia. J Clin Oncol. 1987;5:299-303
9. Somervaille TCP, Hann IM, Harrison G. et al. Intraocular relapse of childhood acute lymphoblastic leukaemia. Br J Haematol. 2003;121:280-8
10. Pflugrath AE, Brar VS. Bilateral optic nerve and retinal infiltration as an initial site of relapse in a child with T-cell acute lymphoblastic leukemia. Am J Ophthalmol Case Rep. 2020;18:100695
11. Puvanachandra N, Goddard K, Lyons CJ. Dramatic visual recovery after prompt radiotherapy and chemotherapy for leukaemic infiltration of the optic nerve in a child. Eye (Lond). 2010;24:927-8
12. Shah P, Yohendran J, Lowe D, McCluskey P. Devastating bilateral optic nerve leukaemic infiltration. Clin Experiment Ophthalmol. 2012;40:e114-115
13. Chocron IM, Morrison DG, Friedman DL, Desai NA, Donahue SP. Ophthalmic manifestations of relapsing acute childhood leukemia. J AAPOS. 2015;19:284-6
14. Kincaid MC, Green WR. Ocular and orbital involvement in leukemia. Surv Ophthalmol. 1983;27:211-32
15. Lawson SE, Harrison G, Richards S. et al. The UK experience in treating relapsed childhood acute lymphoblastic leukaemia: a report on the medical research council UKALLR1 study. Br J Haematol. 2000;108:531-43
16. Einsiedel HG, von Stackelberg A, Hartmann R. et al. Long-term outcome in children with relapsed ALL by risk-stratified salvage therapy: results of trial acute lymphoblastic leukemia-relapse study of the Berlin-Frankfurt-Münster Group 87. J Clin Oncol. 2005;23:7942-50
17. Rizzari C, Valsecchi MG, Aricò M. et al. Outcome of very late relapse in children with acute lymphoblastic leukemia. Haematologica. 2004;89:427-34
18. Lo Curto M, D'Angelo P, Lumia F. et al. Leukemic ophthalmopathy: a report of 21 pediatric cases. Med Pediatr Oncol. 1994;23:8-13
19. Domenech C, Mercier M, Plouvier E. et al. First isolated extramedullary relapse in children with B-cell precursor acute lymphoblastic leukaemia: results of the Cooprall-97 study. Eur J Cancer. 2008;44:2461-9
20. Domenech C, Suciu S, De Moerloose B. et al. Dexamethasone (6 mg/m2/day) and prednisolone (60 mg/m2/day) were equally effective as induction therapy for childhood acute lymphoblastic leukemia in the EORTC CLG 58951 randomized trial. Haematologica. 2014;99:1220-7
21. Pui C-H, Mullighan CG, Evans WE, Relling MV. Pediatric acute lymphoblastic leukemia: where are we going and how do we get there? Blood. 2012;120:1165-74
22. Hon C, Law RW, Shek TW, Au WY. CNS manifestations of malignancies: case 1. Conjunctival relapse of acute lymphoblastic leukemia heralding pituitary and CNS disease. J Clin Oncol. 2005;23:4225-6
23. Robert MPA, Faure C, Reman O, Miocque S. Leopard spot retinopathy: an early clinical marker of leukaemia recurrence? Ann Hematol. 2008;87:927-9
24. Borowitz MJ, Devidas M, Hunger SP. et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: a Children's Oncology Group study. Blood. 2008;111:5477-85
25. Cavé H, van der Werff ten Bosch J, Suciu S. et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia. European Organization for Research and Treatment of Cancer-Childhood Leukemia Cooperative Group. N Engl J Med. 1998;339:591-8
26. Zhou J, Goldwasser MA, Li A. et al. Quantitative analysis of minimal residual disease predicts relapse in children with B-lineage acute lymphoblastic leukemia in DFCI ALL Consortium Protocol 95-01. Blood. 2007;110:1607-11
27. Toft N, Birgens H, Abrahamsson J. et al. Results of NOPHO ALL2008 treatment for patients aged 1-45 years with acute lymphoblastic leukemia. Leukemia. 2018;32:606-15
28. Yeh T-C, Liang D-C, Hou J-Y. et al. Treatment of childhood acute lymphoblastic leukemia with delayed first intrathecal therapy and omission of prophylactic cranial irradiation: Results of the TPOG-ALL-2002 study. Cancer. 2018;124:4538-47
29. Moghrabi A, Levy DE, Asselin B. et al. Results of the Dana-Farber Cancer Institute ALL Consortium Protocol 95-01 for children with acute lymphoblastic leukemia. Blood. 2007;109:896-904
30. Tubergen DG, Gilchrist GS, O'Brien RT. et al. Improved outcome with delayed intensification for children with acute lymphoblastic leukemia and intermediate presenting features: a Childrens Cancer Group phase III trial. J Clin Oncol. 1993;11:527-37
31. Sullivan MP, Chen T, Dyment PG, Hvizdala E, Steuber CP. Equivalence of intrathecal chemotherapy and radiotherapy as central nervous system prophylaxis in children with acute lymphatic leukemia: a pediatric oncology group study. Blood. 1982;60:948-58
32. Larsen EC, Devidas M, Chen S. et al. Dexamethasone and High-Dose Methotrexate Improve Outcome for Children and Young Adults With High-Risk B-Acute Lymphoblastic Leukemia: A Report From Children's Oncology Group Study AALL0232. JCO. 2016;34:2380-8
33. Woo MH, Hak LJ, Storm MC. et al. Cerebrospinal fluid asparagine concentrations after Escherichia coli asparaginase in children with acute lymphoblastic leukemia. J Clin Oncol. 1999;17:1568-73
34. Avramis VI, Sencer S, Periclou AP. et al. A randomized comparison of native Escherichia coli asparaginase and polyethylene glycol conjugated asparaginase for treatment of children with newly diagnosed standard-risk acute lymphoblastic leukemia: a Children's Cancer Group study. Blood. 2002;99:1986-94
35. Lobel JS, O'Brien RT, McIntosh S, Aspnes GT, Capizzi RL. Methotrexate and asparaginase combination chemotherapy in refractory acute lymphoblastic leukemia of childhood. Cancer. 1979;43:1089-94
36. Peters C, Dalle J-H, Locatelli F. et al. Total Body Irradiation or Chemotherapy Conditioning in Childhood ALL: A Multinational, Randomized, Noninferiority Phase III Study. J Clin Oncol. 2021;39:295-307
37. Gupta T, Kannan S, Dantkale V, Laskar S. Cyclophosphamide plus total body irradiation compared with busulfan plus cyclophosphamide as a conditioning regimen prior to hematopoietic stem cell transplantation in patients with leukemia: a systematic review and meta-analysis. Hematol Oncol Stem Cell Ther. 2011;4:17-29
38. Chessells JM, Veys P, Kempski H. et al. Long-term follow-up of relapsed childhood acute lymphoblastic leukaemia. Br J Haematol. 2003;123:396-405
39. Kolb EA, Steinherz PG. A new multidrug reinduction protocol with topotecan, vinorelbine, thiotepa, dexamethasone, and gemcitabine for relapsed or refractory acute leukemia. Leukemia. 2003;17:1967-72
40. Yalcinbayir O, Baytan B, Gelisken O. et al. Spectral domain optical coherence tomography findings of patients under treatment for pediatric acute lymphoblastic leukemia. J AAPOS. 2017;21:131-135.e1
41. Buchmann S, Schrappe M, Baruchel A. et al. Remission, treatment failure, and relapse in pediatric ALL: An international consensus of the Ponte-di-Legno Consortium. Blood. 2021 blood.2021012328
42. Jones B, Freeman AI, Shuster JJ. et al. Lower incidence of meningeal leukemia when prednisone is replaced by dexamethasone in the treatment of acute lymphocytic leukemia. Med Pediatr Oncol. 1991;19:269-75
43. Halsey C, Buck G, Richards S, Vargha-Khadem F, Hill F, Gibson B. The impact of therapy for childhood acute lymphoblastic leukaemia on intelligence quotients; results of the risk-stratified randomized central nervous system treatment trial MRC UKALL XI. J Hematol Oncol. 2011;4:42
44. Vora A, Andreano A, Pui C-H. et al. Influence of Cranial Radiotherapy on Outcome in Children With Acute Lymphoblastic Leukemia Treated With Contemporary Therapy. J Clin Oncol. 2016;34:919-26
45. Pillai V, Maude SL. CART attack. Blood. 2017;130:229
Author contact
![]() Corresponding author: Solenne LE LOUET Address: Institut d'Hématologie et Oncologie Pédiatrique, 1 place Joseph Renaut, 69008 Lyon, France E-mail address: solenne.lelouetunicancer.fr tel: +33 4 69 16 65 67
Corresponding author: Solenne LE LOUET Address: Institut d'Hématologie et Oncologie Pédiatrique, 1 place Joseph Renaut, 69008 Lyon, France E-mail address: solenne.lelouetunicancer.fr tel: +33 4 69 16 65 67